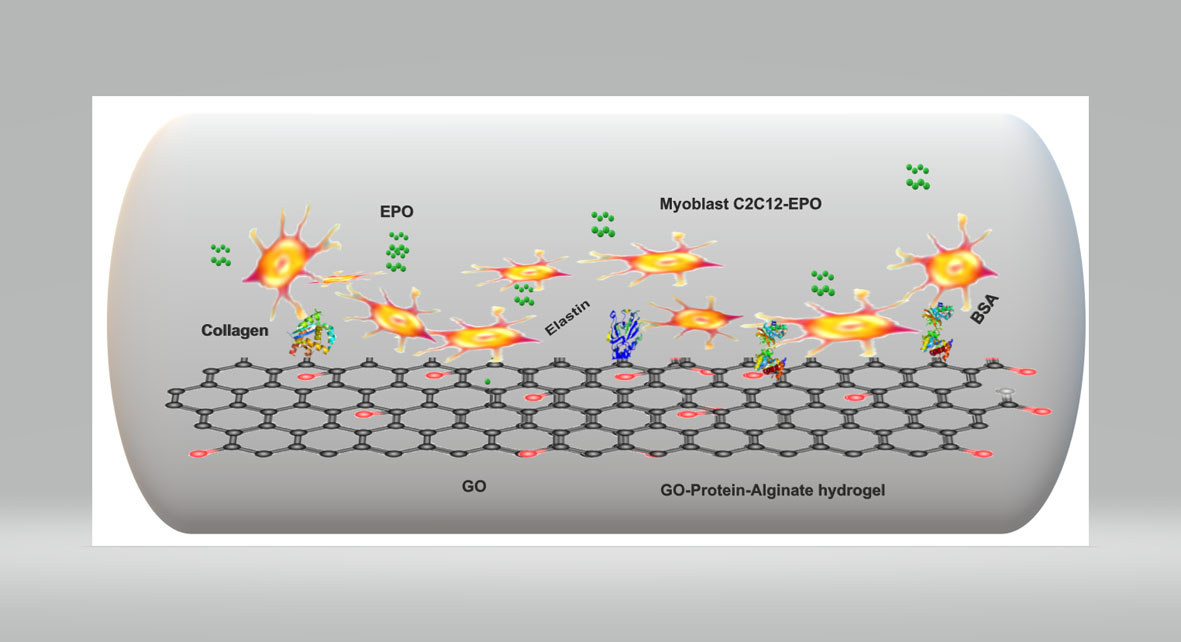
BSA- and Elastin-Coated GO, but Not Collagen-Coated GO, Enhance the Biological Performance of Alginate Hydrogels

 , , , , and
, , , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Characterization of GO-Protein Interactions
2.3. Adsorption Capacity Experiments
2.3.1. Effect of Initial Concentration
2.3.2. Adsorption Isotherms
2.4. Kinetic Study of Protein Adsorption
2.5. Thermodynamic Studies
2.6. Electrochemical Study
2.7. In Vitro Cell Viability Studies
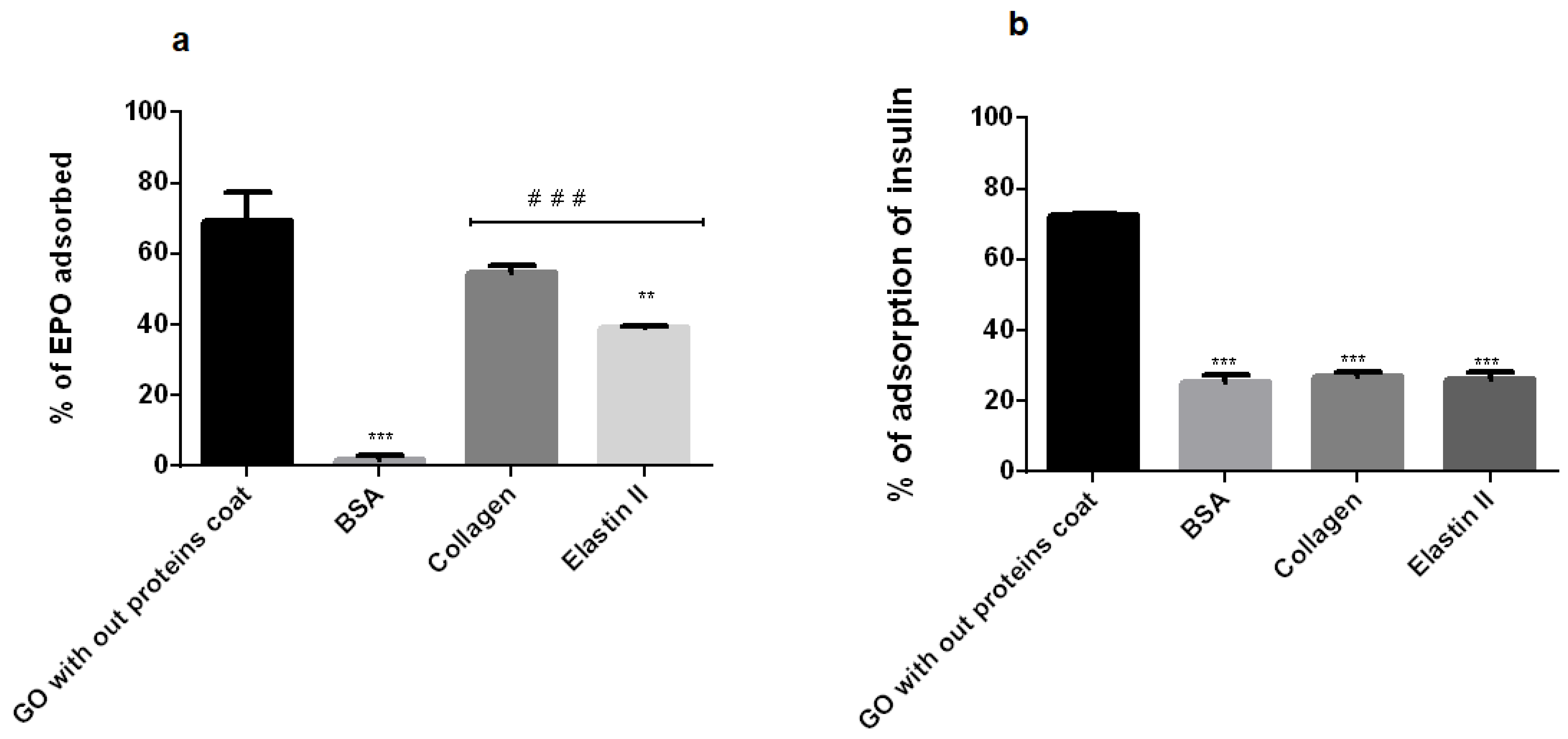
2.8. EPO and Insulin Adsorption Blocking Study
2.9. Statistical Analysis
3. Results and Discussion
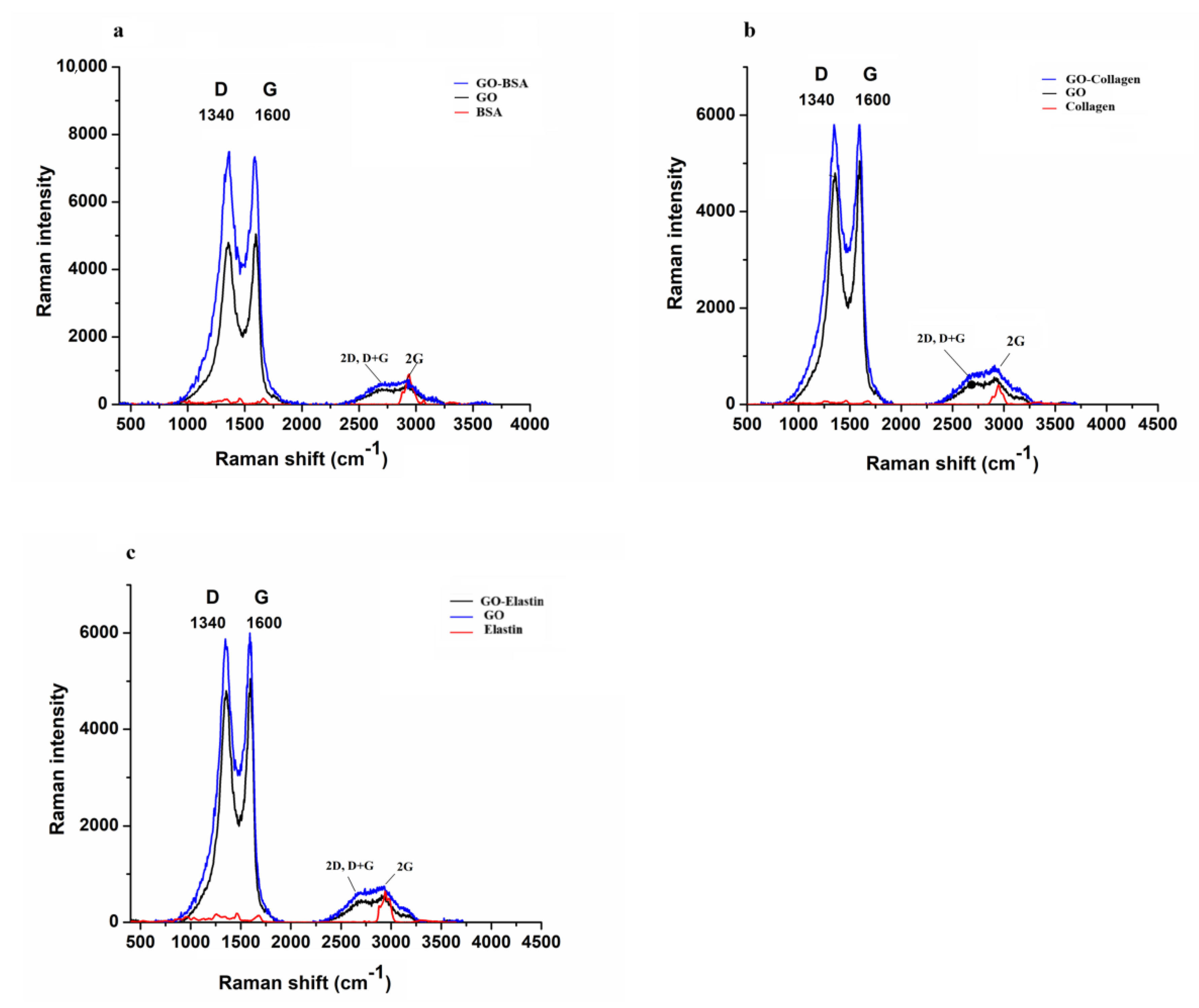
3.1. Raman Spectroscopy Shows the Functionalization of GO by BSA, Collagen and Elastin
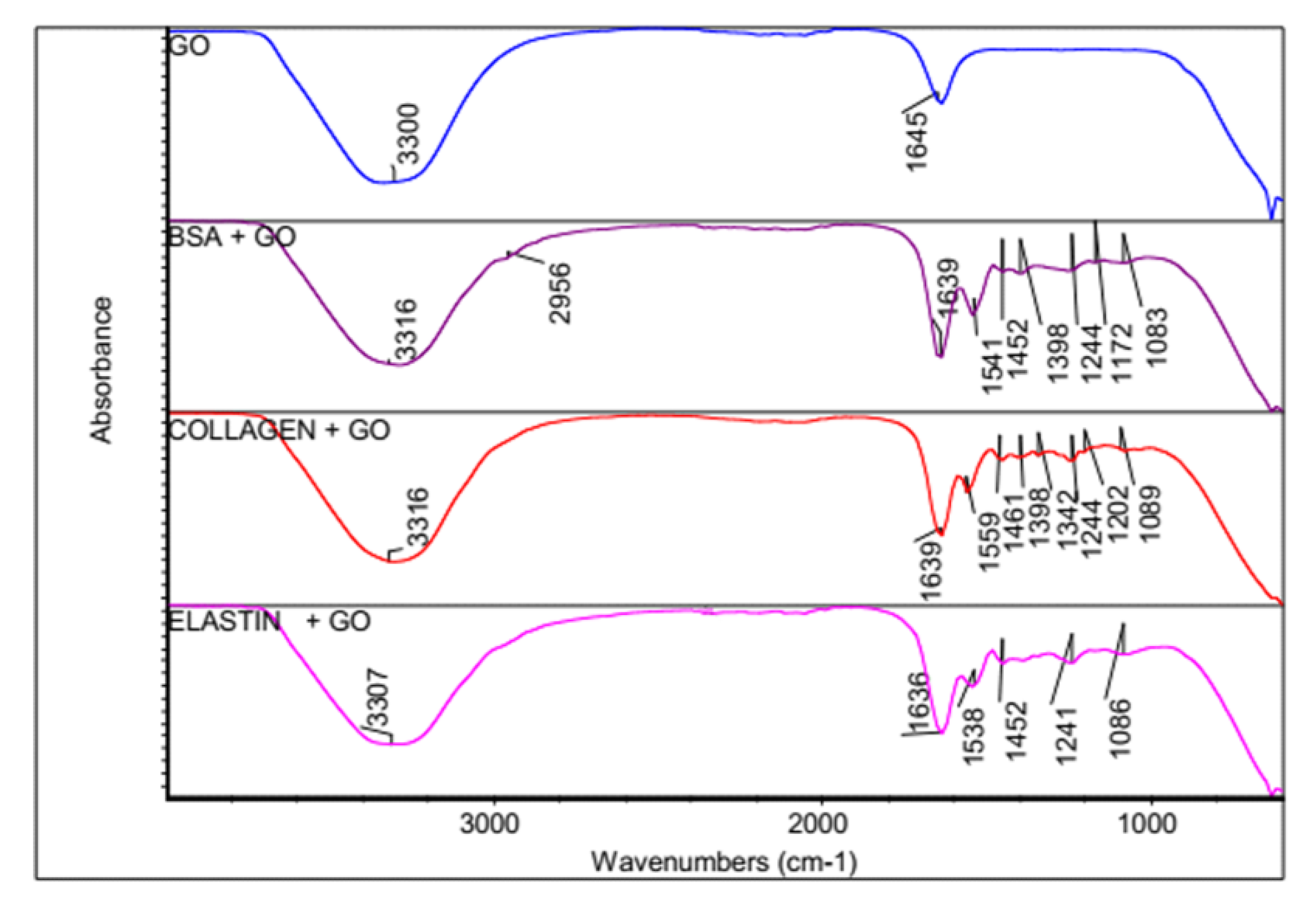
3.2. FTIR Spectroscopy Indicates a Formation of a Bio-Corona on the GO Surface
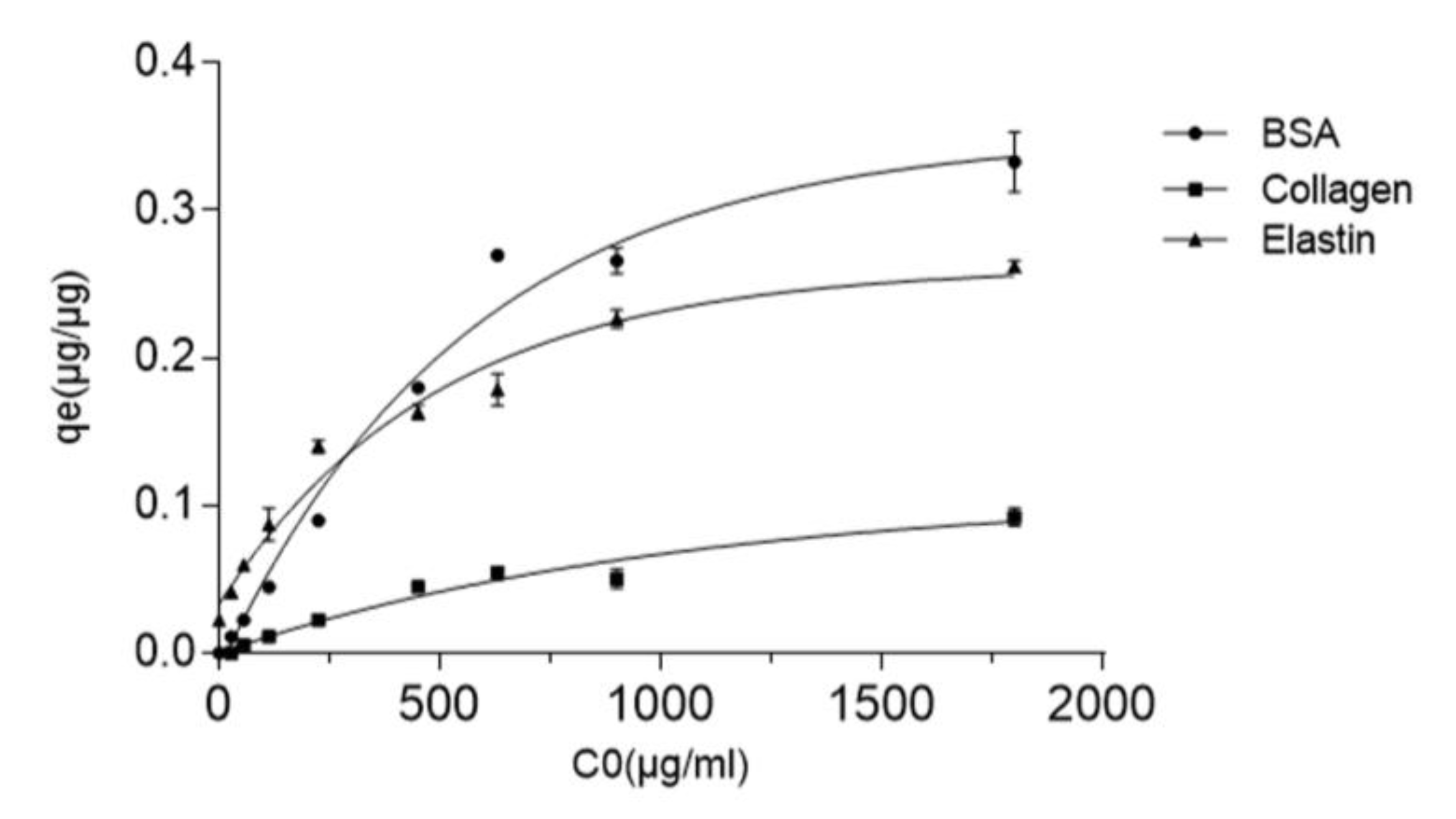
3.3. Protein-GO Adsorption Capacity Is Related to the Protein Molecular Weight
