
Nanostructured Lipid Carriers Loaded with Dexamethasone Prevent Inflammatory Responses in Primary Non-Parenchymal Liver Cells


 , , , ,
, , , ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of DXM Loaded Nanostructured Lipid Carriers (NLC)
2.3. Analytical Detection of DXM
2.4. Determination of Encapsulation Efficiency (EE) and Drug Loading (DL)
2.5. Particle Size, Zeta Potential (Z pot), and Polydispersity Index (PDI)
2.6. Transmission Electron Microscopy (TEM)
2.7. In Vitro Release of DXM
2.8. Hemotoxicity Studies
2.9. In Vitro Interaction with Plasma Proteins
2.10. Isolation of Liver Non-Parenchymal Cells (NPCs)
2.11. Flow Cytometry
2.12. Cytometric Bead Array
2.13. Detection of Nuclear Factor ‘Kappa-Light-Chain-Enhancer’ of Activated B-Cells (NF-κB) in a Raw-Blue™ Reporter Cell Line
2.14. Statistical Analysis
3. Results
3.1. Preparation and Characterization of DXM-Loaded NLCs
3.2. DXM Release from NLCs
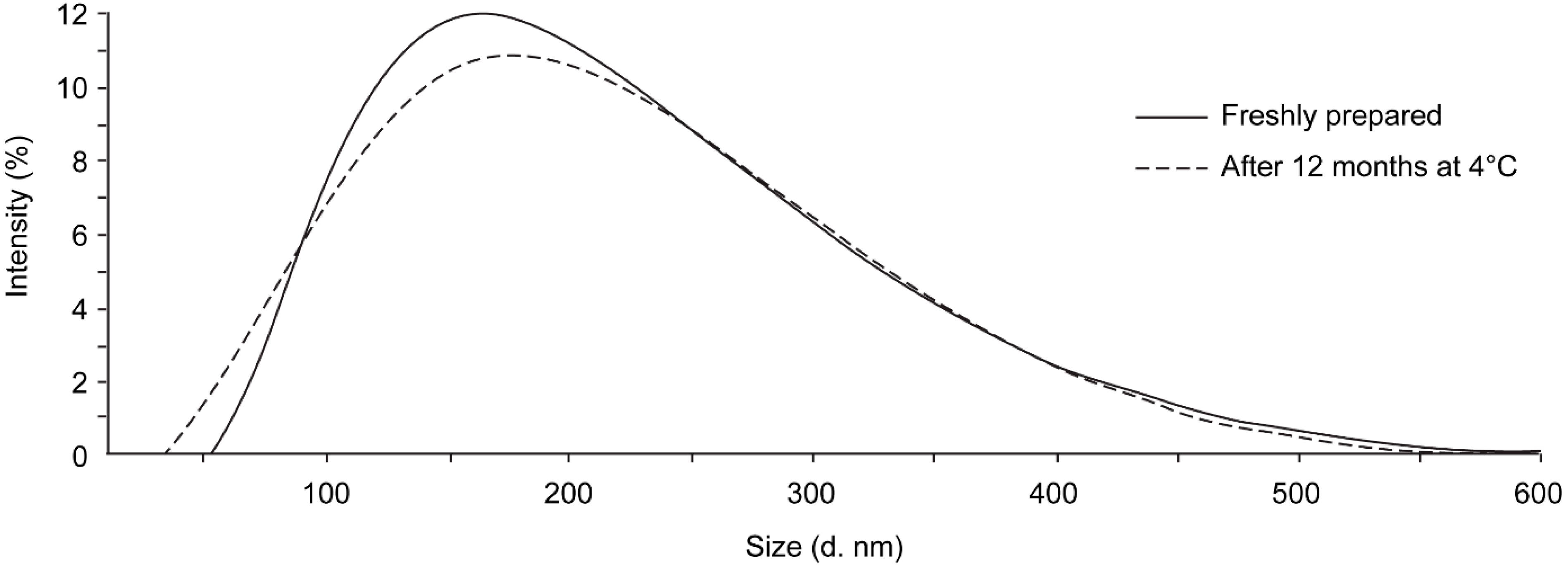
3.3. Stability after Storage
3.4. Hemotoxicity Studies
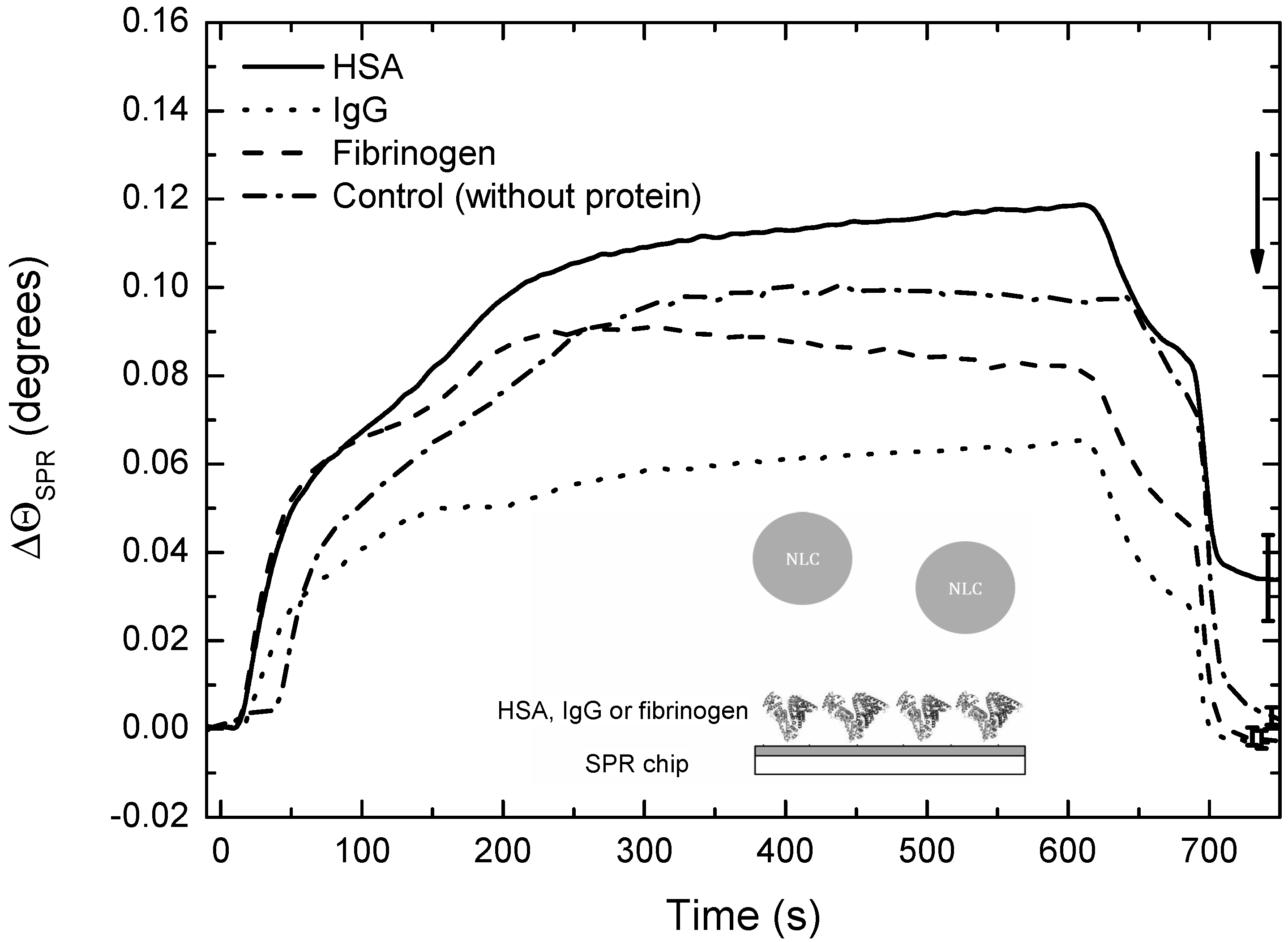
3.5. Interactions of DXM-Loaded NLCs with Serum Proteins
3.6. NLCs Binding to Liver NPC Populations
3.7. DXM Functionality Is Not Affected by Encapsulation into NLCs
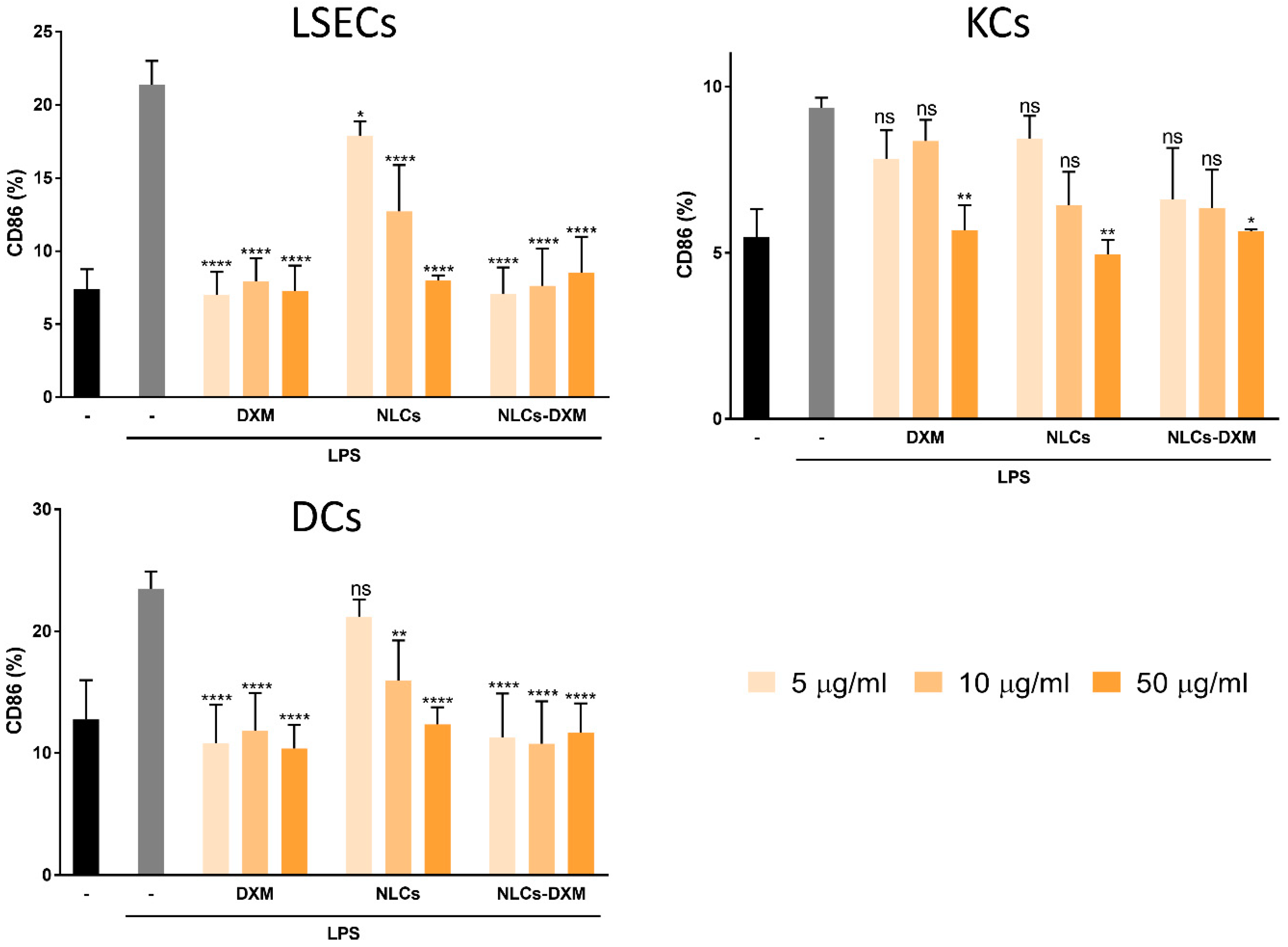
3.8. NLCs Induce Anti-Inflammatory Effects Evidenced by the Reduced Activation State of NPCs Subpopulations
3.9. Delayed Release of DXM from NLCs Affects LPS-Induced NF-kB Activity in a Time-Dependent Manner
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mieli-Vergani, G.; Vergani, D.; Czaja, A.J.; Manns, M.P.; Krawitt, E.L.; Vierling, J.M.; Lohse, A.W.; Montano-Loza, A.J. Autoimmune Hepatitis. Nat. Rev. Dis. Primers 2018, 4, 18017. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.C.; Cheng, P.N.; Kao, J.H. Systematic Review: Chronic Viral Hepatitis and Metabolic Derangement. Aliment. Pharmacol. Ther. 2020, 51, 216–230. [Google Scholar] [CrossRef] [PubMed]
- Czaja, A.J.; Carpenter, H.A. Progressive Fibrosis during Corticosteroid Therapy of Autoimmune Hepatitis. Hepatology 2004, 39, 1631–1638. [Google Scholar] [CrossRef] [PubMed]
- Czaja, A.J. Hepatic Inflammation and Progressive Liver Fibrosis in Chronic Liver Disease. World J. Gastroenterol. 2014, 20, 2515–2532. [Google Scholar] [CrossRef]
- FakhriRavari, A.; Jin, S.; Kachouei, F.H.; Le, D.; Lopez, M. Systemic Corticosteroids for Management of COVID-19: Saving Lives or Causing Harm? Int. J. Immunopathol. Pharmacol. 2021, 35, 20587384211063976. [Google Scholar] [CrossRef]
