
Metabolomics and EMT Markers of Breast Cancer: A Crosstalk and Future Perspective

 , ,
, ,
Abstract
:
1. Introduction
2. Status of Metastasis with Breast Cancer Subtypes, Metabolic Rewiring, EMT and MET—An Insight
2.1. Breast Cancer Subtypes
2.2. Metabolic Rewiring, EMT, MET and Their Impact on Breast Cancer Progression
3. Metabolomics of Breast Cancer in Driving the EMT Marker Expression and Their Blockers
3.1. Glucose Metabolism
3.2. Lipid Metabolism
3.3. Amino Acid Metabolism
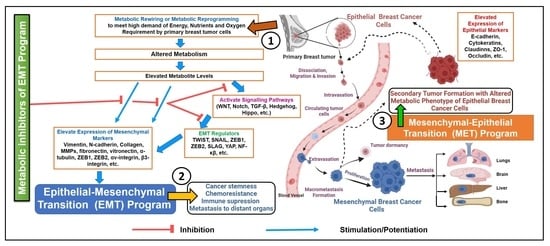
4. Metabolic Inhibitors of EMT Program
EMT-Metabolic Inhibitors at Clinical Levels
5. Conclusions and Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Vargo-Gogola, T.; Rosen, J.M. Modelling breast cancer: One size does not fit all. Nat. Cancer 2007, 7, 659–672. [Google Scholar] [CrossRef] [PubMed]
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buijs, J.T.; van der Pluijm, G. Osteotropic cancers: From primary tumor to bone. Cancer Lett. 2009, 273, 177–193. [Google Scholar] [CrossRef] [PubMed]
- Sporn, M.B. The war on cancer. Lancet 1996, 347, 1377–1381. [Google Scholar] [CrossRef]
- Eckhardt, B.L.; Francis, P.A.; Parker, B.S.; Anderson, R.L. Strategies for the discovery and development of therapies for metastatic breast cancer. Nat. Rev. Drug Discov. 2012, 11, 479–497. [Google Scholar] [CrossRef]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Sørlie, T.; Tibshirani, R.; Parker, J.; Hastie, T.; Marron, J.S.; Nobel, A.; Deng, S.; Johnsen, H.; Pesich, R.; Geisler, S.; et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl. Acad. Sci. USA 2003, 100, 8418–8423. [Google Scholar] [CrossRef] [Green Version]
- Sotiriou, C.; Neo, S.-Y.; McShane, L.M.; Korn, E.L.; Long, P.M.; Jazaeri, A.; Martiat, P.; Fox, S.B.; Harris, A.L.; Liu, E.T. Breast cancer classification and prognosis based on gene expression profiles from a population-based study. Proc. Natl. Acad. Sci. USA 2003, 100, 10393–10398. [Google Scholar] [CrossRef] [Green Version]
- Kast, K.; Link, T.; Friedrich, K.; Petzold, A.; Niedostatek, A.; Schoffer, O.; Werner, C.; Klug, S.J.; Werner, A.; Gatzweiler, A.; et al. Impact of breast cancer subtypes and patterns of metastasis on outcome. Breast Cancer Res. Treat. 2015, 150, 621–629. [Google Scholar] [CrossRef]
- Sethi, S.; Sarkar, F.H.; Ahmed, Q.; Bandyopadhyay, S.; Nahleh, Z.A.; Semaan, A.; Sakr, W.; Munkarah, A.; Ali-Fehmi, R. Molecular markers of epithelial-to-mesenchymal transition are associated with tumor aggressiveness in breast carcinoma. Transl. Oncol. 2011, 4, 222–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blick, T.; Widodo, E.; Hugo, H.; Waltham, M.; Lenburg, M.E.; Neve, R.M.; Thompson, E.W. Epithelial mesenchymal transition traits in human breast cancer cell lines. Clin. Exp. Metastasis 2008, 25, 629–642. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.L.; Cobleigh, M.A.; Tripathy, D.; Gutheil, J.C.; Harris, L.N.; Fehrenbacher, L.; Slamon, D.J.; Murphy, M.; Novotny, W.F.; Burchmore, M.; et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J. Clin. Oncol. 2002, 20, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Montagna, E.; Cancello, G.; D’Agostino, D.; Lauria, R.; Forestieri, V.; Esposito, A.; Silvestro, L.; Accurso, A.; De Placido, S.; De Laurentiis, M. Central nervous system metastases in a cohort of metastatic breast cancer patients treated with trastuzumab. Cancer Chemother. Pharmacol. 2008, 63, 275–280. [Google Scholar] [CrossRef]
- Clayton, A.J.; Danson, S.; Jolly, S.; Ryder, W.D.J.; Burt, P.A.; Stewart, A.L.; Wilkinson, P.M.; Welch, R.S.; Magee, B.; Wilson, G.; et al. Incidence of cerebral metastases in patients treated with trastuzumab for metastatic breast cancer. Br. J. Cancer 2004, 91, 639–643. [Google Scholar] [CrossRef]
- Chang, J.C.; Wooten, E.C.; Tsimelzon, A.; Hilsenbeck, S.G.; Gutierrez, M.C.; Tham, Y.-L.; Kalidas, M.; Elledge, R.; Mohsin, S.; Osborne, C.K.; et al. Patterns of Resistance and Incomplete Response to Docetaxel by Gene Expression Profiling in Breast Cancer Patients. J. Clin. Oncol. 2005, 23, 1169–1177. [Google Scholar] [CrossRef]
