
Development of a Novel Vaginal Drug Delivery System to Control Time of Farrowing and Allow Supervision of Piglet Delivery

 , , ,
, , , 
Abstract
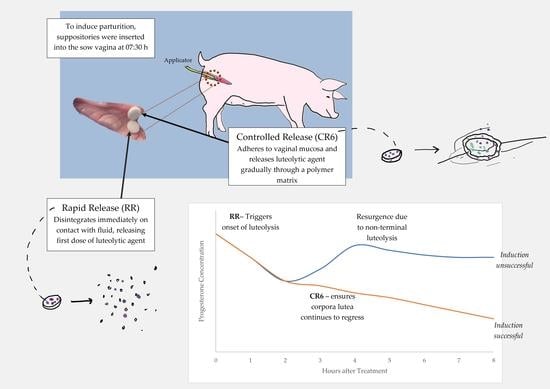
:
1. Introduction
2. Materials and Methods
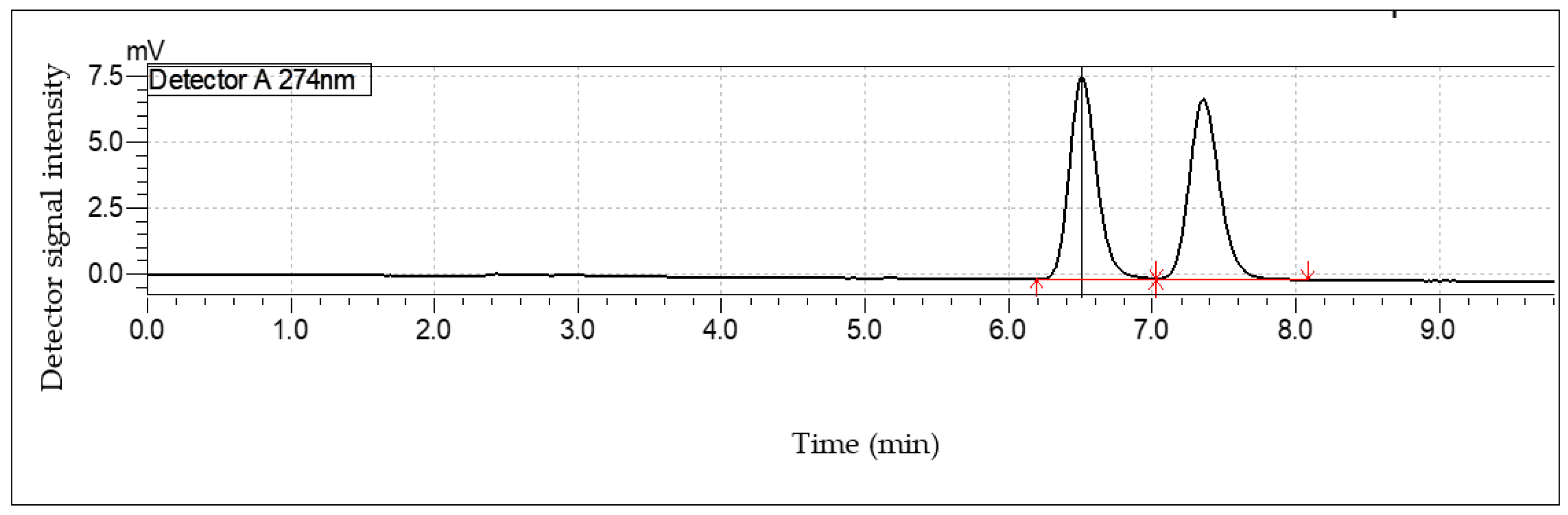
2.1. High Performance Liquid Chromatography (HPLC)
2.2. NVDDS Preparation
2.3. Friability Test
2.4. Drug Content Uniformity
2.5. Dissolution Study
2.6. Polymer Swelling for CR NVDDS
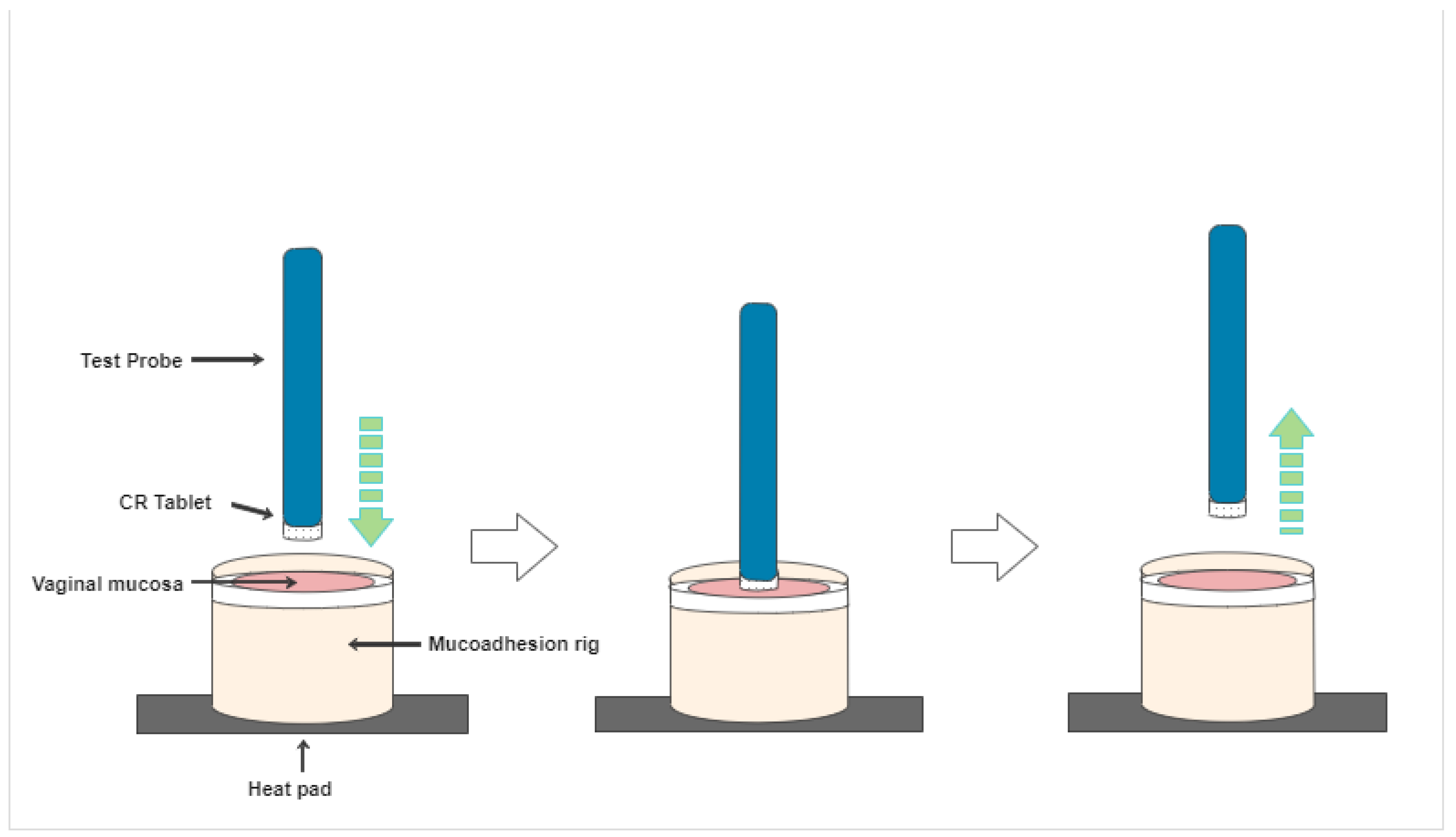
2.7. CR NVDDS Bio-Adhesion Test
2.8. In Vivo Testing of NVDDS
- SI: Injection of 250 µg cloprostenol (Juramate®, Jurox Pty, Ltd., Rutherford, NSW, Australia) into the vulva at 7:00 h;
- SDI: Injection of 125 µg cloprostenol into the vulva at 7:00 h and again at 13:00 h;
- IRT: Insertion of IR tablet at 7:00 h and again at 13:00 h; for each vaginal deposition, the tablet applicator was sanitised (F10SC Veterinary Disinfectant), rinsed with water and lubricated (Obstetrical Lubricant, ZebraVet, Sherwood, QLD, Australia)
- IRCRT: Insertion of IR and CR (formulation CR6) tablets at 7:00 h.
- Control: No cloprostenol administration
2.9. Statistics
3. Results
3.1. Tablet Physical Properties
3.2. Dissolution Studies
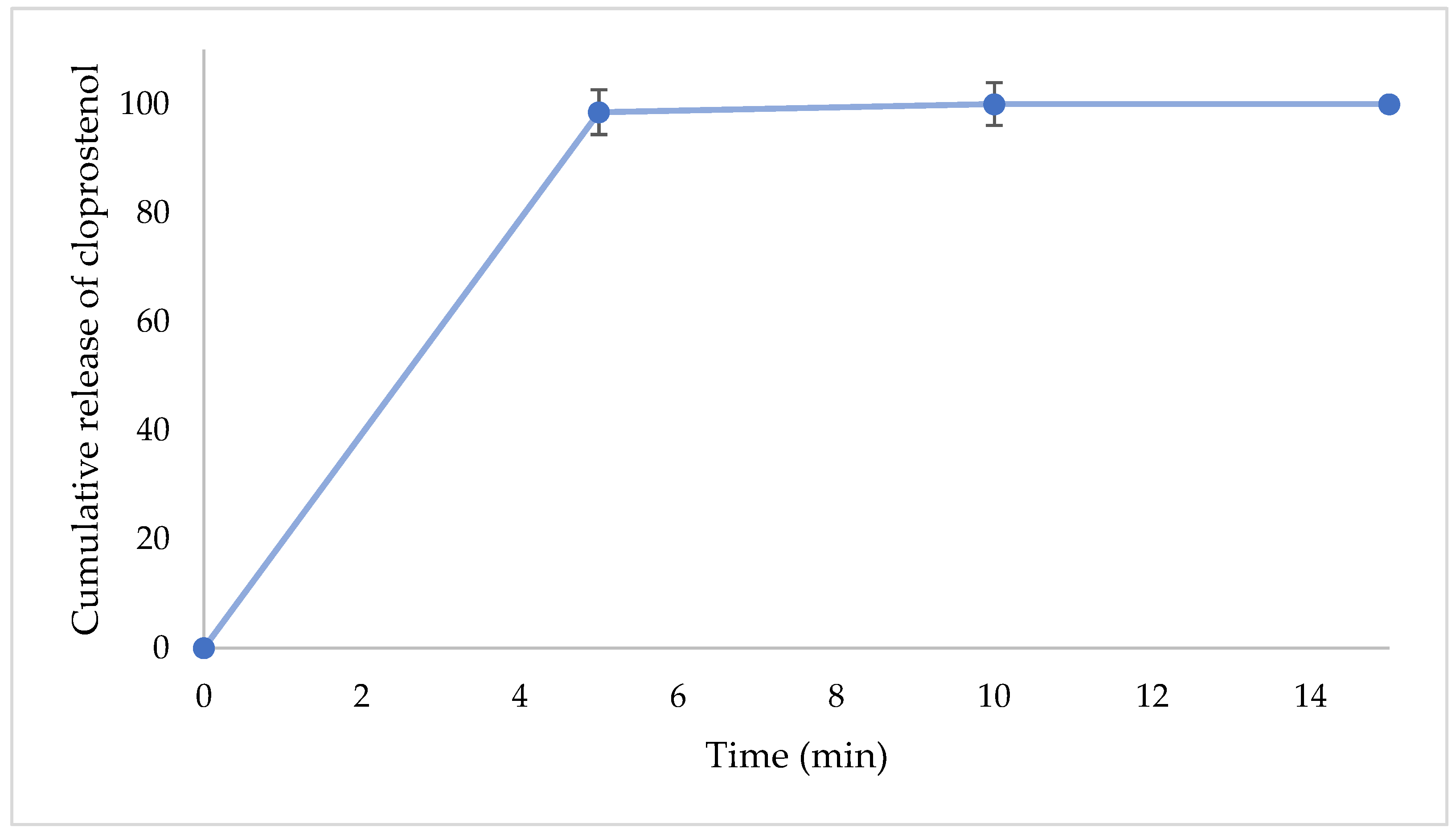
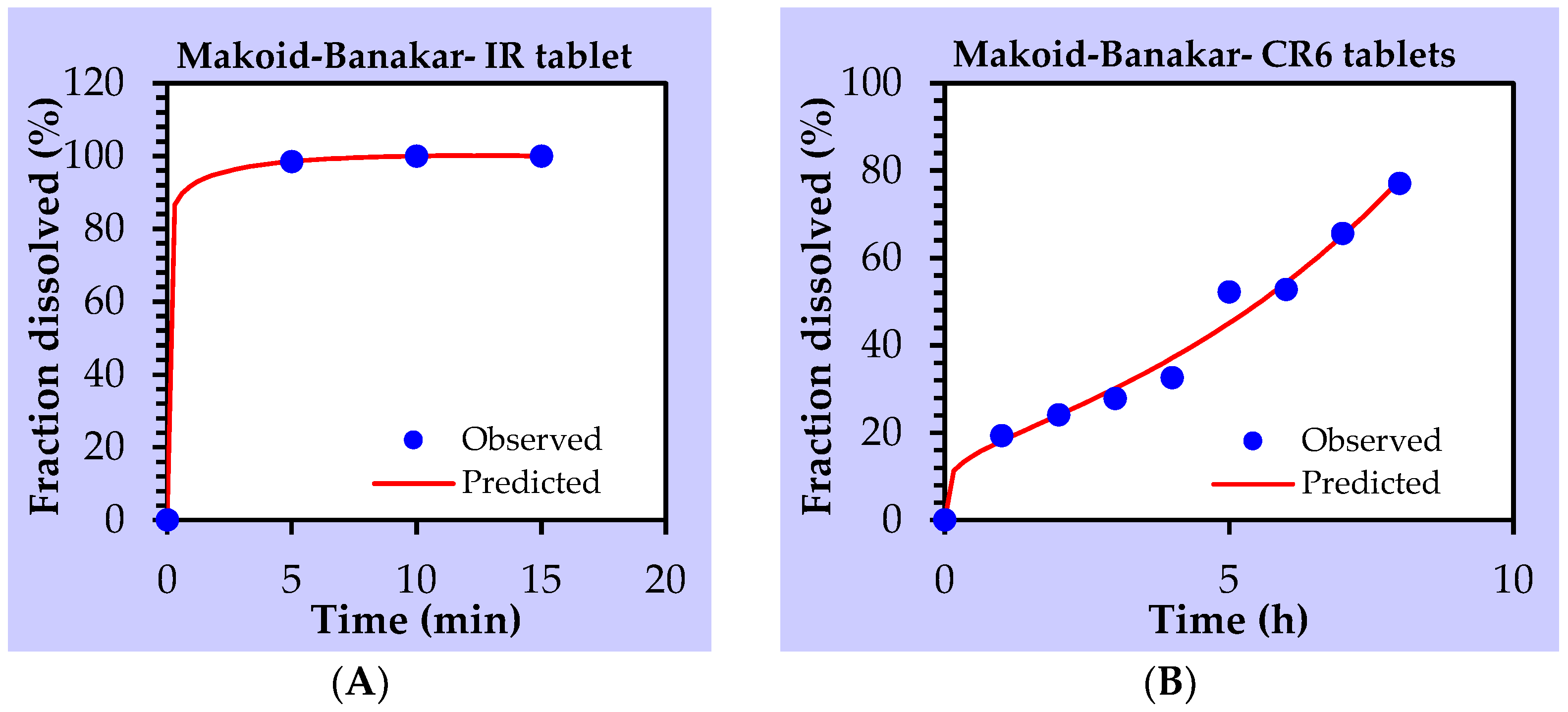
3.2.1. IR-NVDDS
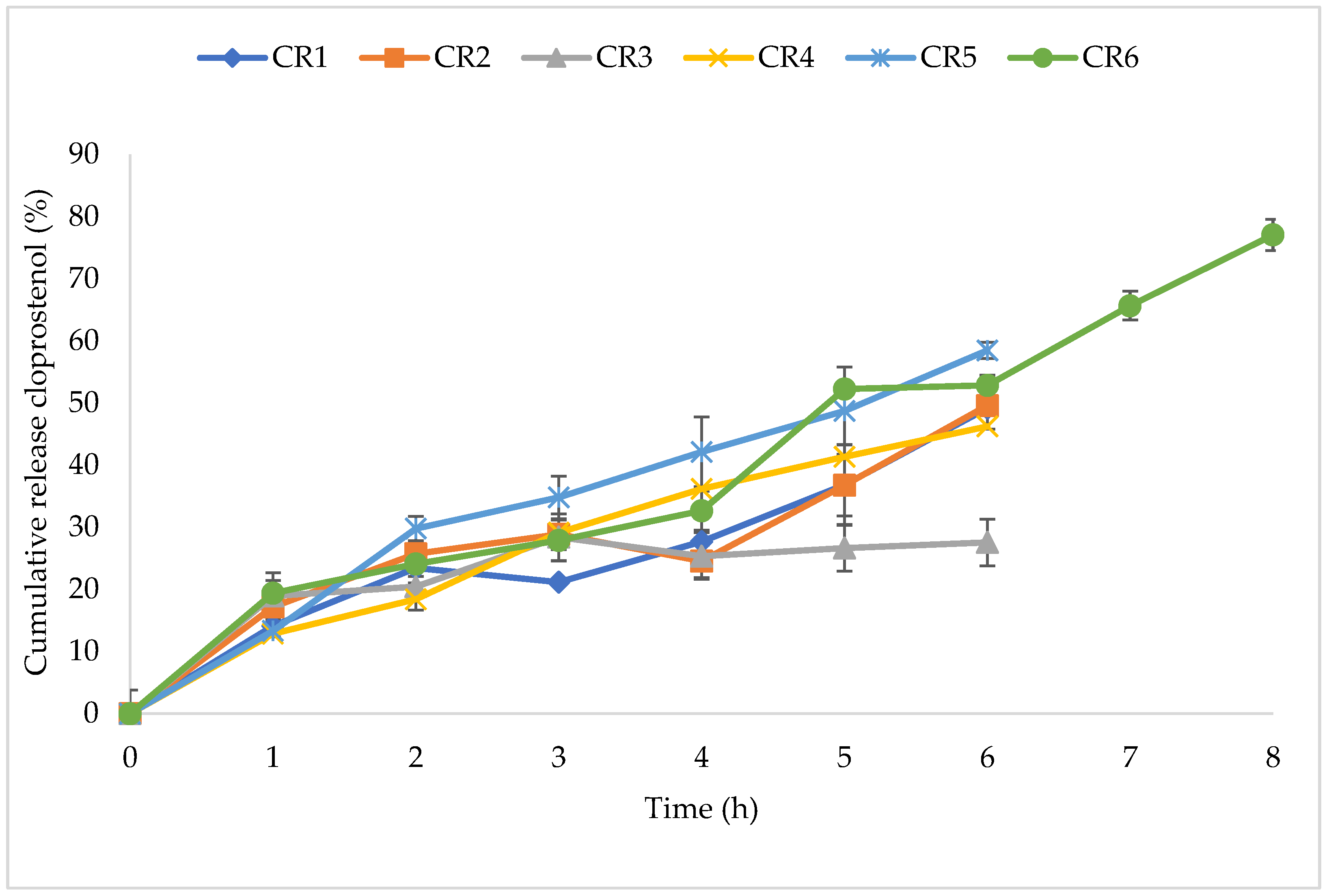
3.2.2. CR-NVDDS
3.3. Mechanism of Drug Release
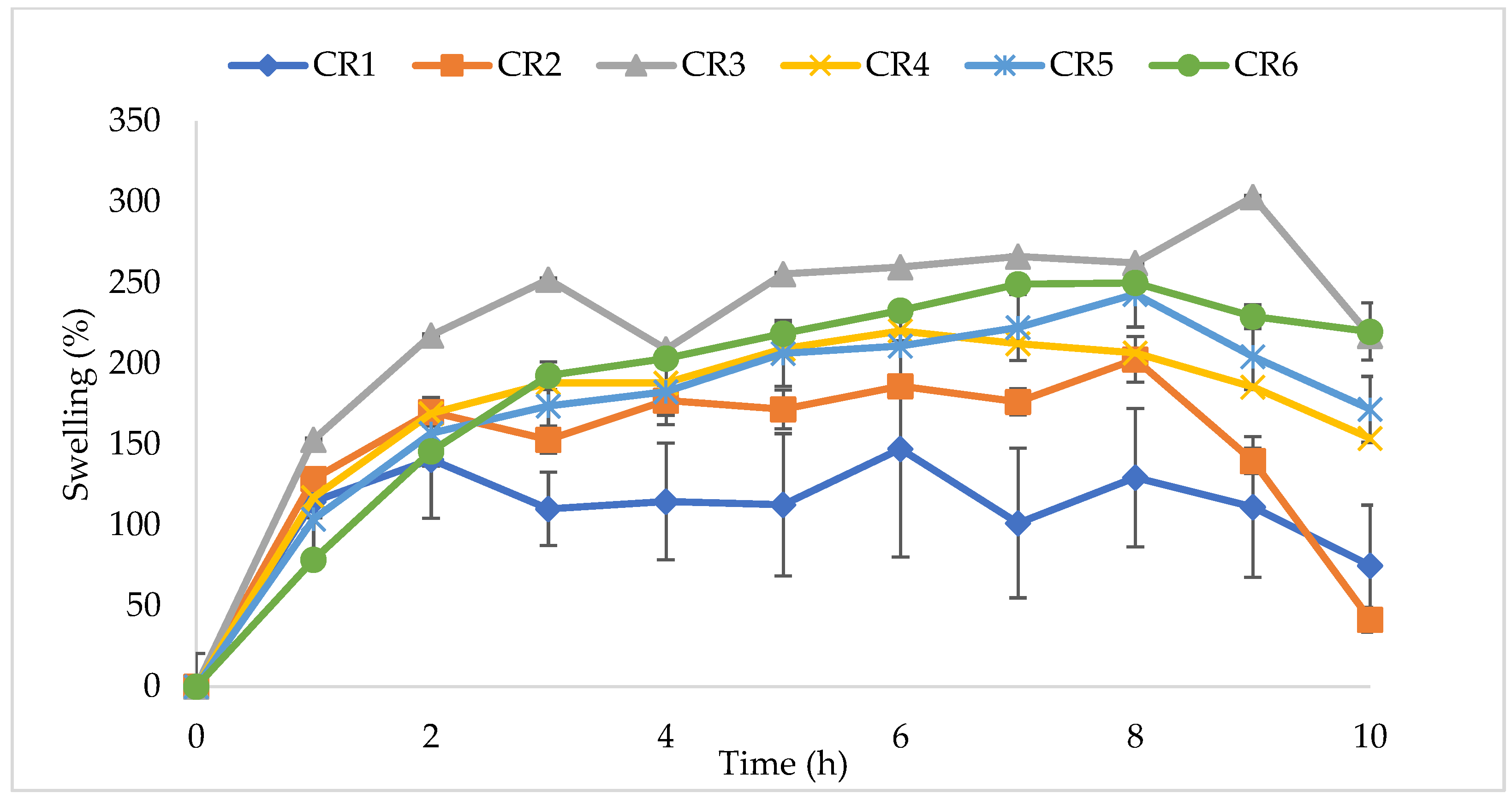
3.4. Swelling Tests
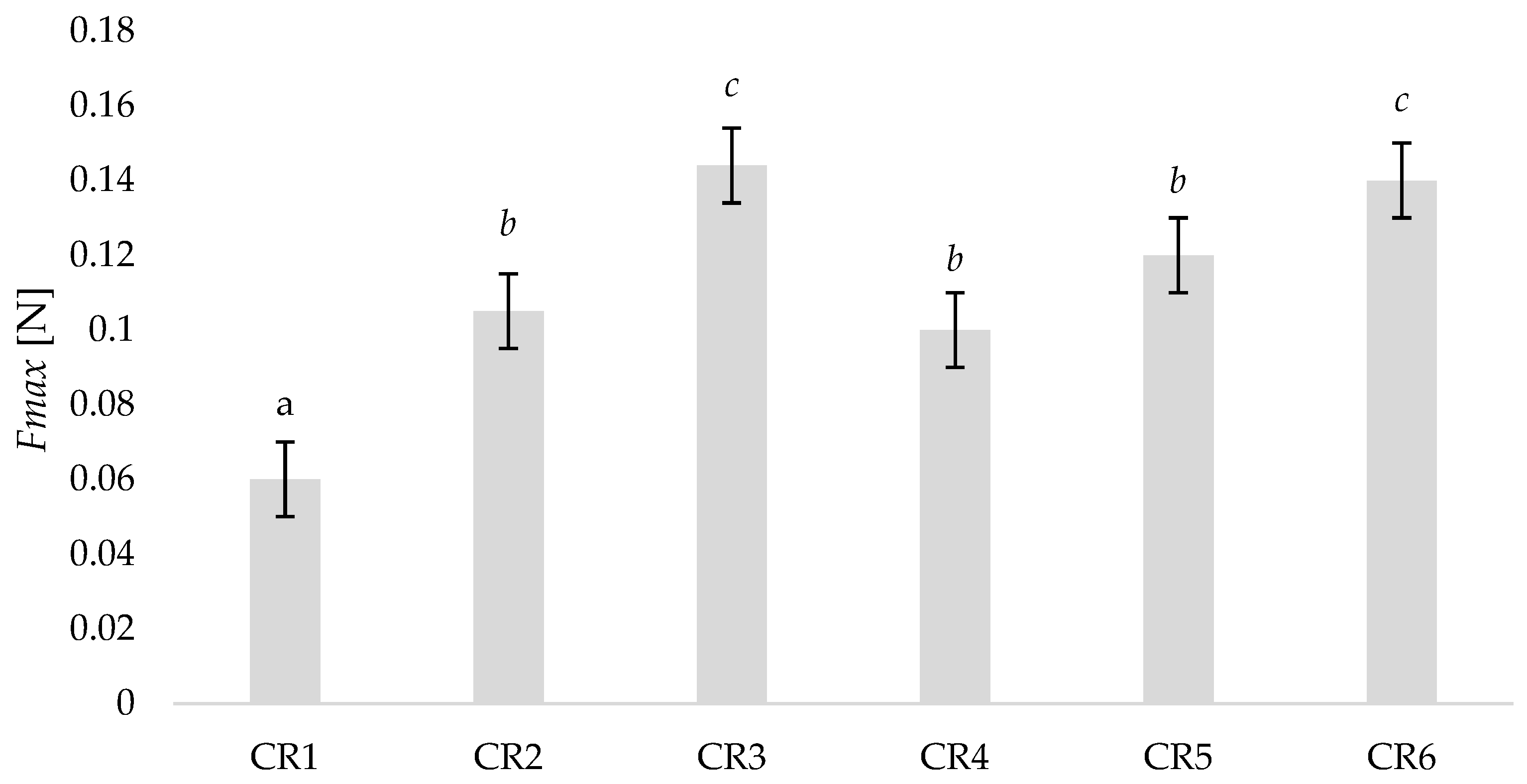
3.5. Bio-Adhesion Tests
3.6. Selected Formulations Uniformity and Friability Tests
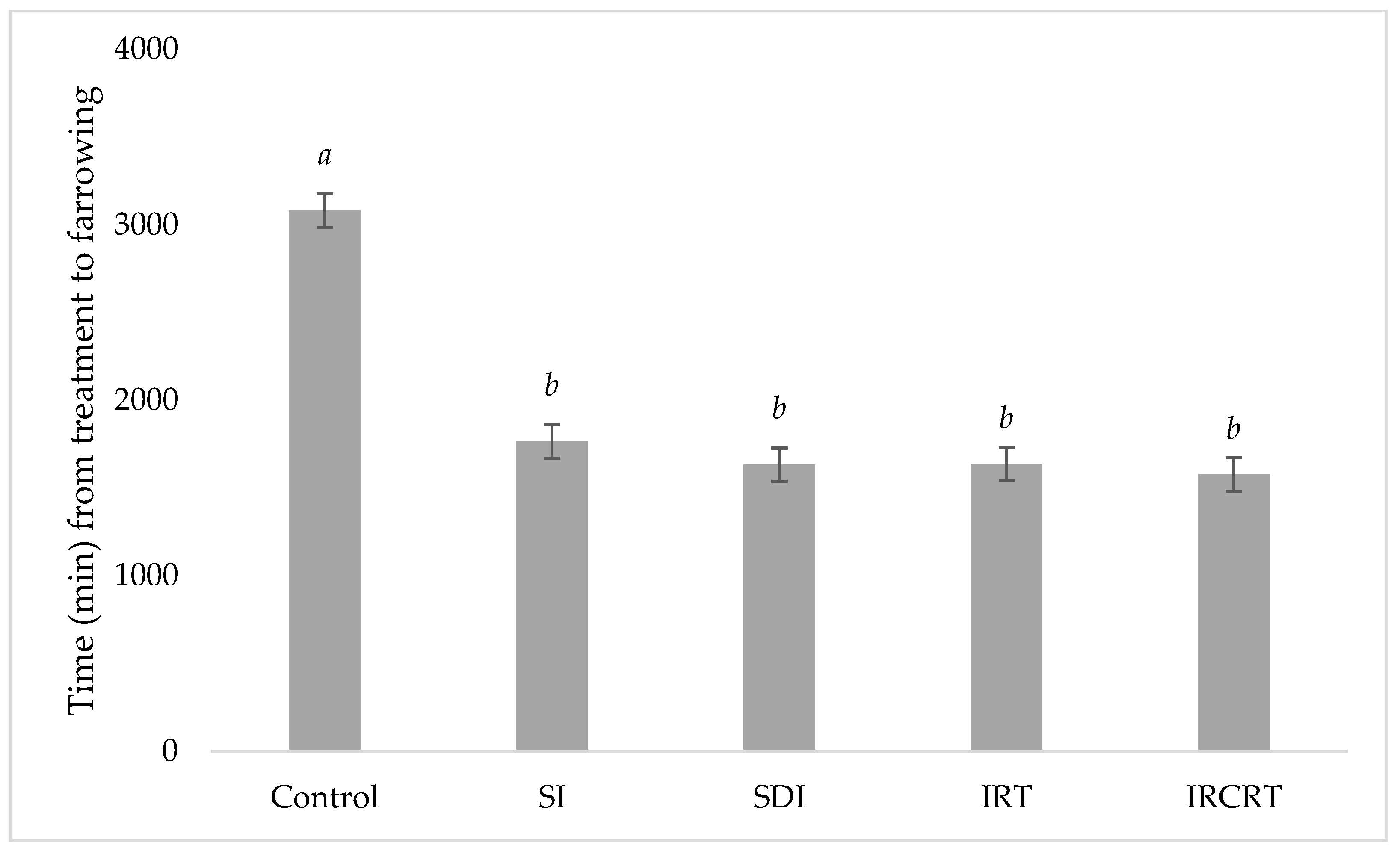
3.7. In Vivo Tests
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model Name | Equation | Goodness of Fit Parameter | IR | CR 6 |
|---|---|---|---|---|
| Zero order | F = k0 × t | R2 adjusted | 0.4457 | 0.9599 |
| RMSE | 1372.1250 | 24.1740 | ||
| AIC | 35.2909 | 49.3825 | ||
| First order | F = 100 × [1 − Exp(−k1 × t)] | R2 adjusted | 1.0000 | 0.9359 |
| RMSE | 0.0002 | 38.5851 | ||
| AIC | −28.3566 | 53.5908 | ||