- Stark, A.; Carlo, W.; Tyson, J.; Papile, A.; Wright, L.; Shankaran, S.; Donovan, E.; Oh, W.; Bauer, C.; Saha, S.; et al. Adverse Effects of Early Dexamethasone Treatment in Extremely-Low-Birth-Weight Infants. N. Engl. J. Med. 2001, 344, 95–101. [Google Scholar] [CrossRef]
- Doyle, L.W.; Ehrenkranz, R.A.; Halliday, H.L. Dexamethasone Treatment in the First Week of Life for Preventing Bronchopulmonary Dysplasia in Preterm Infants: A Systematic Review. Neonatology 2010, 98, 217–224. [Google Scholar] [CrossRef]
- Zhang, Y.N.; Poon, W.; Tavares, A.J.; McGilvray, I.D.; Chan, W.C.W. Nanoparticle-Liver Interactions: Cellular Uptake and Hepatobiliary Elimination. J. Control. Release 2016, 240, 332–348. [Google Scholar] [CrossRef]
- Cacicedo, M.L.; Medina-Montano, C.; Kaps, L.; Kappel, C.; Gehring, S.; Bros, M. Role of Liver-Mediated Tolerance in Nanoparticle-Based Tumor Therapy. Cells 2020, 9, 1985. [Google Scholar] [CrossRef] [PubMed]
- Koyama, Y.; Brenner, D.A.; Koyama, Y.; Brenner, D.A. Liver Inflammation and Fibrosis Find the Latest Version: Liver Inflammation and Fibrosis. J. Clin. Investig. 2017, 127, 55–64. [Google Scholar] [CrossRef]
- Poisson, J.; Lemoinne, S.; Boulanger, C.; Durand, F.; Moreau, R.; Valla, D.; Rautou, P.E. Liver Sinusoidal Endothelial Cells: Physiology and Role in Liver Diseases. J. Hepatol. 2017, 66, 212–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Jiang, S.; Haller, A.; Wirsching, S.; Fichter, M.; Simon, J.; Wagner, M.; Mailänder, V.; Gehring, S.; Crespy, D.; et al. Encapsulation of Polyprodrugs Enables an Efficient and Controlled Release of Dexamethasone. Nanoscale Horiz. 2021, 6, 791–800. [Google Scholar] [CrossRef]
- Bartneck, M.; Scheyda, K.M.; Warzecha, K.T.; Rizzo, L.Y.; Hittatiya, K.; Luedde, T.; Storm, G.; Trautwein, C.; Lammers, T.; Tacke, F. Fluorescent Cell-Traceable Dexamethasone-Loaded Liposomes for the Treatment of Inflammatory Liver Diseases. Biomaterials 2015, 37, 367–382. [Google Scholar] [CrossRef] [PubMed]
- Bartneck, M.; Peters, F.M.; Warzecha, K.T.; Bienert, M.; van Bloois, L.; Trautwein, C.; Lammers, T.; Tacke, F. Liposomal Encapsulation of Dexamethasone Modulates Cytotoxicity, Inflammatory Cytokine Response, and Migratory Properties of Primary Human Macrophages. Nanomed. Nanotechnol. Biol. Med. 2014, 10, 1209–1220. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Prozeller, D.; Pereira, J.; Simon, J.; Han, S.; Wirsching, S.; Fichter, M.; Mottola, M.; Lieberwirth, I.; Morsbach, S.; et al. Controlling Protein Interactions in Blood for Effective Liver Immunosuppressive Therapy by Silica Nanocapsules. Nanoscale 2020, 12, 2626–2637. [Google Scholar] [CrossRef] [Green Version]