- Li, X.; Lewis, M.T.; Huang, J.; Gutierrez, C.; Osborne, C.K.; Wu, M.-F.; Hilsenbeck, S.G.; Pavlick, A.; Zhang, X.; Chamness, G.C.; et al. Intrinsic Resistance of Tumorigenic Breast Cancer Cells to Chemotherapy. JNCI J. Natl. Cancer Inst. 2008, 100, 672–679. [Google Scholar] [CrossRef]
- Eroles, P.; Bosch, A.; Pérez-Fidalgo, J.A.; Lluch, A. Molecular biology in breast cancer: Intrinsic subtypes and signaling pathways. Cancer Treat. Rev. 2012, 38, 698–707. [Google Scholar] [CrossRef]
- Kennecke, H.; Yerushalmi, R.; Woods, R.; Cheang, M.C.U.; Voduc, D.; Speers, C.H.; Nielsen, T.O.; Gelmon, K. Metastatic Behavior of Breast Cancer Subtypes. J. Clin. Oncol. 2010, 28, 3271–3277. [Google Scholar] [CrossRef]
- Thiery, J.P. Epithelial–mesenchymal transitions in tumour progression. Nat. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Velasco-Velázquez, M.A.; Popov, V.M.; Lisanti, M.P.; Pestell, R.G. The Role of Breast Cancer Stem Cells in Metastasis and Therapeutic Implications. Am. J. Pathol. 2011, 179, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Creighton, C.J.; Li, X.; Landis, M.; Dixon, J.M.; Neumeister, V.M.; Sjolund, A.; Rimm, D.L.; Wong, H.; Rodriguez, A.; Herschkowitz, J.I.; et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc. Natl. Acad. Sci. USA 2009, 106, 13820–13825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.Q.; Xu, J.D.; Wang, W.J.; Cao, X.X.; Chen, Q.; Tang, F.; Chen, Z.-Q.; Liu, X.-P.; Xu, Z.-D. Twist1-mediated adriamycin-induced epithelial-mesenchymal transition relates to multidrug resistance and invasive potential in breast cancer cells. Clin. Cancer Res. 2009, 15, 2657–2665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.M.; Dedhar, S.; Kalluri, R.; Thompson, E.W. The epithelial–mesenchymal transition: New insights in signaling, development, and disease. J. Cell Biol. 2006, 172, 973–981. [Google Scholar] [CrossRef]
- Nieto, M.A. Epithelial Plasticity: A Common Theme in Embryonic and Cancer Cells. Science 2013, 342, 1234850. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Zeisberg, M.; Neilson, E.G. Biomarkers for epithelial-mesenchymal transitions. J. Clin. Investig. 2009, 119, 1429–1437. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2010, 120, 1786. [Google Scholar] [CrossRef] [Green Version]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; El-Naggar, S.; Darling, D.S.; Higashi, Y.; Dean, D.C. Zeb1 links epithelial-mesenchymal transition and cellular senescence. Development 2008, 135, 579–588. [Google Scholar] [CrossRef] [Green Version]
- Nakaya, Y.; Sheng, G. EMT in developmental morphogenesis. Cancer Lett. 2013, 341, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Micalizzi, D.S.; Farabaugh, S.M.; Ford, H.L. Epithelial-Mesenchymal Transition in Cancer: Parallels Between Normal Development and Tumor Progression. J. Mammary Gland Biol. Neoplasia 2010, 15, 117–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ewald, A.J.; Brenot, A.; Duong, M.; Chan, B.S.; Werb, Z. Collective Epithelial Migration and Cell Rearrangements Drive Mammary Branching Morphogenesis. Dev. Cell 2008, 14, 570–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ewald, A.J.; Huebner, R.J.; Palsdottir, H.; Lee, J.K.; Perez, M.J.; Jorgens, D.M.; Tauscher, A.N.; Cheung, K.J.; Werb, Z.; Auer, M. Mammary collective cell migration involves transient loss of epithelial features and individual cell migration within the epithelium. J. Cell Sci. 2012, 125, 2638–2654. [Google Scholar] [PubMed] [Green Version]
- Foubert, E.; De Craene, B.; Berx, G. Key signalling nodes in mammary gland development and cancer. The Snail1-Twist1 conspiracy in malignant breast cancer progression. Breast Cancer Res. 2010, 12, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouros-Mehr, H.; Werb, Z. Candidate regulators of mammary branching morphogenesis identified by genome-wide transcript analysis. Dev. Dyn. 2006, 235, 3404–3412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosário, M.; Birchmeier, W. How to make tubes: Signaling by the Met receptor tyrosine kinase. Trends Cell Biol. 2003, 13, 328–335. [Google Scholar] [CrossRef]