3.4. Proteins Are Adsorbed in GO Platelets as a Monolayer
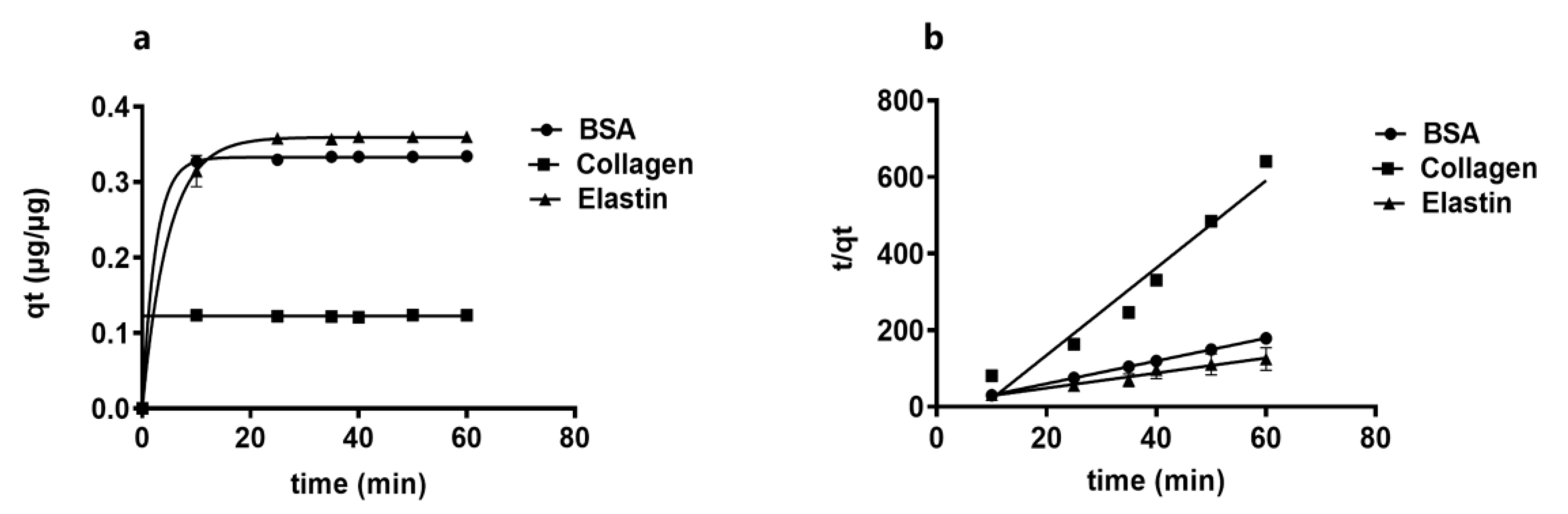
3.5. Kinetic Study of the Protein Adsorption into GO Shows that Lower Molecular Weight Proteins Are Adsorbed Faster
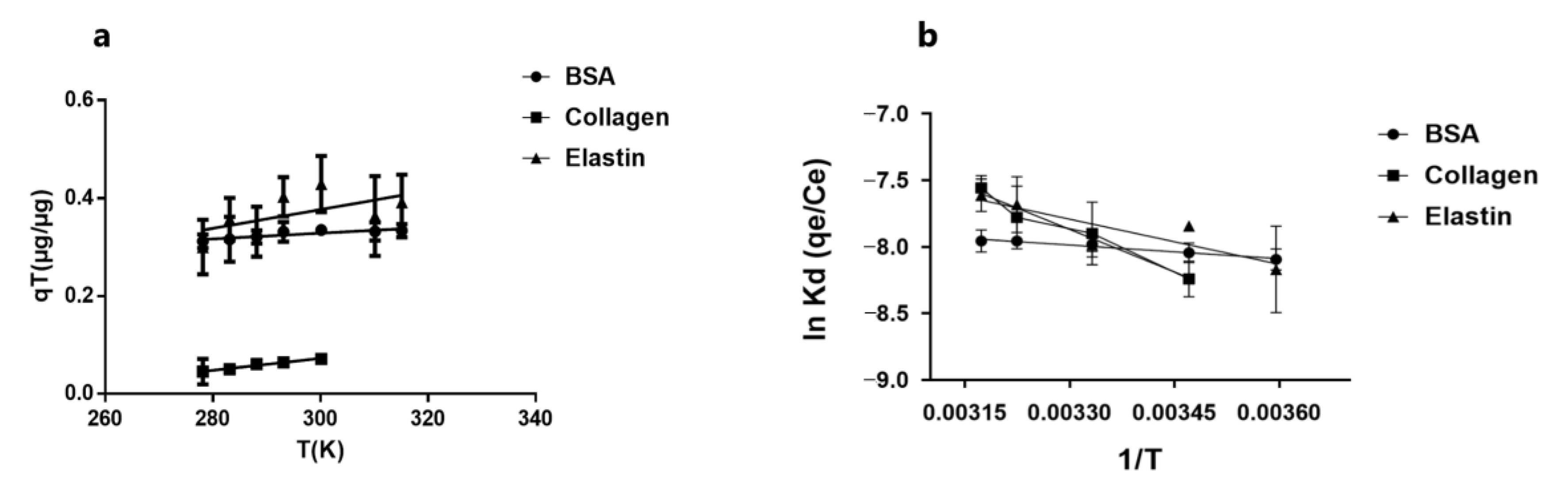
3.6. Protein Molecules Determine Thermodynamic Behavior in Their Adsorption by GO
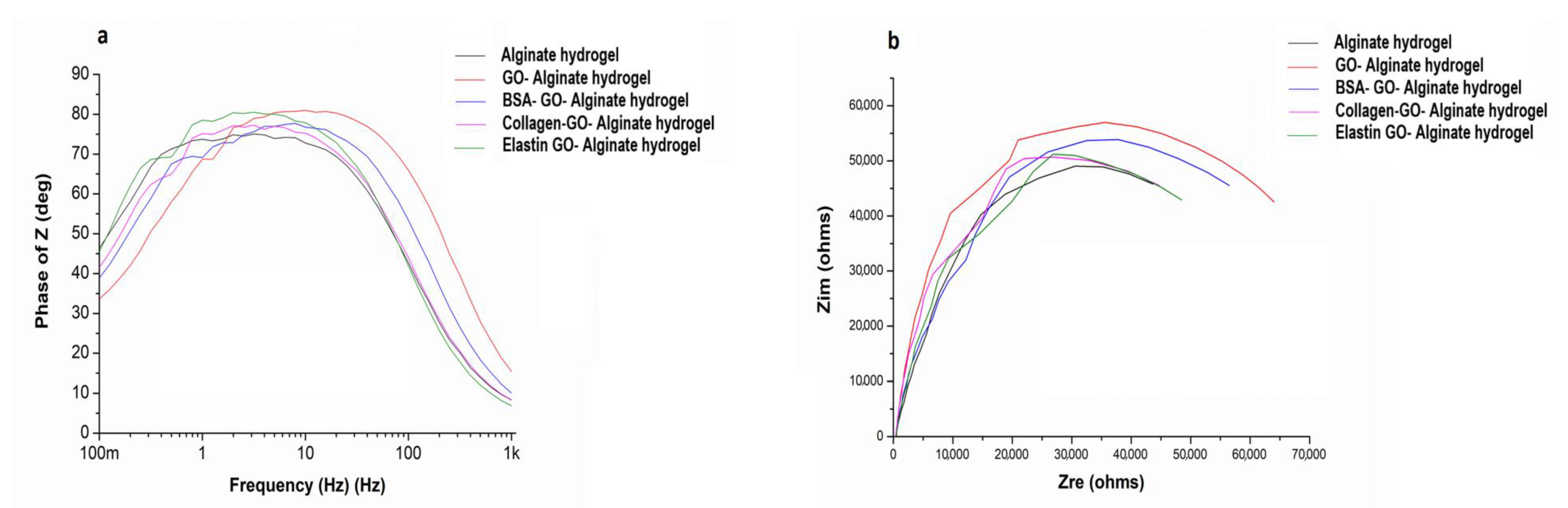
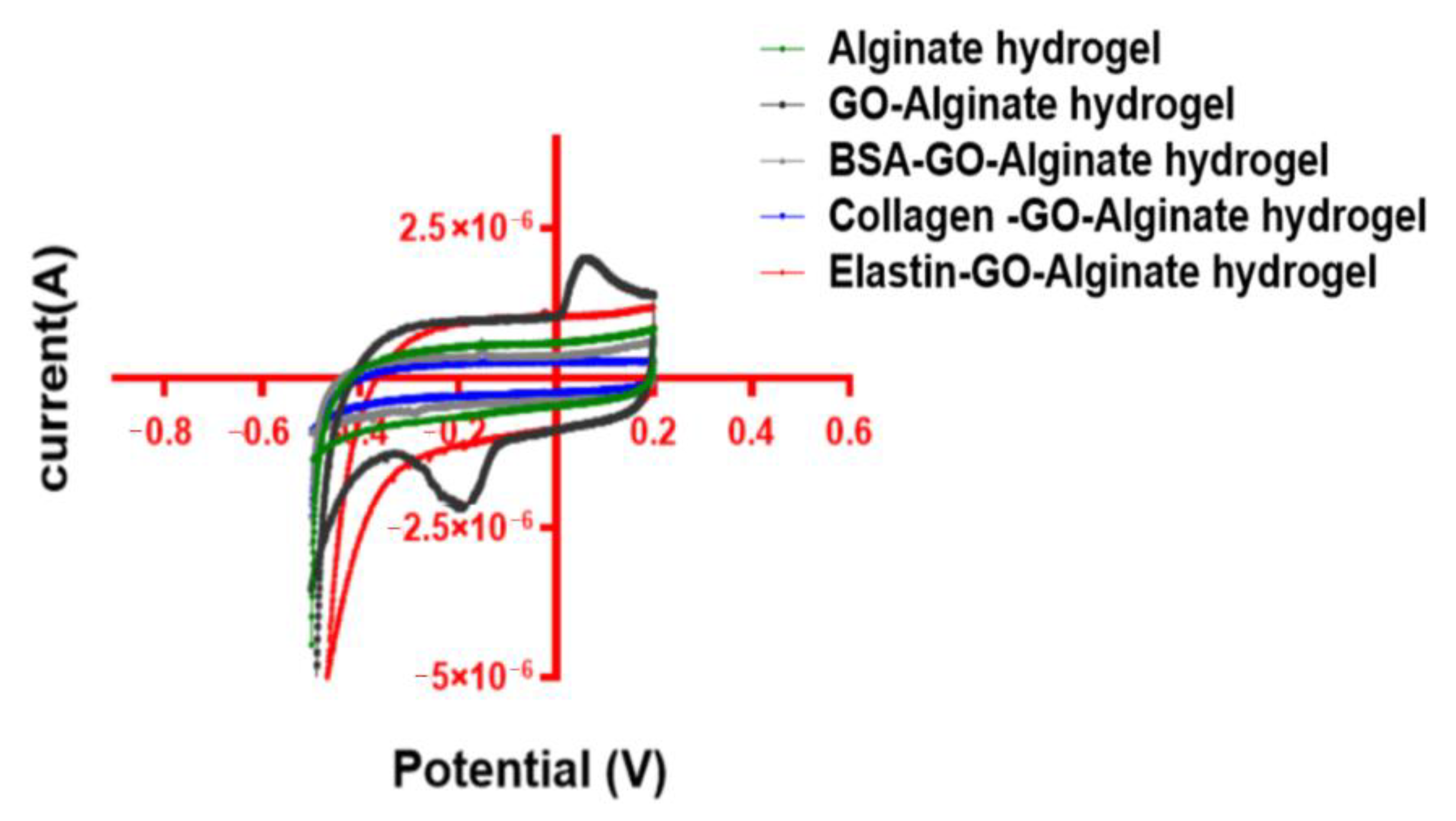
3.7. Conductivity Is Improved after Coating GO with Proteins within Alginate Matrixes
3.8. Capacitance Is Reduced after GO Protein Coating
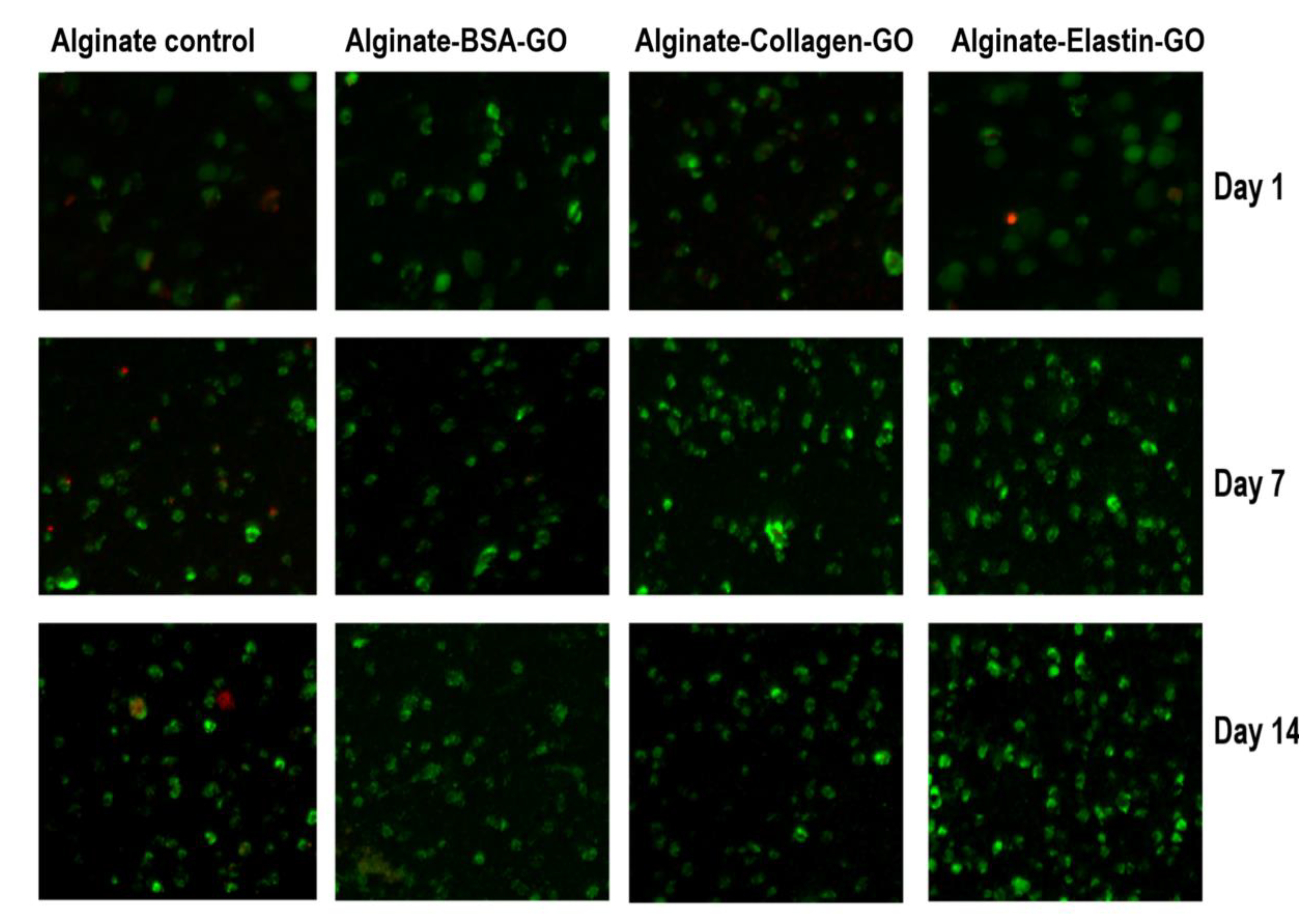
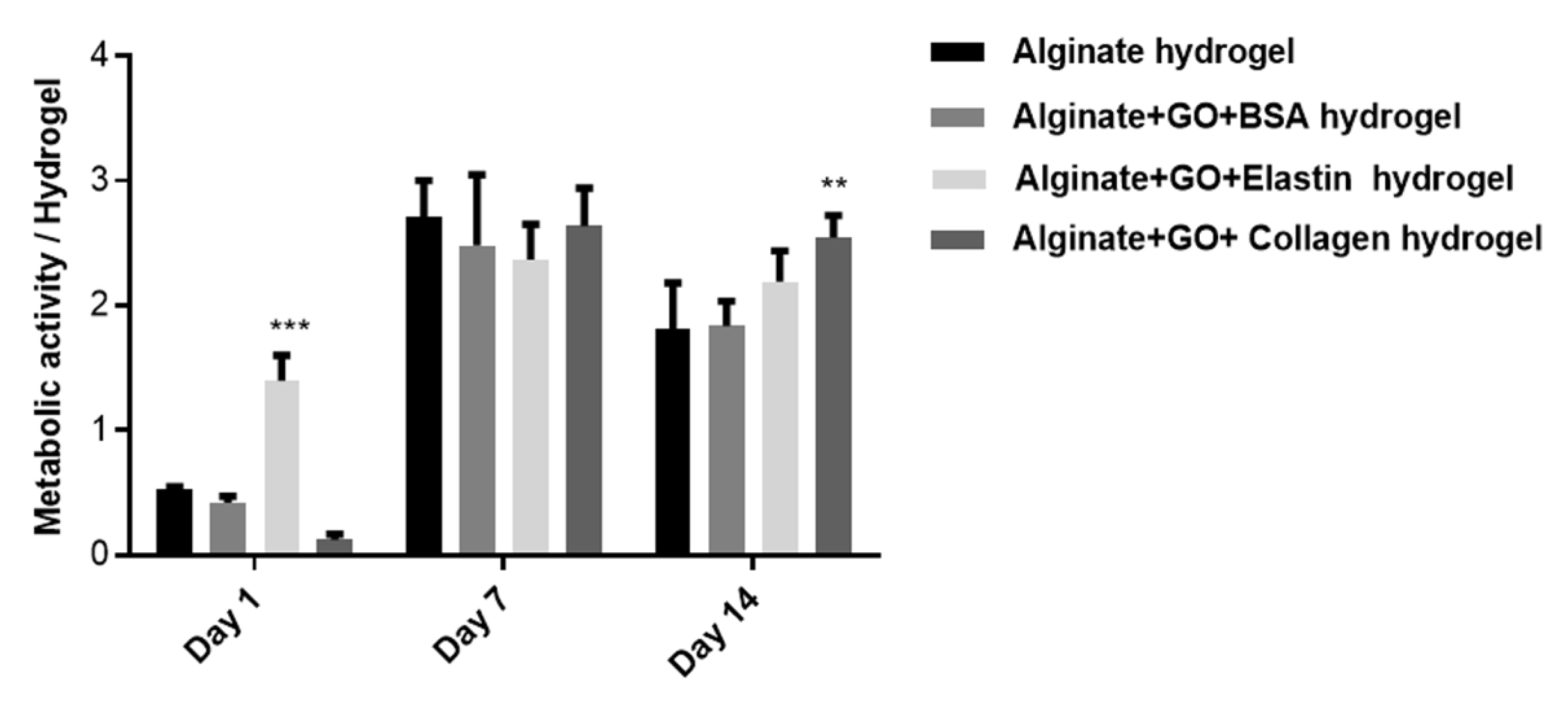
3.9. Collagen and Elastin Coated GO Improves Alginate Hydrogel-Embedded C2C12 Cell Viability
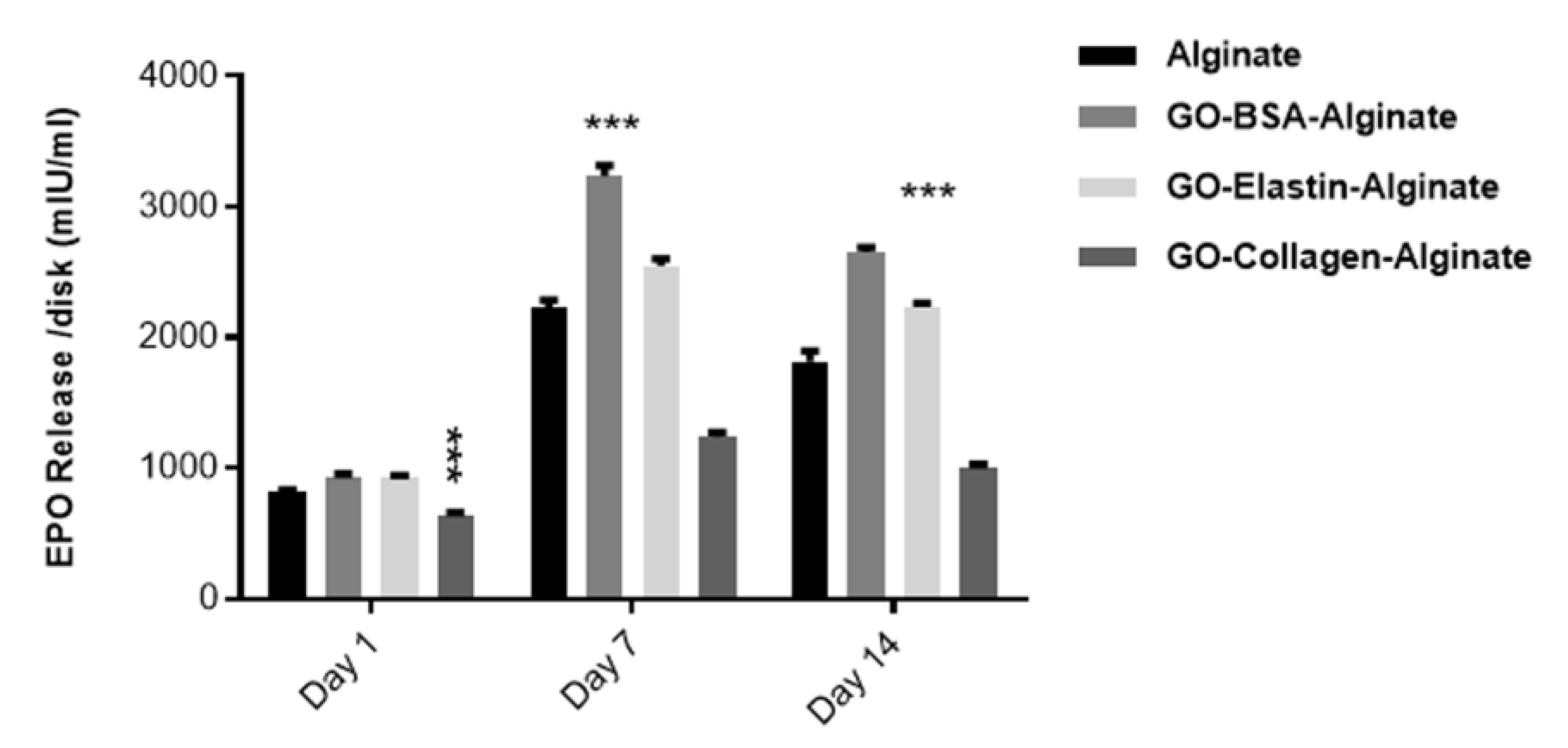
3.10. Protein Release by Embedded Cells Is Influenced by the Type of Protein-Coated GO