| Higuchi | F = kH × t0.5 | R2 adjusted | 0.8308 | 0.8815 |
| RMSE | 418.8915 | 71.3687 | ||
| AIC | 30.5449 | 59.1257 | ||
| Hixson–Crowell | F = 100 × [1 − (1 − kHC × t)3] | R2 adjusted | 0.9695 | 0.9494 |
| RMSE | 75.5287 | 30.4404 | ||
| AIC | 23.6925 | 51.4569 | ||
| Hopfenberg | F = 100 × [1 − (1 − kHB × t)n] | R2 adjusted | 1.0000 | 0.9548 |
| RMSE | 0.0000 | 27.1928 | ||
| AIC | −245.5330 | 51.2398 | ||
| Makoid–Banakar | F = kMB × tn × Exp(−k × t) | R2 adjusted | 1.0000 | 0.9776 |
| RMSE | 0.0000 | 13.4826 | ||
| AIC | Perfect fit | 45.5384 | ||
| Baker–Lonsdale | 3/2 × [1 − (1 − F/100)2/3] − F/100 = kBL × t | R2 adjusted | 0.9330 | 0.8418 |
| 165.8327 | 95.2837 | 95.2837 | ||
| AIC | 26.8384 | 61.7267 | ||
| Peppas–Sahlin | F = k1 × tm + k2 × t2m | R2 adjusted | 1.0000 | 0.9602 |
| RMSE | 0.0000 | 23.9593 | ||
| AIC | −249.0782 | 50.7130 |
Appendix B
| Sow Parity | Days Until Due Date | Vaginal pH | Temperature |
|---|---|---|---|
| 1 | 1 | 7 | 37.8 |
| 1 | 2 | 8 | 38.1 |
| 1 | 0 | 7 | 39.1 |
| 1 | 2 | 7.5 | 37.7 |
| 3 | 3 | 7 | 38.1 |
| 4 | 3 | 7 | 37.9 |
| 0 | 3 | 7.5 | 38 |
| 0 | 1 | 7 | 38.2 |
| 2 | 0 | 8 | 37.7 |
| 3 | 0 | 7.5 | 37.7 |
| 4 | 0 | 7 | 36.5 |
| 2 | 0 | 6.5 | 38.2 |
| 1 | 1 | 7.5 | 38.2 |
| 2 | 0 | 7 | 38 |
| 4 | 0 | 8 | 38.5 |
| 1 | 0 | 7 | 38 |
| 6 | 2 | 7.5 | 38 |
| 4 | 1 | 6.5 | 38.1 |
| 4 | 0 | 7.2 | 37.5 |
| 3 | 1 | 6.5 | 37.6 |
| 1 | 2 | 7 | 38.2 |
| 1 | 1 | 7.5 | 37.5 |
| 4 | 0 | 7 | 37.2 |
| 3 | 1 | 6.5 | 38.5 |
| Average | 7.1 | 37.9 |
References
- KilBride, A.; Mendl, M.; Statham, P.; Held, S.; Harris, M.; Cooper, S.; Green, L. A cohort study of preweaning piglet mortality and farrowing accommodation on 112 commercial pig farms in England. Prev. Vet. Med. 2012, 104, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Muns, R.; Nuntapaitoon, M.; Tummaruk, P. Non-infectious causes of pre-weaning mortality in piglets. Livest. Sci. 2016, 184, 46–57. [Google Scholar] [CrossRef]
- Baxter, E.M.; Edwards, S.A. Piglet mortality and morbidity: Inevitable or unacceptable? In Advances in Piglet Welfare; Elsevier: Amsterdam, The Netherlands, 2018; pp. 73–100. [Google Scholar]