- Chakrabarti, R.; Hwang, J.; Andres Blanco, M.; Wei, Y.; Lukačišin, M.; Romano, R.A.; Kang, Y. Elf5 inhibits the epithelial–mesenchymal transition in mammary gland development and breast cancer metastasis by transcriptionally repressing Snail2. Nat. Cell Biol. 2012, 14, 1212–1222. [Google Scholar] [CrossRef]
- Choi, Y.S.; Chakrabarti, R.; Escamilla-Hernandez, R.; Sinha, S. Elf5 conditional knockout mice reveal its role as a master regulator in mammary alveolar development: Failure of Stat5 activation and functional differentiation in the absence of Elf5. Dev. Biol. 2009, 329, 227–241. [Google Scholar] [CrossRef] [Green Version]
- Oakes, S.R.; Naylor, M.J.; Asselin-Labat, M.-L.; Blazek, K.D.; Gardiner-Garden, M.; Hilton, H.N.; Kazlauskas, M.; Pritchard, M.A.; Chodosh, L.A.; Pfeffer, P.L.; et al. The Ets transcription factor Elf5 specifies mammary alveolar cell fate. Genes Dev. 2008, 22, 581–586. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Weinberg, R.A. Epithelial–Mesenchymal Plasticity: A Central Regulator of Cancer Progression. Trends Cell Biol. 2015, 25, 675–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaffer, C.L.; San Juan, B.P.; Lim, E.; Weinberg, R.A. EMT, cell plasticity and metastasis. Cancer Metastasis Rev. 2016, 35, 645. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Parker, J.S.; Karginova, O.; Fan, C.; Livasy, C.; Herschkowitz, J.I.; He, X.; Perou, C.M. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010, 12, R68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Keckesova, Z.; Donaher, J.L.; Shibue, T.; Tischler, V.; Reinhardt, F.; Itzkovitz, S.; Noske, A.; Zürrer-Härdi, U.; Bell, G.; et al. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell 2012, 148, 1015–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, S.S.; Gonugunta, V.K.; Bandyopadhyay, A.; Rao, M.K.; Goodall, G.J.; Sun, L.; Vadlamudi, R.K. Significance of PELP1/HDAC2/miR-200 regulatory network in EMT and metastasis of breast cancer. Oncogene 2014, 33, 3707–3716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, H.D.; Luitel, K.; Kim, M.; Zhang, K.; Longmore, G.D.; Tran, D.D. Transient SNAIL1 Expression Is Necessary for Metastatic Competence in Breast Cancer. Cancer Res. 2014, 74, 6330–6340. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a Master Regulator of Morphogenesis, Plays an Essential Role in Tumor Metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Corsa, C.; Ponik, S.; Prior, J.L.; Piwnica-Worms, D.; Eliceiri, K.; Keely, P.J.; Longmore, G.D. The collagen receptor discoidin domain receptor 2 stabilizes SNAIL1 to facilitate breast cancer metastasis. Nat. Cell Biol. 2013, 15, 677–687. [Google Scholar] [CrossRef]
- Weinberg, R.A. Leaving Home Early: Reexamination of the Canonical Models of Tumor Progression. Cancer Cell 2008, 14, 283–284. [Google Scholar] [CrossRef] [Green Version]
- Chao, Y.L.; Shepard, C.R.; Wells, A. Breast carcinoma cells re-express E-cadherin during mesenchymal to epithelial reverting transition. Mol. Cancer 2010, 9, 179. [Google Scholar] [CrossRef] [Green Version]
- Chaffer, C.L.; Brennan, J.P.; Slavin, J.L.; Blick, T.; Thompson, E.W.; Williams, E. Mesenchymal-to-Epithelial Transition Facilitates Bladder Cancer Metastasis: Role of Fibroblast Growth Factor Receptor-2. Cancer Res. 2006, 66, 11271–11278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaffer, C.L.; Thompson, E.W.; Williams, E.D. Mesenchymal to epithelial transition in development and disease. Cells Tissues Organs 2007, 185, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Hugo, H.; Ackland, M.L.; Blick, T.; Lawrence, M.G.; Clements, J.A.; Williams, E.D.; Thompson, E.W. Epithelial—mesenchymal and mesenchymal—epithelial transitions in carcinoma progression. J. Cell. Physiol. 2007, 213, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Bernards, R.; Weinberg, R.A. Metastasis genes: A progression puzzle. Nature 2002, 418, 823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [Green Version]