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Paredes Juárez, G.A.; Spasojevic, M.; Faas, M.M.; de Vos, P. Immunological and technical considerations in application of alginate-based microencapsulation systems. Front. Bioeng. Biotechnol. 2014, 2, 106–126. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.Y.; Mooney, D.J. Alginate: Properties and biomedical applications. Prog. Polym. Sci. 2012, 37, 106–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orive, G.; De Castro, M.; Kong, H.; Hernández, R.M.A.; Ponce, S.; Mooney, D.J.; Pedraz, J.L. Bioactive cell-hydrogel microcapsules for cell-based drug delivery. J. Control. Release 2009, 135, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Garate, A.; Ciriza, J.; Casado, J.G.; Blazquez, R.; Pedraz, J.L.; Orive, G.; Hernandez, R.M. Assessment of the Behavior of Mesenchymal Stem Cells Immobilized in Biomimetic Alginate Microcapsules. Mol. Pharm. 2015, 12, 3953–3962. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Yin, Z.; Wu, S.; Qi, X.; He, Q.; Zhang, Q.; Yan, Q.; Boey, F.; Zhang, H. Graphene-based materials: Synthesis, characterization, properties, and applications. Small 2011, 7, 1876–1902. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Fal’ko, V.I.; Colombo, L.; Gellert, P.R.; Schwab, M.G.; Kim, K. A roadmap for graphene. Nature 2012, 490, 192. [Google Scholar] [CrossRef]
- Kuila, T.; Bose, S.; Khanra, P.; Mishra, A.K.; Kim, N.H.; Lee, J.H. Recent advances in graphene-based biosensors. Biosens. Bioelectron. 2011, 26, 4637–4648. [Google Scholar] [CrossRef]
- Nguyen, D.A.; Lee, Y.R.; Raghu, A.V.; Jeong, H.M.; Shin, C.M.; Kim, B.K. Morphological and physical properties of a thermoplastic polyurethane reinforced with functionalized graphene sheet. Polym. Int. 2009, 58, 412–417. [Google Scholar] [CrossRef]
- Wang, L.; Fan, H.; Zhang, Z.; Lou, A.; Pei, G.; Jiang, S.; Mu, T.; Qin, J.; Chen, S.; Jin, D. Osteogenesis and angiogenesis of tissue-engineered bone constructed by prevascularized β-tricalcium phosphate scaffold and mesenchymal stem cells. Biomaterials 2010, 31, 9452–9461. [Google Scholar] [CrossRef]