- Kaeoket, K. The effect of dose and route of administration of R-cloprostenol on the parturient response of sows. Reprod. Domest. Anim. 2006, 41, 472–476. [Google Scholar] [CrossRef]
- Alonso-Spilsbury, M.; Ramirez-Necoechea, R.; Gonzlez-Lozano, M.; Mota-Rojas, D.; Trujillo-Ortega, M. Piglet survival in early lactation: A review. J. Anim. Vet. Adv. 2007, 6, 76–86. [Google Scholar]
- Villanueva-García, D.; Mota-Rojas, D.; Martínez-Burnes, J.; Olmos-Hernández, A.; Mora-Medina, P.; Salmerón, C.; Gómez, J.; Boscato, L.; Gutiérrez-Pérez, O.; Cruz, V. Hypothermia in newly born piglets: Mechanisms of thermoregulation and pathophysiology of death. J. Anim. Behav. Biometeorol. 2020, 9, 2101. [Google Scholar] [CrossRef]
- Ward, S.A.; Kirkwood, R.N.; Plush, K.J. Are larger litters a concern for piglet survival or an effectively manageable trait? Animals 2020, 10, 309. [Google Scholar] [CrossRef] [Green Version]
- Kirkden, R.; Broom, D.; Andersen, I. Invited review: Piglet mortality: Management solutions. J. Anim. Sci. 2013, 91, 3361–3389. [Google Scholar] [CrossRef] [PubMed]
- Houben, M.A.; Tobias, T.J.; Holstege, M.M. The effect of double nursing, an alternative nursing strategy for the hyper-prolific sow herd, on herd performance. Porc. Health Manag. 2017, 3, 2. [Google Scholar] [CrossRef] [Green Version]
- Alexopoulos, J.G.; Lines, D.S.; Hallett, S.; Plush, K.J. A review of success factors for piglet fostering in lactation. Animals 2018, 8, 38. [Google Scholar] [CrossRef] [Green Version]
- Vande Pol, K.D.; Tolosa, A.F.; Bautista, R.O.; Willard, N.C.; Gates, R.S.; Shull, C.M.; Brown, C.B.; Alencar, S.A.; Lents, C.A.; Ellis, M. Effects of drying and providing supplemental oxygen to piglets at birth on rectal temperature over the first 24 h after birth. Transl. Anim. Sci. 2021, 5, txab095. [Google Scholar] [CrossRef]
- Kirkwood, R.N. Induction of parturition in sows. Thai J. Vet. Med. 2015, 45, 487. [Google Scholar]