- Korpal, M.; Ell, B.J.; Buffa, F.; Ibrahim, T.; Blanco, M.A.; Celià-Terrassa, T.; Mercatali, L.; Khan, Z.; Goodarzi, H.; Hua, Y.; et al. Direct targeting of Sec23a by miR-200s influences cancer cell secretome and promotes metastatic colonization. Nat. Med. 2011, 17, 1101–1108. [Google Scholar] [CrossRef] [Green Version]
- Hurteau, G.J.; Carlson, J.A.; Spivack, S.D.; Brock, G.J. Overexpression of the microRNA hsa-miR-200c leads to reduced expression of transcription factor 8 and increased expression of E-cadherin. Cancer Res. 2007, 67, 7972–7976. [Google Scholar] [CrossRef] [Green Version]
- Bendoraite, A.; Knouf, E.C.; Garg, K.S.; Parkin, R.K.; Kroh, E.M.; O’Briant, K.C.; Ventura, A.P.; Godwin, A.K.; Karlan, B.Y.; Drescher, C.W.; et al. Regulation of miR-200 family microRNAs and ZEB transcription factors in ovarian cancer: Evidence supporting a mesothelial-to-epithelial transition. Gynecol. Oncol. 2010, 116, 117–125. [Google Scholar] [CrossRef] [Green Version]
- Brabletz, S.; Brabletz, T. The ZEB/miR-200 feedback loop—a motor of cellular plasticity in development and cancer? EMBO Rep. 2010, 11, 670–677. [Google Scholar] [CrossRef] [Green Version]
- Gregory, P.A.; Bracken, C.P.; Smith, E.; Bert, A.G.; Wright, J.A.; Roslan, S.; Morris, M.; Wyatt, L.; Farshid, G.; Lim, Y.Y.; et al. An autocrine TGF-β/ZEB/miR-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition. Mol. Biol. Cell 2011, 22, 1686–1698. [Google Scholar] [CrossRef]
- Burk, U.; Schubert, J.; Wellner, U.; Schmalhofer, O.; Vincan, E.; Spaderna, S.; Brabletz, T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008, 9, 582–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celià-Terrassa, T.; Meca-Cortés, Ó.; Mateo, F.; De Paz, A.M.; Rubio, N.; Arnal-Estapé, A.; Ell, B.J.; Bermudo, R.; Díaz, A.; Guerra-Rebollo, M.; et al. Epithelial-mesenchymal transition can suppress major attributes of human epithelial tumor-initiating cells. J. Clin. Investig. 2012, 122, 1849–1868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittal, V. Epithelial Mesenchymal Transition in Tumor Metastasis. Annu. Rev. Pathol. 2018, 13, 395–412. [Google Scholar] [CrossRef] [PubMed]
- Pastushenko, I.; Blanpain, C. Transition States during Tumor Progression, E.M.T. and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Sarkissyan, M.; Vadgama, J.V. Epithelial-mesenchymal transition and breast cancer. J. Clin. Med. 2016, 5, 13. [Google Scholar] [CrossRef] [Green Version]
- Felipe Lima, J.; Nofech-Mozes, S.; Bayani, J.; Bartlett, J. EMT in breast carcinoma—A review. J. Clin. Med. 2016, 5, 65. [Google Scholar] [CrossRef] [Green Version]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.C.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef]
- Van Staalduinen, J.; Baker, D.; Dijke, P.T.; van Dam, H. Epithelial–mesenchymal-transition-inducing transcription factors: New targets for tackling chemoresistance in cancer? Oncogene 2018, 37, 6195–6211. [Google Scholar] [CrossRef]
- Dongre, A.; Rashidian, M.; Reinhardt, F.; Bagnato, A.; Keckesova, Z.; Ploegh, H.L.; Weinberg, R.A. Epithelial-to-mesenchymal transition contributes to immunosuppression in breast carcinomas. Cancer Res. 2017, 77, 3982–3989. [Google Scholar] [CrossRef] [Green Version]
- Akalay, I.; Janji, B.; Hasmim, M.; Noman, M.Z.; Andre, F.; De Cremoux, P.; Bertheau, P.; Badoual, C.; Vielh, P.; Larsen, A.K.; et al. Epithelial-to-Mesenchymal Transition and Autophagy Induction in Breast Carcinoma Promote Escape from T-cell–Mediated Lysis. Cancer Res. 2013, 73, 2418–2427. [Google Scholar] [CrossRef] [Green Version]