- Fichter, M.; Baier, G.; Dedters, M.; Pretsch, L.; Pietrzak-Nguyen, A.; Landfester, K.; Gehring, S. Nanocapsules Generated out of a Polymeric Dexamethasone Shell Suppress the Inflammatory Response of Liver Macrophages. Nanomed. Nanotechnol. Biol. Med. 2013, 9, 1223–1234. [Google Scholar] [CrossRef]
- Lammers, T.; Sofias, A.M.; van der Meel, R.; Schiffelers, R.; Storm, G.; Tacke, F.; Koschmieder, S.; Brümmendorf, T.H.; Kiessling, F.; Metselaar, J.M. Dexamethasone Nanomedicines for COVID-19. Nat. Nanotechnol. 2020, 15, 622–624. [Google Scholar] [CrossRef]
- Simón-Vázquez, R.; Tsapis, N.; Lorscheider, M.; Rodríguez, A.; Calleja, P.; Mousnier, L.; de Miguel Villegas, E.; González-Fernández, Á.; Fattal, E. Improving Dexamethasone Drug Loading and Efficacy in Treating Arthritis through a Lipophilic Prodrug Entrapped into PLGA-PEG Nanoparticles. Drug Deliv. Transl. Res. 2022, 12, 1270–1284. [Google Scholar] [CrossRef]
- Wang, H.; Zhou, Y.; Sun, Q.; Zhou, C.; Hu, S.; Lenahan, C.; Xu, W.; Deng, Y.; Li, G.; Tao, S. Update on Nanoparticle-Based Drug Delivery System for Anti-Inflammatory Treatment. Front. Bioeng. Biotechnol. 2021, 9, 630352. [Google Scholar] [CrossRef]
- Mirchandani, Y.; Patravale, V.B.; Brijesh, S. Solid Lipid Nanoparticles for Hydrophilic Drugs. J. Control. Release 2021, 335, 457–464. [Google Scholar] [CrossRef]
- Haider, M.; Abdin, S.M.; Kamal, L.; Orive, G. Nanostructured Lipid Carriers for Delivery of Chemotherapeutics: A Review. Pharmaceutics 2020, 12, 288. [Google Scholar] [CrossRef] [Green Version]
- Pardeike, J.; Hommoss, A.; Müller, R.H. Lipid Nanoparticles (SLN, NLC) in Cosmetic and Pharmaceutical Dermal Products. Int. J. Pharm. 2009, 366, 170–184. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Dhar, A.; Patel, C.; Khimani, M.; Neogi, S.; Sharma, P.; Siva Kumar, N.; Vekariya, R.L. A Brief Review on Solid Lipid Nanoparticles: Part and Parcel of Contemporary Drug Delivery Systems. RSC Adv. 2020, 10, 26777–26791. [Google Scholar] [CrossRef] [PubMed]
- Mishra, V.; Bansal, K.K.; Verma, A.; Yadav, N.; Thakur, S.; Sudhakar, K.; Rosenholm, J.M. Solid Lipid Nanoparticles: Emerging Colloidal Nano Drug Delivery Systems. Pharmaceutics 2018, 10, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodenak-Kladniew, B.; Noacco, N.; Pérez de Berti, I.; Stewart, S.J.; Cabrera, A.F.; Alvarez, V.A.; García de Bravo, M.; Durán, N.; Castro, G.R.; Islan, G.A. Design of Magnetic Hybrid Nanostructured Lipid Carriers Containing 1,8-Cineole as Delivery Systems for Anticancer Drugs: Physicochemical and Cytotoxic Studies. Colloids Surf. B Biointerfaces 2021, 202, 111710. [Google Scholar] [CrossRef]