- Kirkwood, R.N.; Aherne, F.X. Increasing the predictability of cloprostenol-induced farrowing in sows. J. Swine Health Prod. 1998, 6, 57–59. [Google Scholar]
- Decaluwe, R.; Janssens, G.; Declerck, I.; de Kruif, A.; Maes, D. Induction of parturition in the sow. Vlaams Diergeneeskd. Tijdschr. 2012, 81, 158–165. [Google Scholar] [CrossRef]
- Robert, S.; Passille, A.-M.D.; St-Pierre, N.; Pelletier, G.; Petitclerc, D.; Dubreuil, P.; Brazeau, P. Effect of the stress of injections on the serum concentration of cortisol, prolactin, and growth hormone in gilts and lactating sows. Can. J. Anim. Sci. 1989, 69, 663–672. [Google Scholar] [CrossRef]
- Ougi, K.; Okada, K.; Leong, K.H.; Hayashi, Y.; Kumada, S.; Onuki, Y. Effect of the molecular mobility of water adsorbed by disintegrants on storage-induced hydrolytic degradation of acetylsalicylic acid incorporated into tablets under humid conditions. Eur. J. Pharm. Sci. 2020, 154, 105502. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, R.; Hong, E.; Villafuerte, L. Influence of admixed citric acid on the release profile of pelanserin hydrochloride from HPMC matrix tablets. Int. J. Pharm. 2000, 201, 165–173. [Google Scholar] [CrossRef]
- Nokhodchi, A.; Norouzi-Sani, S.; Siahi-Shadbad, M.-R.; Lotfipoor, F.; Saeedi, M. The effect of various surfactants on the release rate of propranolol hydrochloride from hydroxypropylmethylcellulose (HPMC)-Eudragit matrices. Eur. J. Pharm. Biopharm. 2002, 54, 349–356. [Google Scholar] [CrossRef]
- Siepmann, J.; Peppas, N.A. Modeling of drug release from delivery systems based on hydroxypropyl methylcellulose (HPMC). Adv. Drug Deliv. Rev. 2012, 64, 163–174. [Google Scholar] [CrossRef]
- Caccavo, D.; Lamberti, G.; Barba, A.A.; Abrahmsén-Alami, S.; Viridén, A.; Larsson, A. Effects of HPMC substituent pattern on water up-take, polymer and drug release: An experimental and modelling study. Int. J. Pharm. 2017, 528, 705–713. [Google Scholar] [CrossRef]
- Tospitakkul, P.; Kraomkaew, K.; Thammasin, K.; Uttarak, P.; Nuntapaitoon, M.; De Rensis, F.; Tummaruk, P. Induction of parturition by double administration of prostaglandin F2alpha in sows reduces the variation of gestation length without affecting the colostrum yield and piglet performance. J. Vet. Med. Sci. 2019, 81, 1334–1340. [Google Scholar] [CrossRef] [Green Version]