- Akalay, I.; Janji, B.; Hasmim, M.; Noman, M.Z.; Thiery, J.P.; Mami-Chouaib, F.; Chouaib, S. EMT impairs breast carcinoma cell susceptibility to CTL-mediated lysis through autophagy induction. Autophagy 2013, 9, 1104–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sciacovelli, M.; Frezza, C. Metabolic reprogramming and epithelial-to-mesenchymal transition in cancer. FEBS J. 2017, 284, 3132–3144. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Thompson, C.B. Metabolic regulation of cell growth and proliferation. Nat. Rev. Mol. Cell Biol. 2019, 20, 436–450. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Zong, X. Aberrant cancer metabolism in epithelial–mesenchymal transition and cancer metastasis: Mechanisms in cancer progression. Crit. Rev. Oncol. Hematol. 2017, 115, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Shaul, Y.D.; Freinkman, E.; Comb, W.C.; Cantor, J.R.; Tam, W.L.; Thiru, P.; Kim, D.; Kanarek, N.; Pacold, M.E.; Chen, W.W.; et al. Dihydropyrimidine accumulation is required for the epithelial-mesenchymal transition. Cell 2014, 158, 1094–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, A.L.; Walker, A.K.; Sloan, E.K.; Creek, D.J. Optimized Method for Untargeted Metabolomics Analysis of MDA-MB-231 Breast Cancer Cells. Metabolites 2016, 6, 30. [Google Scholar] [CrossRef]
- Shima, H.; Yamada, A.; Ishikawa, T.; Endo, I. Are breast cancer stem cells the key to resolving clinical issues in breast cancer therapy? Gland Surg. 2017, 6, 82–88. [Google Scholar] [CrossRef] [Green Version]
- Judes, G.; Rifaï, K.; Daures, M.; Dubois, L.; Bignon, Y.-J.; Penault-Llorca, F.; Bernard-Gallon, D. High-throughput «Omics» technologies: New tools for the study of triple-negative breast cancer. Cancer Lett. 2016, 382, 77–85. [Google Scholar] [CrossRef]
- Budczies, J.; Denkert, C. Tissue-Based Metabolomics to Analyze the Breast Cancer Metabolome. Metab. Cancer 2016, 207, 157–175. [Google Scholar]
- Gowda, G.A.N.; Zhang, S.; Gu, H.; Asiago, V.; Shanaiah, N.; Raftery, D. Metabolomics-based methods for early disease diagnostics. Expert Rev. Mol. Diagn. 2008, 8, 617–633. [Google Scholar] [CrossRef] [Green Version]
- Shajahan-Haq, A.N.; Cheema, M.S.; Clarke, R. Application of Metabolomics in Drug Resistant Breast Cancer Research. Metabolites 2015, 5, 100–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penkert, J.; Ripperger, T.; Schieck, M.; Schlegelberger, B.; Steinemann, D.; Illig, T. On metabolic reprogramming and tumor biology: A comprehensive survey of metabolism in breast cancer. Oncotarget 2016, 7, 67626–67649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, P.P.; Sabatini, D.M. Cancer Cell Metabolism: Warburg and Beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samudio, I.; Fiegl, M.; Andreeff, M. Mitochondrial uncoupling and the Warburg effect: Molecular basis for the reprogramming of cancer cell metabolism. Cancer Res. 2009, 69, 2163–2166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otto, W. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Marchiq, I.; Pouysségur, J. Hypoxia, cancer metabolism and the therapeutic benefit of targeting lactate/H+ symporters. Klin. Wochenschr. 2015, 94, 155–171. [Google Scholar] [CrossRef] [Green Version]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef]
- Kondaveeti, Y.; Reed, I.G.; White, B.A. Epithelial–mesenchymal transition induces similar metabolic alterations in two independent breast cancer cell lines. Cancer Lett. 2015, 364, 44–58. [Google Scholar] [CrossRef]