- Ciriza, J.; Saenz del Burgo, L.; Virumbrales-Muñoz, M.; Ochoa, I.; Fernandez, L.J.; Orive, G.; Hernandez, R.M.; Pedraz, J.L. Graphene oxide increases the viability of C2C12 myoblasts microencapsulated in alginate. Int. J. Pharm. 2015, 493, 260–270. [Google Scholar] [CrossRef]
- Goenka, S.; Sant, V.; Sant, S. Graphene-based nanomaterials for drug delivery and tissue engineering. J. Control. Release 2014, 173, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Cui, L.; Losic, D. Graphene and graphene oxide as new nanocarriers for drug delivery applications. Acta Biomater. 2013, 9, 9243–9257. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Liu, Z.; Welsher, K.; Robinson, J.; Goodwin, A.; Zaric, S.; Dai, H. Nano-graphene oxide for cellular imaging and drug delivery. Nano Res. 2008, 1, 203–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zeng, X.; Ren, T.; van der Heide, E. The preparation of graphene oxide and its derivatives and their application in bio-tribological systems. Lubricants 2014, 2, 137–161. [Google Scholar] [CrossRef] [Green Version]
- Bradder, P.; Ling, S.K.; Wang, S.; Liu, S. Dye adsorption on layered graphite oxide. J. Chem. Eng. Data 2011, 56, 138–141. [Google Scholar] [CrossRef]
- Suk, J.W.; Piner, R.D.; An, J.; Ruoff, R.S. Mechanical properties of monolayer graphene oxide. ACS Nano 2010, 4, 6557–6564. [Google Scholar] [CrossRef]
- Konkena, B.; Vasudevan, S. Understanding aqueous dispersibility of graphene oxide and reduced graphene oxide through pKa measurements. J. Phys. Chem. Lett. 2012, 3, 867. [Google Scholar] [CrossRef]
- Park, S.; An, J.; Jung, I.; Piner, R.D.; An, S.J.; Li, X.; Velamakanni, A.; Ruoff, R.S. Colloidal suspensions of highly reduced graphene oxide in a wide variety of organic solvents. Nano Lett. 2009, 9, 1593–1597. [Google Scholar] [CrossRef]
- Anonymous. Graphene Oxide: Preparation, Functionalization, and Electrochemical Applications. Available online: https://pubs.acs.org/doi/ipdf/10.1021/cr300115g (accessed on 7 September 2018).