- Kalíková, K.; Tesařová, E.; Bosáková, Z. HPLC method for enantioselective analysis of cloprostenol. J. Pharm. Biomed. Anal. 2008, 46, 892–897. [Google Scholar] [CrossRef] [PubMed]
- Bhat, S.; Shivakumar, H. Bioadhesive controlled release clotrimazole vaginal tablets. Trop. J. Pharm. Res. 2010, 9, 339–346. [Google Scholar] [CrossRef] [Green Version]
- Owen, D.H.; Katz, D.F. A vaginal fluid simulant. Contraception 1999, 59, 91–95. [Google Scholar] [CrossRef]
- Lorenzen, E.; Follmann, F.; Jungersen, G.; Agerholm, J.S. A review of the human vs. porcine female genital tract and associated immune system in the perspective of using minipigs as a model of human genital Chlamydia infection. Vet. Res. 2015, 46, 116. [Google Scholar] [CrossRef] [PubMed]
- Chaibva, F.A.; Khamanga, S.M.; Walker, R.B. Swelling, erosion and drug release characteristics of salbutamol sulfate from hydroxypropyl methylcellulose-based matrix tablets. Drug Dev. Ind. Pharm. 2010, 36, 1497–1510. [Google Scholar] [CrossRef]
- Berginc, K.; Škalko-Basnet, N.; Basnet, P.; Kristl, A. Development and evaluation of an in vitro vaginal model for assessment of drug’s biopharmaceutical properties: Curcumin. AAPS PharmSciTech 2012, 13, 1045–1053. [Google Scholar] [CrossRef] [Green Version]
- Hiorth, M.; Nilsen, S.; Tho, I. Bioadhesive mini-tablets for vaginal drug delivery. Pharmaceutics 2014, 6, 494–511. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Yang, Y.; Xie, J.; Xu, J.; Yue, P.; Yang, M. Novel nanocrystal-based solid dispersion with high drug loading, enhanced dissolution, and bioavailability of andrographolide. Int. J. Nanomed. 2018, 13, 3763. [Google Scholar] [CrossRef] [Green Version]
- Kucera, S.U.; DiNunzio, J.C.; Kaneko, N.; McGinity, J.W. Evaluation of Ceolus™ microcrystalline cellulose grades for the direct compression of enteric-coated pellets. Drug Dev. Ind. Pharm. 2012, 38, 341–350. [Google Scholar] [CrossRef]