- Scioli Montoto, S.; Muraca, G.; di Ianni, M.; Couyoupetrou, M.; Pesce, G.; Islan, G.A.; Chain, C.Y.; Vela, M.E.; Ruiz, M.E.; Talevi, A.; et al. Preparation, Physicochemical and Biopharmaceutical Characterization of Oxcarbazepine-Loaded Nanostructured Lipid Carriers as Potential Antiepileptic Devices. J. Drug Deliv. Sci. Technol. 2021, 63, 102470. [Google Scholar] [CrossRef]
- Muraca, G.S.; Soler-Arango, J.; Castro, G.R.; Islan, G.A.; Brelles-Mariño, G. Improving Ciprofloxacin Antimicrobial Activity through Lipid Nanoencapsulation or Non-Thermal Plasma on Pseudomonas Aeruginosa Biofilms. J. Drug Deliv. Sci. Technol. 2021, 64, 102644. [Google Scholar] [CrossRef]
- Rodenak-Kladniew, B.; Scioli Montoto, S.; Sbaraglini, M.L.; di Ianni, M.; Ruiz, M.E.; Talevi, A.; Alvarez, V.A.; Durán, N.; Castro, G.R.; Islan, G.A. Hybrid Ofloxacin/Eugenol Co-Loaded Solid Lipid Nanoparticles with Enhanced and Targetable Antimicrobial Properties. Int. J. Pharm. 2019, 569, 118575. [Google Scholar] [CrossRef]
- Islan, G.A.; Gonçalves, L.M.D.; Marto, J.; Duarte, A.; Alvarez, V.A.; Castro, G.R.; Almeida, A.J. Effect of α-Tocopherol on the Physicochemical, Antioxidant and Antibacterial Properties of Levofloxacin Loaded Hybrid Lipid Nanocarriers. New J. Chem. 2021, 45, 1029–1042. [Google Scholar] [CrossRef]
- Rodenak-Kladniew, B.; Islan, G.A.; de Bravo, M.G.; Durán, N.; Castro, G.R. Design, Characterization and in Vitro Evaluation of Linalool-Loaded Solid Lipid Nanoparticles as Potent Tool in Cancer Therapy. Colloids Surf. B Biointerfaces 2017, 154, 123–132. [Google Scholar] [CrossRef]
- Islan, G.A.; Tornello, P.C.; Abraham, G.A.; Duran, N.; Castro, G.R. Smart Lipid Nanoparticles Containing Levofloxacin and DNase for Lung Delivery. Design and Characterization. Colloids Surf. B Biointerfaces 2016, 143, 168–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cacicedo, M.L.; Ruiz, M.C.; Scioli-Montoto, S.; Ruiz, M.E.; Fernández, M.A.; Torres-Sanchez, R.M.; Baran, E.J.; Castro, G.R.; León, I.E. Lipid Nanoparticles-Metvan: Revealing a Novel Way to Deliver a Vanadium Compound to Bone Cancer Cells. New J. Chem. 2019, 43, 17726–17734. [Google Scholar] [CrossRef]
- Scioli Montoto, S.; Sbaraglini, M.L.; Talevi, A.; Couyoupetrou, M.; di Ianni, M.; Pesce, G.O.; Alvarez, V.A.; Bruno-Blanch, L.E.; Castro, G.R.; Ruiz, M.E.; et al. Carbamazepine-Loaded Solid Lipid Nanoparticles and Nanostructured Lipid Carriers: Physicochemical Characterization and in Vitro/in Vivo Evaluation. Colloids Surf. B Biointerfaces 2018, 167, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Salgarella, A.R.; Zahoranová, A.; Šrámková, P.; Majerčíková, M.; Pavlova, E.; Luxenhofer, R.; Kronek, J.; Lacík, I.; Ricotti, L. Investigation of Drug Release Modulation from Poly(2-Oxazoline) Micelles through Ultrasound. Sci. Rep. 2018, 8, 9893. [Google Scholar] [CrossRef] [PubMed]