- Park, S.; Ruoff, R.S. Chemical methods for the production of graphenes. Nat. Nanotechnol. 2009, 4, 217–224. [Google Scholar] [CrossRef]
- Zhang, L.; Lu, Z.; Zhao, Q.; Huang, J.; Shen, H.; Zhang, Z. Enhanced chemotherapy efficacy by sequential delivery of siRNA and anticancer drugs using PEI-grafted graphene oxide. Small 2011, 7, 460–464. [Google Scholar] [CrossRef]
- Liu, Z.; Robinson, J.T.; Sun, X.; Dai, H. PEGylated nanographene oxide for delivery of water-insoluble cancer drugs. J. Am. Chem. Soc. 2008, 130, 10876–10877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, C.; Kim, Y.; Shin, D.; Ryoo, S.; Hong, B.H.; Min, D. Biomedical applications of graphene and graphene oxide. Acc. Chem. Res. 2013, 46, 2211–2224. [Google Scholar] [CrossRef]
- Simsikova, M.; Šikola, T. Interaction of graphene oxide with proteins and applications of their conjugates. J. Nanomed. Res. 2017, 5. [Google Scholar] [CrossRef]
- Emadi, F.; Amini, A.; Gholami, A.; Ghasemi, Y. Functionalized graphene oxide with chitosan for protein nanocarriers to protect against enzymatic cleavage and retain collagenase activity. Sci. Rep. 2017, 7, 42258. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.; Liu, Q.; Liu, J.; Zhang, H.; Li, Z.; Li, R.; Liu, L.; Wang, J. Bovine serum albumin-coated graphene oxide for effective adsorption of uranium(VI) from aqueous solutions. Ind. Eng. Chem. Res. 2017, 56, 3588–3598. [Google Scholar] [CrossRef]
- Hassan, M.; Walter, M.; Moseler, M. Interactions of polymers with reduced graphene oxide: Van der Waals binding energies of benzene on graphene with defects. Phys. Chem. Chem. Phys. 2014, 16, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Wang, X.; Ai, Y.; Liang, Y.; Ji, Y.; Li, J.; Hayat, T.; Alsaedi, A.; Wang, X. Spectroscopic and theoretical studies on the counterion effect of Cu(ii) ion and graphene oxide interaction with titanium dioxide. Environ. Sci. Nano 2016, 3, 1361–1368. [Google Scholar] [CrossRef]
- Xu, X.; Mao, X.; Wang, Y.; Li, D.; Du, Z.; Wu, W.; Jiang, L.; Yang, J.; Li, J. Study on the interaction of graphene oxide-silver nanocomposites with bovine serum albumin and the formation of nanoparticle-protein corona. Int. J. Biol. Macromol. 2018, 116, 492–501. [Google Scholar] [CrossRef]
- Lv, M.; Yan, L.; Liu, C.; Su, C.; Zhou, Q.; Zhang, X.; Lan, Y.; Zheng, Y.; Lai, L.; Liu, X.; et al. Non-covalent functionalized graphene oxide (GO) adsorbent with an organic gelator for co-adsorption of dye, endocrine-disruptor, pharmaceutical and metal ion. Chem. Eng. J. 2018, 349, 791–799. [Google Scholar] [CrossRef]
- Kuchlyan, J.; Kundu, N.; Banik, D.; Roy, A.; Sarkar, N. Spectroscopy and fluorescence lifetime imaging microscopy to probe the interaction of bovine serum albumin with graphene oxide. Langmuir ACS J. Surf. Colloids 2015, 31, 13793–13801. [Google Scholar] [CrossRef]
- Šimšíková, M. Interaction of graphene oxide with albumins: Effect of size, pH, and temperature. Arch. Biochem. Biophys. 2016, 593, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Cao, Z. Adsorption of nitrogen oxides on graphene and graphene oxides: Insights from density functional calculations. J. Chem. Phys. 2011, 134, 044710. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, S.F.; Bisker, G.; Bakh, N.A.; Gibbs, S.L.; Landry, M.P.; Strano, M.S. Protein functionalized carbon nanomaterials for biomedical applications. Carbon 2015, 95, 767–779. [Google Scholar] [CrossRef] [Green Version]
- Carrier-bound Immobilized Enzymes: Principles, Application and Design. Available online: https://www.wiley.com/enus/Carrier+bound+Immobilized+Enzymes%3A+Principles%2C+Application+and+Design-p-9783527312320 (accessed on 26 March 2019).
- Feng, L.; Liu, Z. Graphene in biomedicine: Opportunities and challenges. Nanomedicine (London) 2011, 6, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Zhao, F.; Li, S.; Hu, Z.; Zhao, Y. Low-toxic and safe nanomaterials by surface-chemical design, carbon nanotubes, fullerenes, metallofullerenes, and graphenes. Nanoscale 2011, 3, 362–382. [Google Scholar] [CrossRef]
- Sasidharan, A.; Panchakarla, L.S.; Sadanandan, A.R.; Ashokan, A.; Chandran, P.; Girish, C.M.; Menon, D.; Nair, S.V.; Rao, C.N.R.; Koyakutty, M. Hemocompatibility and macrophage response of pristine and functionalized graphene. Small 2012, 8, 1251–1263. [Google Scholar] [CrossRef]
- Yue, H.; Wei, W.; Yue, Z.; Wang, B.; Luo, N.; Gao, Y.; Ma, D.; Ma, G.; Su, Z. The role of the lateral dimension of graphene oxide in the regulation of cellular responses. Biomaterials 2012, 33, 4013–4021. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Singh, M.K.; Kulkarni, P.P.; Sonkar, V.K.; Grácio, J.J.A.; Dash, D. Amine-modified graphene: Thrombo-protective safer alternative to graphene oxide for biomedical applications. ACS Nano 2012, 6, 2731–2740. [Google Scholar] [CrossRef]
- Hu, W.; Peng, C.; Luo, W.; Lv, M.; Li, X.; Li, D.; Huang, Q.; Fan, C. Graphene-based antibacterial paper. ACS Nano 2010, 4, 4317–4323. [Google Scholar] [CrossRef]
- Chang, Y.; Yang, S.; Liu, J.; Dong, E.; Wang, Y.; Cao, A.; Liu, Y.; Wang, H. In vitro toxicity evaluation of graphene oxide on A549 cells. Toxicol. Lett. 2011, 200, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ali, S.F.; Dervishi, E.; Xu, Y.; Li, Z.; Casciano, D.; Biris, A.S. Cytotoxicity effects of graphene and single-wall carbon nanotubes in neural phaeochromocytoma-derived PC12 cells. ACS Nano 2010, 4, 3181–3186. [Google Scholar] [CrossRef] [PubMed]
- Srikanth, K.; Sundar, L.S.; Pereira, E.; Duarte, A.C. Graphene oxide induces cytotoxicity and oxidative stress in bluegill sunfish cells. J. Appl. Toxicol. 2018, 38, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Raslan, A.; Saenz del Burgo, L.; Ciriza, J.; Luis Pedraz, J. Graphene oxide and reduced graphene oxide-based scaffolds in regenerative medicine. Int. J. Pharm. 2020, 580, 119–226. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Ruan, J.; Song, H.; Zhang, J.; Wo, Y.; Guo, S.; Cui, D. Biocompatibility of graphene oxide. Nanoscale Res. Lett. 2011, 6, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Ciriza, J.; Saenz Del Burgo, L.; Gurruchaga, H.; Borras, F.E.; Franquesa, M.; Orive, G.; Hernández, R.M.; Pedraz, J.L. Graphene oxide enhances alginate encapsulated cells viability and functionality while not affecting the foreign body response. Drug Deliv. 2018, 25, 1147–1160. [Google Scholar] [CrossRef] [Green Version]
- Saenz del Burgo, L.; Ciriza, J.; Acarregui, A.; Gurruchaga, H.; Blanco, F.J.; Orive, G.; Hernández, R.M.; Pedraz, J.L. Hybrid alginate–protein-coated graphene oxide microcapsules enhance the functionality of erythropoietin secreting C2C12 myoblasts. Mol. Pharm. 2017, 14, 885–898. [Google Scholar] [CrossRef]
- Simonin, J. On the comparison of pseudo-first order and pseudo-second order rate laws in the modeling of adsorption kinetics. Chem. Eng. J. 2016, 300, 254–263. [Google Scholar] [CrossRef] [Green Version]
- Fierro, V.; Torné-Fernández, V.; Montané, D.; Celzard, A. Adsorption of phenol onto activated carbons having different textural and surface properties. Microporous Mesoporous Mater. 2008, 111, 276–284. [Google Scholar] [CrossRef]