- Ghimire, M.; Hodges, L.A.; Band, J.; O’Mahony, B.; McInnes, F.J.; Mullen, A.B.; Stevens, H.N. In-vitro and in-vivo erosion profiles of hydroxypropylmethylcellulose (HPMC) matrix tablets. J. Control. Release 2010, 147, 70–75. [Google Scholar] [CrossRef]
- Mohamed, F.A.; Roberts, M.; Seton, L.; Ford, J.L.; Levina, M.; Rajabi-Siahboomi, A.R. The influence of HPMC concentration on release of theophylline or hydrocortisone from extended release mini-tablets. Drug Dev. Ind. Pharm. 2013, 39, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Vueba, M.; Batista de Carvalho, L.; Veiga, F.; Sousa, J.; Pina, M. In vitro release of ketoprofen from hydrophilic matrix tablets containing cellulose polymer mixtures. Drug Dev. Ind. Pharm. 2013, 39, 1651–1662. [Google Scholar] [CrossRef] [PubMed]
- Levina, M.; Rajabi-Siahboomi, A.R. An Industrial Perspective on Hydrophilic Matrix Tablets Based on Hyproxypropyl Methylcellulose (Hypromellose). In Hydrophilic Matrix Tablets for Oral Controlled Release; Springer: Berlin/Heidelberg, Germany, 2014; pp. 53–85. [Google Scholar]
- Gonçalves-Araújo, T.; Rajabi-Siahboomi, A.R.; Caraballo, I. Application of percolation theory in the study of an extended release Verapamil hydrochloride formulation. Int. J. Pharm. 2008, 361, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Heng, P.W.S.; Chan, L.W.; Easterbrook, M.G.; Li, X. Investigation of the influence of mean HPMC particle size and number of polymer particles on the release of aspirin from swellable hydrophilic matrix tablets. J. Control. Release 2001, 76, 39–49. [Google Scholar] [CrossRef]
- Zarmpi, P.; Flanagan, T.; Meehan, E.; Mann, J.; Fotaki, N. Biopharmaceutical understanding of excipient variability on drug apparent solubility based on drug physicochemical properties. Case study: Superdisintegrants. AAPS J. 2020, 22, 46. [Google Scholar] [CrossRef] [Green Version]
- Kouchak, M.; Mahmoodzadeh, M.; Farrahi, F. Designing of a pH-triggered Carbopol®/HPMC in situ gel for ocular delivery of dorzolamide HCl: In vitro, in vivo, and ex vivo evaluation. AAPS PharmSciTech 2019, 20, 210. [Google Scholar] [CrossRef]