- Thote, A.J.; Chappell, J.T.; Gupta, R.B.; Kumar, R. Reduction in the Initial-Burst Release by Surface Crosslinking of PLGA Microparticles Containing Hydrophilic or Hydrophobic Drugs. Drug Dev. Ind. Pharm. 2005, 31, 43–57. [Google Scholar] [CrossRef]
- Abbina, S.; Takeuchi, L.E.; Anilkumar, P.; Yu, K.; Rogalski, J.C.; Shenoi, R.A.; Constantinescu, I.; Kizhakkedathu, J.N. Blood Circulation of Soft Nanomaterials Is Governed by Dynamic Remodeling of Protein Opsonins at Nano-Biointerface. Nat. Commun. 2020, 11, 3048. [Google Scholar] [CrossRef]
- Rampado, R.; Crotti, S.; Caliceti, P.; Pucciarelli, S.; Agostini, M. Recent Advances in Understanding the Protein Corona of Nanoparticles and in the Formulation of “Stealthy” Nanomaterials. Front. Bioeng. Biotechnol. 2020, 8, 166. [Google Scholar] [CrossRef] [PubMed]
- Hermanson, G. Bioconjugate Techniques, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Medina-Montano, C.; Cacicedo, M.L.; Svensson, M.; Limeres, M.J.; Zeyn, Y.; Chaves-Giraldo, J.E.; Röhrig, N.; Grabbe, S.; Gehring, S.; Bros, M. Enrichment Methods for Murine Liver Non-Parenchymal Cells Differentially Affect Their Immunophenotype and Responsiveness towards Stimulation. Int. J. Mol. Sci. 2022, 23, 6543. [Google Scholar] [CrossRef]
- Mansour, H.M.; Sohn, M.J.; Al-Ghananeem, A.; DeLuca, P.P. Materials for Pharmaceutical Dosage Forms: Molecular Pharmaceutics and Controlled Release Drug Delivery Aspects. Int. J. Mol. Sci. 2010, 11, 3298–3322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murdock, R.C.; Braydich-Stolle, L.; Schrand, A.M.; Schlager, J.J.; Hussain, S.M. Characterization of Nanomaterial Dispersion in Solution Prior to in Vitro Exposure Using Dynamic Light Scattering Technique. Toxicol. Sci. 2008, 101, 239–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cacicedo, M.L.; Islan, G.A.; León, I.E.; Álvarez, V.A.; Chourpa, I.; Allard-Vannier, E.; García-Aranda, N.; Díaz-Riascos, Z.V.; Fernández, Y.; Schwartz, S.; et al. Bacterial Cellulose Hydrogel Loaded with Lipid Nanoparticles for Localized Cancer Treatment. Colloids Surf. B Biointerfaces 2018, 170, 596–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruschi, M.L. Mathematical Models of Drug Release. In Strategies to Modify the Drug Release from Pharmaceutical Systems; Elsevier: Amsterdam, The Netherlands, 2015; pp. 63–86. [Google Scholar]
- Permanadewi, I.; Kumoro, A.C.; Wardhani, D.H.; Aryanti, N. Modelling of Controlled Drug Release in Gastrointestinal Tract Simulation. In Journal of Physics: Conference Series; Institute of Physics Publishing: Bristol, UK, 2019; Volume 1295. [Google Scholar]