- Dong, C.; Yuan, T.; Wu, Y.; Wang, Y.; Fan, T.W.; Miriyala, S.; Lin, Y.; Yao, J.; Shi, J.; Kang, T.; et al. Loss of FBP1 by Snail-Mediated Repression Provides Metabolic Advantages in Basal-like Breast Cancer. Cancer Cell 2013, 23, 316–331. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Li, K.; Pang, X.; Guo, B.; Su, M.; Huang, Y.; Wang, N.; Ji, F.; Zhong, C.; Yang, J.; et al. Leptin promotes epithelial-mesenchymal transition of breast cancer via the upregulation of pyruvate kinase M2. J. Exp. Clin. Cancer Res. 2016, 35, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Wang, S.; Wei, X.; Zhang, S.; Song, Z.; Chen, X.; Zhang, J. Role of inhibitor of yes-associated protein 1 in triple-negative breast cancer with taxol-based chemoresistance. Cancer Sci. 2019, 110, 561–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Li, M.; Zhang, L.; Zhao, H.; Şahin, Ö.; Chen, J.; Zhao, J.J.; Songyang, Z.; Yu, D. Oncogenic Kinase–Induced PKM2 Tyrosine 105 Phosphorylation Converts Nononcogenic PKM2 to a Tumor Promoter and Induces Cancer Stem–like Cells. Cancer Res. 2018, 78, 2248–2261. [Google Scholar] [CrossRef] [Green Version]
- Dupuy, F.; Tabariès, S.; Andrzejewski, S.; Dong, Z.; Blagih, J.; Annis, M.G.; Omeroglu, A.; Gao, D.; Leung, S.; Amir, E.; et al. PDK1-Dependent Metabolic Reprogramming Dictates Metastatic Potential in Breast Cancer. Cell Metab. 2015, 22, 577–589. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Yang, M.; Chen, S.; Li, D.; Chang, Z.; Dong, Z. PDK1 promotes tumor growth and metastasis in a spontaneous breast cancer model. Oncogene 2015, 35, 3314–3323. [Google Scholar] [CrossRef]
- Peng, F.; Wang, J.-H.; Fan, W.-J.; Meng, Y.-T.; Li, M.-M.; Li, T.-T.; Cui, B.; Wang, H.-F.; Zhao, Y.; An, F.; et al. Glycolysis gatekeeper PDK1 reprograms breast cancer stem cells under hypoxia. Oncogene 2017, 37, 1062–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funasaka, T.; Hogan, V.; Raz, A. Phosphoglucose isomerase/autocrine motility factor mediates epithelial and mesenchymal phenotype conversions in breast cancer. Cancer Res. 2009, 69, 5349–5356. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, A.; Aboukameel, A.; Kong, D.; Wang, Z.; Sethi, S.; Chen, W.; Sarkar, F.H.; Raz, A. Phosphoglucose Isomerase/Autocrine Motility Factor Mediates Epithelial-Mesenchymal Transition Regulated by miR-200 in Breast Cancer Cells. Cancer Res. 2011, 71, 3400–3409. [Google Scholar] [CrossRef] [Green Version]
- Avagliano, A.; Ruocco, M.R.; Aliotta, F.; Belviso, I.; Accurso, A.; Masone, S.; Montagnani, S.; Arcucci, A. Mitochondrial Flexibility of Breast Cancers: A Growth Advantage and a Therapeutic Opportunity. Cells 2019, 8, 401. [Google Scholar] [CrossRef] [Green Version]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef] [Green Version]
- Guha, M.; Srinivasan, S.; Ruthel, G.; Kashina, A.K.; Carstens, R.P.; Mendoza, A.; Khanna, C.; Van Winkle, T.; Avadhani, N.G. Mitochondrial retrograde signaling induces epithelial–mesenchymal transition and generates breast cancer stem cells. Oncogene 2013, 33, 5238–5250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, M.; Brooks, M.; Wicha, M.S. Asparagine and Glutamine: Co-conspirators Fueling Metastasis. Cell Metab. 2018, 27, 947–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: Regulation and biological consequences. Antioxid. Redox Signal. 2008, 10, 179–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Zhu, G.; Dong, B.; Piao, J.; Chen, L.; Lin, Z. The NQO1/PKLR axis promotes lymph node metastasis and breast cancer progression by modulating glycolytic reprogramming. Cancer Lett. 2019, 453, 170–183. [Google Scholar] [CrossRef] [PubMed]
- Di, L.-J.; Byun, J.S.; Wong, M.M.; Wakano, C.; Taylor, T.; Bilke, S.; Baek, S.; Hunter, K.; Yang, H.; Lee, M.; et al. Genome-wide profiles of CtBP link metabolism with genome stability and epithelial reprogramming in breast cancer. Nat. Commun. 2013, 4, 1449. [Google Scholar] [CrossRef] [Green Version]
- Feng, R.; Morine, Y.; Ikemoto, T.; Imura, S.; Iwahashi, S.; Saito, Y.; Shimada, M. Nrf2 activation drive macrophages polarization and cancer cell epithelial-mesenchymal transition during interaction. Cell Commun. Signal. 2018, 16, 54. [Google Scholar] [CrossRef] [Green Version]
- Xiong, G.; Stewart, R.L.; Chen, J.; Gao, T.; Scott, T.L.; Samayoa, L.M.; O’Connor, K.; Lane, A.N.; Xu, R. Collagen prolyl 4-hydroxylase 1 is essential for HIF-1α stabilization and TNBC chemoresistance. Nat. Commun. 2018, 9, 4456. [Google Scholar] [CrossRef] [Green Version]