- Nuengmatcha, P.; Mahachai, R.; Chanthai, S. Adsorption capacity of the as-synthetic graphene oxide for the removal of alizarin red S dye from aqueous solution. Orient. J. Chem. 2016, 32, 1399–1410. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, M.A.; Ahmad Puad, N.A.; Bello, O.S. Kinetic, equilibrium and thermodynamic studies of synthetic dye removal using pomegranate peel activated carbon prepared by microwave-induced KOH activation. Water Resour. Ind. 2014, 6, 18–35. [Google Scholar] [CrossRef] [Green Version]
- Hong, F.; Yue, B.; Hirao, N.; Liu, Z.; Chen, B. Significant improvement in Mn2O3 transition metal oxide electrical conductivity via high pressure. Sci. Rep. 2017, 7, 44078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Li, D.; Mu, L. Biotransformation of graphene oxide nanosheets in blood plasma affects their interactions with cells. Environ. Sci. Nano 2017, 4, 1569–1578. [Google Scholar] [CrossRef]
- Peng, B.; Chen, L.; Que, C.; Yang, K.; Deng, F.; Deng, X.; Shi, G.; Xu, G.; Wu, M. Adsorption of antibiotics on graphene and biochar in aqueous solutions induced by π-π interactions. Sci. Rep. 2016, 6, 31920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, X.; Wei, Y.; Chen, S.; Wang, P.; Liu, L. Bio-based graphene/sodium alginate aerogels for strain sensor. RSC Adv. 2016, 6, 64056–64064. [Google Scholar] [CrossRef]
- Sarfert, F.T.; Etzel, M.R. Mass transfer limitations in protein separations using ion-exchange membranes. J. Chromatogr. A 1997, 764, 3–20. [Google Scholar] [CrossRef]
- Manzi, B.M.; Werner, M.; Ivanova, E.P.; Crawford, R.J.; Baulin, V.A. Simulations of protein adsorption on nanostructured surfaces. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef]
- Foo, K.Y.; Hameed, B.H. Insights into the modeling of adsorption isotherm systems. Chem. Eng. J. 2010, 156, 2–10. [Google Scholar] [CrossRef]
- Chen, P.; Li, H.; Song, S.; Weng, X.; He, D.; Zhao, Y. Adsorption of dodecylamine hydrochloride on graphene oxide in water. Results Phys. 2017, 7, 2281–2288. [Google Scholar] [CrossRef]
- Wu, F.; Tseng, R.; Juang, R. Initial behavior of intraparticle diffusion model used in the description of adsorption kinetics. Chem. Eng. J. 2009, 153, 1–8. [Google Scholar] [CrossRef]
- Vijayakumar, G.; Tamilarasan, R.; Dharmendirakumar, M. Adsorption, Kinetic, Equilibrium and Thermodynamic studies on the removal of basic dye Rhodamine-B from aqueous solution by the use of natural adsorbent perlite. J. Mater. Environ. Sci. 2012, 3, 157–170. [Google Scholar]
- Dreyer, D.R.; Park, S.; Bielawski, C.W.; Ruoff, R.S. The chemistry of graphene oxide. Chem. Soc. Rev. 2009, 39, 228–240. [Google Scholar] [CrossRef]
- Mohan Kumar, T.M.; Praveen, D. Impedance analysis of sodium alginate: Graphene oxide composite. In IOP Conference Series: Materials Science and Engineering; IOP Science: Bengaluru, India, 2018; Volume 310, p. 12150. [Google Scholar] [CrossRef]
- Kostarelos, K.; Vincent, M.; Hebert, C.; Garrido, J.A. Graphene in the design and engineering of next-generation neural interfaces. Adv. Mater. Weinheim. 2017, 29. [Google Scholar] [CrossRef]
- Reina, G.; González-Domínguez, J.M.; Criado, A.; Vázquez, E.; Bianco, A.; Prato, M. Promises, facts and challenges for graphene in biomedical applications. Chem. Soc. Rev. 2017, 46, 4400–4416. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.R.; Li, Y.; Jang, H.L.; Khoshakhlagh, P.; Akbari, M.; Nasajpour, A.; Zhang, Y.S.; Tamayol, A.; Khademhosseini, A. Graphene-based materials for tissue engineering. Adv. Drug Deliv. Rev. 2016, 105, 255–274. [Google Scholar] [CrossRef] [Green Version]
- Qhatani, M.; Fadl-allah, S.; El-Shenawy, N. Electrochemical impedance spectroscopy study of the adsorption behavior of bovine serum albumin at biomimetic calcium–phosphate coating. Int. J. Electrochem. Sci. 2012, 7, 4510–4527. [Google Scholar]
- Ero-Tolliver, I.A.; Hudson, B.G.; Bhave, G. The ancient immunoglobulin domains of peroxidasin are required to form sulfilimine cross-links in collagen IV. J. Biol. Chem. 2015, 290, 21741–21748. [Google Scholar] [CrossRef] [Green Version]
- Bhave, G.; Cummings, C.F.; Vanacore, R.M.; Kumagai-Cresse, C.; Ero-Tolliver, I.A.; Rafi, M.; Kang, J.; Pedchenko, V.; Fessler, L.I.; Fessler, J.H.; et al. Peroxidasin forms sulfilimine chemical bonds using hypohalous acids in tissue genesis. Nat. Chem. Biol. 2012, 8, 784–790. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Xu, H.H.K. The fast release of stem cells from alginate-fibrin microbeads in injectable scaffolds for bone tissue engineering. Biomaterials 2011, 32, 7503–7513. [Google Scholar] [CrossRef] [Green Version]
- Hernández, R.M.A.; Orive, G.; Murua, A.; Pedraz, J.L. Microcapsules and microcarriers for in situ cell delivery. Adv. Drug Deliv. Rev. 2010, 62, 711–730. [Google Scholar] [CrossRef]
- Toyoda, T.; Arakawa, T.; Yamaguchi, H. N-Glycans stabilize human erythropoietin through hydrophobic interactions with the hydrophobic protein surface: Studies by surface plasmon resonance analysis. J. Biochem. 2002, 131, 511–515. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Wavenumbers (cm−1) at 532 nm Excitation Wavelength | GO | GO + Protein |
|---|---|---|
| (I) | 1245 | 1230–1250 |
| (D) | 1354 | 1352–1354 |
| (G1) | 1569 | 1530–1570 |
| (G2) | 1603 | 1595–1605 |
| Band Intensity Ratio | GO | GO + Protein |
| I/D | 0.25 | 0.45–0.48 |
| D/(G1 + G2) | 1.3 | 1.1–1.5 |
| (D + I)/(G1 + G2) | 1.7 | 1.6–2.2 |
| Langmuir Model | Freundlich Model | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| qe (µg/µg) | qmax (µg/µg) | KL (mL/µg) | RL | R2 | 1/n | N | KF | R2 | |
| BSA | 0.332 | 0.330 | 0.057 | 0.009 | 0.99 | 0.050 | 22.030 | 0.225 | 0.750 |
| Elastin | 0.262 | 0.380 | 0.002 | 0.249 | 0.97 | 0.380 | 2.600 | 0.014 | 0.950 |
| Collagen | 0.122 | 0.071 | 0.023 | 0.044 | 0.97 | 0.200 | 4.780 | 0.019 | 0.700 |
| qe (µg/µg) | K2(µg/µg.min) | R2 | qt (µg/µg) | |
|---|---|---|---|---|
| BSA | 0.336 | 7.220 | 0.990 | 0.332 |
| Collagen | 0.362 | 12.860 | 0.990 | 0.123 |
| Elastin | 0.125 | 4.158 | 0.990 | 0.352 |
| Kp (µg/µg.min1/2) | C | R2 | |
|---|---|---|---|
| BSA | 0.00014 | 0.163 | 0.91 |
| Collagen | 1.15 × 10−5 | 0.060 | 0.37 |
| Elastin | 0.00078 | 0.161 | 0.70 |
| ΔH° (kJ/mol) | ΔS° (kJ/mol.K) | ΔG° (kJ/mol) | R2 | |
|---|---|---|---|---|
| BSA | 2.598 | −0.057887 | 20.543 | 0.981 |
| Elastin | 16.270 | −0.008881 | 19.031 | 0.779 |
| Collagen | 17.363 | −0.006935 | 19.513 | 0.935 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raslan, A.; Saenz del Burgo, L.; Espona-Noguera, A.; Ochoa de Retana, A.M.; Sanjuán, M.L.; Cañibano-Hernández, A.; Gálvez-Martín, P.; Ciriza, J.; Pedraz, J.L. BSA- and Elastin-Coated GO, but Not Collagen-Coated GO, Enhance the Biological Performance of Alginate Hydrogels. Pharmaceutics 2020, 12, 543. https://doi.org/10.3390/pharmaceutics12060543
Raslan A, Saenz del Burgo L, Espona-Noguera A, Ochoa de Retana AM, Sanjuán ML, Cañibano-Hernández A, Gálvez-Martín P, Ciriza J, Pedraz JL. BSA- and Elastin-Coated GO, but Not Collagen-Coated GO, Enhance the Biological Performance of Alginate Hydrogels. Pharmaceutics. 2020; 12(6):543. https://doi.org/10.3390/pharmaceutics12060543
Chicago/Turabian StyleRaslan, Ahmed, Laura Saenz del Burgo, Albert Espona-Noguera, Ana María Ochoa de Retana, María Luisa Sanjuán, Alberto Cañibano-Hernández, Patricia Gálvez-Martín, Jesús Ciriza, and Jose Luis Pedraz. 2020. "BSA- and Elastin-Coated GO, but Not Collagen-Coated GO, Enhance the Biological Performance of Alginate Hydrogels" Pharmaceutics 12, no. 6: 543. https://doi.org/10.3390/pharmaceutics12060543