- Kamaly, N.; Yameen, B.; Wu, J.; Farokhzad, O.C. Degradable Controlled-Release Polymers and Polymeric Nanoparticles: Mechanisms of Controlling Drug Release. Chem. Rev. 2016, 116, 2602–2663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordillo-Galeano, A.; Mora-Huertas, C.E. Solid Lipid Nanoparticles and Nanostructured Lipid Carriers: A Review Emphasizing on Particle Structure and Drug Release. Eur. J. Pharm. Biopharm. 2018, 133, 285–308. [Google Scholar] [CrossRef]
- Xiang, Q.Y.; Wang, M.T.; Chen, F.; Gong, T.; Jian, Y.L.; Zhang, Z.R.; Huang, Y. Lung-Targeting Delivery of Dexamethasone Acetate Loaded Solid Lipid Nanoparticles. Arch. Pharm. Res. 2007, 30, 519–525. [Google Scholar] [CrossRef]
- Wang, M.T.; Jin, Y.; Yang, Y.X.; Zhao, C.Y.; Yang, H.Y.; Xu, X.F.; Qin, X.; Wang, Z.D.; Zhang, Z.R.; Jian, Y.L.; et al. In Vivo Biodistribution, Anti-Inflammatory, and Hepatoprotective Effects of Liver Targeting Dexamethasone Acetate Loaded Nanostructured Lipid Carrier System. Int. J. Nanomed. 2010, 5, 487–497. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Zhao, C.; Yang, H.; Jian, Y.; Zhang, Z.; Huang, Y. Anti-Inflammatory Activity of Injectable Dexamethasone Acetate-Loaded Nanostructured Lipid Carriers. Drug Deliv. 2011, 18, 485–492. [Google Scholar] [CrossRef]
- Doktorovova, S.; Souto, E.B. Nanostructured Lipid Carrier-Based Hydrogel Formulations for Drug Delivery: A Comprehensive Review. Expert Opin. Drug Deliv. 2009, 6, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Lourenco, C.; Teixeira, M.; Simoes, S.; Gaspar, R. Steric Stabilization of Nanoparticles: Size and Surface Properties. Int. J. Pharm. 1996, 138, 1–12. [Google Scholar] [CrossRef]
- Santander-Ortega, M.J.; Jódar-Reyes, A.B.; Csaba, N.; Bastos-González, D.; Ortega-Vinuesa, J.L. Colloidal Stability of Pluronic F68-Coated PLGA Nanoparticles: A Variety of Stabilisation Mechanisms. J. Colloid Interface Sci. 2006, 302, 522–529. [Google Scholar] [CrossRef]
- Kovacevic, A.; Savic, S.; Vuleta, G.; Müller, R.H.; Keck, C.M. Polyhydroxy Surfactants for the Formulation of Lipid Nanoparticles (SLN and NLC): Effects on Size, Physical Stability and Particle Matrix Structure. Int. J. Pharm. 2011, 406, 163–172. [Google Scholar] [CrossRef] [Green Version]
- Shah, R.; Eldridge, D.; Palombo, E.; Harding, I. Optimisation and Stability Assessment of Solid Lipid Nanoparticles Using Particle Size and Zeta Potential. J. Phys. Sci. 2014, 25, 59–75. [Google Scholar]
- Richtering, W.; Alberg, I.; Zentel, R. Nanoparticles in the Biological Context: Surface Morphology and Protein Corona Formation. Small 2020, 16, 2002162. [Google Scholar] [CrossRef]