- Radisky, D.C.; Levy, D.D.; Littlepage, L.E.; Liu, H.; Nelson, C.M.; Fata, J.E.; Leake, D.; Godden, E.L.; Albertson, D.G.; Nieto, M.A.; et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature 2005, 436, 123–127. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.-M.; Giltnane, J.M.; Balko, J.M.; Schwarz, L.J.; Guerrero-Zotano, A.L.; Hutchinson, K.E.; Nixon, M.J.; Estrada, M.V.; Sánchez, V.; Sanders, M.E.; et al. MYC and MCL1 Cooperatively Promote Chemotherapy-Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell Metab. 2017, 26, 633–647.e7. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Zhang, H.; Liu, Y.; Su, P.; Zhang, J.; Wang, X.; Sun, M.; Chen, B.; Zhao, W.; Wang, L.; et al. SREBP1, targeted by miR-18a-5p, modulates epithelial-mesenchymal transition in breast cancer via forming a co-repressor complex with Snail and HDAC1/2. Cell Death Differ. 2019, 26, 843–859. [Google Scholar] [CrossRef]
- Li, J.; Dong, L.; Wei, D.; Wang, X.; Zhang, S.; Li, H. Fatty Acid Synthase Mediates the Epithelial-Mesenchymal Transition of Breast Cancer Cells. Int. J. Biol. Sci. 2014, 10, 171–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zielinska, H.A.; Holly, J.M.P.; Bahl, A.; Perks, C.M. Inhibition of FASN and ERα signalling during hyperglycaemia-induced matrix-specific EMT promotes breast cancer cell invasion via a caveolin-1-dependent mechanism. Cancer Lett. 2018, 419, 187–202. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Xiao, L.; Sugiura, H.; Huang, X.; Ali, A.; Kuro-O, M.; DeBerardinis, R.J.; Boothman, D.A. Metabolic reprogramming during TGFβ1-induced epithelial-to-mesenchymal transition. Oncogene 2015, 34, 3908–3916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, M.R.; Steinbauer, B.; Srivastava, K.; Singhal, M.; Mattijssen, F.; Maida, A.; Christian, S.; Hess-Stumpp, H.; Augustin, H.G.; Müller-Decker, K.; et al. Acetyl-CoA carboxylase 1-dependent protein acetylation controls breast cancer metastasis and recurrence. Cell Metab. 2017, 26, 842–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Li, X.; Fu, Q.; Cao, Q.; Chen, X.; Wang, M.; Yu, J.; Long, J.; Yao, J.; Liu, H.; et al. AKR1B1 promotes basal-like breast cancer progression by a positive feedback loop that activates the EMT program. J. Exp. Med. 2017, 214, 1065–1079. [Google Scholar] [CrossRef] [Green Version]
- Keinan, O.; Kedan, A.; Gavert, N.; Selitrennik, M.; Kim, S.; Karn, T.; Becker, S.; Lev, S. The lipid-transfer protein Nir2 enhances epithelial-mesenchymal transition and facilitates breast cancer metastasis. J. Cell Sci. 2014, 127, 4740–4749. [Google Scholar] [CrossRef] [Green Version]
- Battula, V.L.; Shi, Y.; Evans, K.W.; Wang, R.-Y.; Spaeth, E.; Jacamo, R.O.; Guerra, R.; Sahin, A.A.; Marini, F.C.; Hortobagyi, G.; et al. Ganglioside GD2 identifies breast cancer stem cells and promotes tumorigenesis. J. Clin. Investig. 2012, 122, 2066–2078. [Google Scholar] [CrossRef]
- Sarkar, T.R.; Battula, V.L.; Werden, S.J.; Vijay, G.V.; Ramirez-Peña, E.Q.; Taube, J.H.; Chang, J.T.; Miura, N.; Porter, W.; Sphyris, N.; et al. GD3 synthase regulates epithelial–mesenchymal transition and metastasis in breast cancer. Oncogene 2014, 34, 2958–2967. [Google Scholar] [CrossRef] [Green Version]
- Giudetti, A.M.; De Domenico, S.; Ragusa, A.; Lunetti, P.; Gaballo, A.; Franck, J.; Simeone, P.; Nicolardi, G.; De Nuccio, F.; Santino, A.; et al. A specific lipid metabolic profile is associated with the epithelial mesenchymal transition program. Biochim. Biophys. Acta BBA-Mol. Cell Biol. Lipids 2018, 1864, 344–357. [Google Scholar] [CrossRef]
- Eiriksson, F.F.; Rolfsson, O.; Ogmundsdottir, H.M.; Haraldsson, G.G.; Thorsteinsdottir, M.; Halldorsson, S. Altered plasmalogen content and fatty acid saturation following epithelial to mesenchymal transition in breast epithelial cell lines. Int. J. Biochem. Cell Biol. 2018, 103, 99–104. [Google Scholar] [CrossRef]