- Aguilar-de-Leyva, Á.; Campiñez, M.D.; Jost, F.; Gavira, M.; Caraballo, I. Study of the critical points in combined matrix tablets containing both inert and swelling excipients. J. Drug Deliv. Sci. Technol. 2019, 52, 885–894. [Google Scholar] [CrossRef]
- Caraballo, I.; Fernández-Arévalo, M.; Holgado, M.; Rabasco, A. Percolation theory: Application to the study of the release behaviour from inert matrix systems. Int. J. Pharm. 1993, 96, 175–181. [Google Scholar] [CrossRef]
- Viridén, A.; Wittgren, B.; Larsson, A. Investigation of critical polymer properties for polymer release and swelling of HPMC matrix tablets. Eur. J. Pharm. Sci. 2009, 36, 297–309. [Google Scholar] [CrossRef]
- Akbari, J.; Saeedi, M.; Enayatifard, R.; Doost, M. Development and evaluation of mucoadhesive chlorhexidine tablet formulations. Trop. J. Pharm. Res. 2010, 9, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Gunvaldsen, R.; Waldner, C.; Harding, J. Effects of farrowing induction on suckling piglet performance. J. Swine Health Prod. 2007, 15, 84–91. [Google Scholar]
- Straw, B.; Bates, R.; May, G. Influence of method of administration of prostaglandin on farrowing and relationship between gestation length and piglet performance. J. Swine Health Prod. 2008, 16, 138–143. [Google Scholar]











| Formulation | Lactose (mg) | HPMC K100 (mg) | HPMC K15 (mg) | HPMC K4 (mg) | HPMC E50 (mg) | HPMC % (w/w) | Lactose % (w/w) |
|---|---|---|---|---|---|---|---|
| CR1 | 85 | 20 | - | - | - | 10 | 46 |
| CR2 | 85 | 15 | 5 | - | - | 10 | 46 |
| CR3 | 60 | - | 15 | 30 | - | 24 | 32 |
| CR4 | 55 | - | - | 50 | 27 | 29 | |
| CR5 | 60 | - | 20 | - | 20 | 23 | 32 |
| CR6 | 55 | - | - | 30 | 20 | 26 | 29 |
| Formulation | Thickness (cm) | Hardness (kgf) |
|---|---|---|
| IR | 2.54 ± 0.02 | 4.39 ± 0.36 |
| CR1 | 2.44 ± 0.03 | 12.96± 1.21 |
| CR2 | 2.47 ± 0.02 | 13.25 ± 1.41 |
| CR3 | 2.53 ± 0.03 | 13.96 ± 0.47 |
| CR4 | 2.56 ± 0.27 | 13.67 ± 0.37 |
| CR5 | 2.55 ± 0.031 | 13.24 ± 0.37 |