- de la Harpe, K.; Kondiah, P.; Choonara, Y.; Marimuthu, T.; du Toit, L.; Pillay, V. The Hemocompatibility of Nanoparticles: A Review of Cell–Nanoparticle Interactions and Hemostasis. Cells 2019, 8, 1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mekkawy, I.A.; Mahmoud, U.M.; Hana, M.N.; Sayed, A.E.D.H. Cytotoxic and Hemotoxic Effects of Silver Nanoparticles on the African Catfish, Clarias Gariepinus (Burchell, 1822). Ecotoxicol. Environ. Saf. 2019, 171, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Chinnaiyan, S.K.; Karthikeyan, D.; Gadela, V.R. Development and Characterization of Metformin Loaded Pectin Nanoparticles for T2 Diabetes Mellitus. Pharm. Nanotechnol. 2018, 6, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Chain, C.Y.; Daza Millone, M.A.; Cisneros, J.S.; Ramirez, E.A.; Vela, M.E. Surface Plasmon Resonance as a Characterization Tool for Lipid Nanoparticles Used in Drug Delivery. Front. Chem. 2021, 8, 605307. [Google Scholar] [CrossRef]
- Rahim, M.N.; Liberal, R.; Miquel, R.; Heaton, N.D.; Heneghan, M.A. Acute Severe Autoimmune Hepatitis: Corticosteroids or Liver Transplantation? Liver Transpl. 2019, 25, 946–959. [Google Scholar] [CrossRef] [PubMed]
- Melgert, B.N.; Olinga, P.; van der Laan, J.M.S.; Weert, B.; Cho, J.; Schuppan, D.; Groothuis, G.M.M.; Meijer, D.K.F.; Poelstra, K. Targeting Dexamethasone to Kupffer Cells: Effects on Liver Inflammation and Fibrosis in Rats. Hepatology 2001, 34, 719–728. [Google Scholar] [CrossRef]
- Bale, S.S.; Geerts, S.; Jindal, R.; Yarmush, M.L. Isolation and Co-Culture of Rat Parenchymal and Non-Parenchymal Liver Cells to Evaluate Cellular Interactions and Response. Sci. Rep. 2016, 6, 25329. [Google Scholar] [CrossRef] [PubMed]
- Molinaro, R.; Pastò, A.; Corbo, C.; Taraballi, F.; Giordano, F.; Martinez, J.O.; Zhao, P.; Wang, X.; Zinger, A.; Boada, C.; et al. Macrophage-Derived Nanovesicles Exert Intrinsic Anti-Inflammatory Properties and Prolong Survival in Sepsis through a Direct Interaction with Macrophages. Nanoscale 2019, 11, 13576–13586. [Google Scholar] [CrossRef] [PubMed]
- Corbo, C.; Cromer, W.E.; Molinaro, R.; Toledano Furman, N.E.; Hartman, K.A.; de Rosa, E.; Boada, C.; Wang, X.; Zawieja, D.C.; Agostini, M.; et al. Engineered Biomimetic Nanovesicles Show Intrinsic Anti-Inflammatory Properties for the Treatment of Inflammatory Bowel Diseases. Nanoscale 2017, 9, 14581–14591. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Rong, Y.; Teng, Y.; Mu, J.; Zhuang, X.; Tseng, M.; Samykutty, A.; Zhang, L.; Yan, J.; Miller, D.; et al. Broccoli-Derived Nanoparticle Inhibits Mouse Colitis by Activating Dendritic Cell AMP-Activated Protein Kinase. Mol. Ther. 2017, 25, 1641–1654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.W.; Bäumer, W.; Monteiro-Riviere, N.A. Cellular Uptake Mechanisms and Toxicity of Quantum Dots in Dendritic Cells. Nanomedicine 2011, 6, 777–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomić, S.; Dokić, J.; Vasilijić, S.; Ogrinc, N.; Rudolf, R.; Pelicon, P.; Vučević, D.; Milosavljević, P.; Janković, S.; Anžel, I.; et al. Size-Dependent Effects of Gold Nanoparticles Uptake on Maturation and Antitumor Functions of Human Dendritic Cells in Vitro. PLoS ONE 2014, 9, e96584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallhov, H.; Gabrielsson, S.; Strømme, M.; Scheynius, A.; Garcia-Bennett, A.E. Mesoporous Silica Particles Induce Size Dependent Effects on Human Dendritic Cells. Nano Lett. 2007, 7, 3576–3582. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Composition | EE a (%) | DL b | Mean Diameter (nm) | PDI | Z Pot c (mV) | ||
|---|---|---|---|---|---|---|---|---|
| MM (mg) | DXM (mg) | Oil (µL) | ||||||
| F1 | 400 | 10 | 0 | 86.4 ± 3.0 | 21.6 | 152.2 ± 2.8 | 0.24 ± 0.01 | −15.9 ± 1.0 |
| F2 | 400 | 10 | 25 | 97.7 ± 3.1 * | 24.4 | 144.9 + 1.0 * | 0.22 ± 0.01 | −10.4 ± 0.8 |
| F3 | 400 | 10 | 50 | 94.3 ± 2.9 * | 23.6 | 142.1 + 1.9 * | 0.20 ± 0.01 | −9.1 ± 0.4 |
| F4 | 400 | 10 | 100 | 94.2 ± 3.0 * | 23.6 | 145.0 + 0.8 * | 0.20 ± 0.01 | −9.3 ± 1.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Medina-Montano, C.; Rivero Berti, I.; Gambaro, R.C.; Limeres, M.J.; Svensson, M.; Padula, G.; Chain, C.Y.; Cisneros, J.S.; Castro, G.R.; Grabbe, S.; et al. Nanostructured Lipid Carriers Loaded with Dexamethasone Prevent Inflammatory Responses in Primary Non-Parenchymal Liver Cells. Pharmaceutics 2022, 14, 1611. https://doi.org/10.3390/pharmaceutics14081611
Medina-Montano C, Rivero Berti I, Gambaro RC, Limeres MJ, Svensson M, Padula G, Chain CY, Cisneros JS, Castro GR, Grabbe S, et al. Nanostructured Lipid Carriers Loaded with Dexamethasone Prevent Inflammatory Responses in Primary Non-Parenchymal Liver Cells. Pharmaceutics. 2022; 14(8):1611. https://doi.org/10.3390/pharmaceutics14081611
Chicago/Turabian StyleMedina-Montano, Carolina, Ignacio Rivero Berti, Rocío C. Gambaro, María José Limeres, Malin Svensson, Gisel Padula, Cecilia Y. Chain, José Sebastián Cisneros, Guillermo R. Castro, Stephan Grabbe, and et al. 2022. "Nanostructured Lipid Carriers Loaded with Dexamethasone Prevent Inflammatory Responses in Primary Non-Parenchymal Liver Cells" Pharmaceutics 14, no. 8: 1611. https://doi.org/10.3390/pharmaceutics14081611