| CR6 | 2.55 ± 0.022 | 13.74 ± 0.40 |
| NVDDS Formulation | Desired Drug Content (µg) | Average Drug Content (µg) | Standard Deviation | Uniformity Value Score (L1 ≤ 15) |
|---|---|---|---|---|
| Immediate release (IR) | 125 | 127.4 | 4.87 | 10.45 |
| Controlled release Formulation 6 (CR6) | 125 | 126.7 | 6.52 | 13.92 |
| NVDDS Formulation | Original Weight (g) | Weight after 100 Rotations (g) | Percentage Lost (%) ≤ 1% |
|---|---|---|---|
| Immediate release (IR) | 6.591 | 6.586 | 0.075 |
| Controlled release formulation (CR6) | 6.503 | 6.495 | 0.123 |
| Measurements on Sow Litter Performance | Control | SI | SDI | IRT | IRCRT | p-Value |
|---|---|---|---|---|---|---|
| Parity | 2.63 ± 1.6 | 2.56 ± 1.6 | 2.25 ± 1.6 | 2.30 ± 1.6 | 2.03 ± 1.6 | 0.585 |
| Total born | 11.94 ± 0.5 | 12.47 ± 0.6 | 13.38 ± 0.7 | 12.48 ± 0.3 | 12.6 ± 0.2 | 0.486 |
| Born alive | 11.25 ± 0.4 | 11.66 ± 0.5 | 12.28 ± 0.6 | 11.73 ± 0.3 | 11.94 ± 0.4 | 0.730 |
| Birthweight (kg) | 1.38 ± 0.05 | 1.44 ± 0.04 | 1.28 ± 0.05 | 1.36 ± 0.05 | 1.30 ± 0.02 | 0.272 |
| Preweaning survival (%) | 90.9 ± 1.99 | 91.43 ± 1.58 | 91.61 ± 1.51 | 90.04 ± 2.67 | 89.89 ± 2.05 | 0.962 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ward, S.A.; Kirkwood, R.N.; Plush, K.J.; Abdella, S.; Song, Y.; Garg, S. Development of a Novel Vaginal Drug Delivery System to Control Time of Farrowing and Allow Supervision of Piglet Delivery. Pharmaceutics 2022, 14, 340. https://doi.org/10.3390/pharmaceutics14020340
Ward SA, Kirkwood RN, Plush KJ, Abdella S, Song Y, Garg S. Development of a Novel Vaginal Drug Delivery System to Control Time of Farrowing and Allow Supervision of Piglet Delivery. Pharmaceutics. 2022; 14(2):340. https://doi.org/10.3390/pharmaceutics14020340
Chicago/Turabian StyleWard, Sophia A., Roy N. Kirkwood, Kate J. Plush, Sadikalmahdi Abdella, Yunmei Song, and Sanjay Garg. 2022. "Development of a Novel Vaginal Drug Delivery System to Control Time of Farrowing and Allow Supervision of Piglet Delivery" Pharmaceutics 14, no. 2: 340. https://doi.org/10.3390/pharmaceutics14020340