- Martinez-Orozco, R.; Navarro-Tito, N.; Soto-Guzman, A.; Castro-Sanchez, L.; Salazar, E.P. Arachidonic acid promotes epithelial-to-mesenchymal-like transition in mammary epithelial cells MCF10A. Eur. J. Cell Biol. 2010, 89, 476–488. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Neira, R.; Mejia-Rangel, J.; Cortes-Reynosa, P.; Salazar, E.P. Linoleic acid induces an EMT-like process in mammary epithelial cells MCF10A. Int. J. Biochem. Cell Biol. 2011, 43, 1782–1791. [Google Scholar] [CrossRef] [PubMed]
- Bertero, T.; Oldham, W.M.; Grasset, E.M.; Bourget, I.; Boulter, E.; Pisano, S.; Hofman, P.; Bellvert, F.; Meneguzzi, G.; Bulavin, D.V.; et al. Tumor-Stroma Mechanics Coordinate Amino Acid Availability to Sustain Tumor Growth and Malignancy. Cell Metab. 2018, 29, 124–140.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halldorsson, S.; Rohatgi, N.; Magnusdottir, M.; Choudhary, K.S.; Gudjonsson, T.; Knutsen, E.; Barkovskaya, A.; Hilmarsdottir, B.; Perander, M.; Mælandsmo, G.M.; et al. Metabolic re-wiring of isogenic breast epithelial cell lines following epithelial to mesenchymal transition. Cancer Lett. 2017, 396, 117–129. [Google Scholar] [CrossRef] [Green Version]
- Xie, G.; Zhou, B.; Zhao, A.; Qiu, Y.; Zhao, X.; Garmire, L.; Shvetsov, Y.B.; Yu, H.; Yen, Y.; Jia, W. Lowered circulating aspartate is a metabolic feature of human breast cancer. Oncotarget 2015, 6, 33369–33381. [Google Scholar] [CrossRef] [Green Version]
- Knott, S.R.V.; Wagenblast, E.; Khan, S.; Kim, S.Y.; Soto, M.; Wagner, M.; Turgeon, M.-O.; Fish, L.; Erard, N.; Gable, A.L.; et al. Asparagine bioavailability governs metastasis in a model of breast cancer. Nature 2018, 554, 378–381. [Google Scholar] [CrossRef]
- Luo, M.; Shang, L.; Brooks, M.D.; Jiagge, E.; Zhu, Y.; Buschhaus, J.M.; Conley, S.; Fath, M.A.; Davis, A.; Gheordunescu, E.; et al. Targeting Breast Cancer Stem Cell State Equilibrium through Modulation of Redox Signaling. Cell Metab. 2018, 28, 69.e6–86.e6. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Ding, C.-K.; Wu, J.; Sjol, J.; Wardell, S.; Spasojevic, I.; George, D.; McDonnell, D.P.; Hsu, D.S.; Chang, J.T.; et al. Cystine addiction of triple-negative breast cancer associated with EMT augmented death signaling. Oncogene 2016, 36, 4235–4242. [Google Scholar] [CrossRef] [Green Version]
- Timmerman, L.A.; Holton, T.; Yuneva, M.; Louie, R.J.; Padró, M.; Daemen, A.; Hu, M.; Chan, D.A.; Ethier, S.P.; Veer, L.J.V.; et al. Glutamine Sensitivity Analysis Identifies the xCT Antiporter as a Common Triple-Negative Breast Tumor Therapeutic Target. Cancer Cell 2013, 24, 450–465. [Google Scholar] [CrossRef] [Green Version]
- Dias, M.M.; Adamoski, D.; Dos Reis, L.M.; Ascenção, C.F.; de Oliveira, K.R.; Mafra, A.C.P.; da Silva Bastos, A.C.; Quintero, M.; de Cassago, G.; Ferreira, I.M.; et al. GLS2 is protumorigenic in breast cancers. Oncogene 2020, 39, 690–702. [Google Scholar] [CrossRef]
- Ramirez-Peña, E.; Arnold, J.; Shivakumar, V.; Joseph, R.; Vidhya Vijay, G.; den Hollander, P.; Bhangre, N.; Allegakoen, P.; Prasad, R.; Conley, Z.; et al. The epithelial to mesenchymal transition promotes glutamine independence by suppressing GLS2 expression. Cancers 2019, 11, 1610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Sharma, R.K.; Chagtoo, M.; Agarwal, G.; George, N.; Sinha, N.; Godbole, M.M. 1H NMR Metabolomics Reveals Association of High Expression of Inositol 1, 4, 5 Trisphosphate Receptor and Metabolites in Breast Cancer Patients. PLoS ONE 2017, 12, e0169330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, M.; Nilsson, R.; Sharma, S.; Madhusudhan, N.; Kitami, T.; Souza, A.L.; Kafri, R.; Kirschner, M.W.; Clish, C.B.; Mootha, V.K. Metabolite Profiling Identifies a Key Role for Glycine in Rapid Cancer Cell Proliferation. Science 2012, 336, 1040–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadi, N.I.; Jamal, Q.; Iqbal, A.; Shaikh, F.; Somroo, S.; Musharraf, S.G. Serum Metabolomic Profiles for Breast Cancer Diagnosis, Grading and Staging by Gas Chromatography-Mass Spectrometry. Sci. Rep. 2017, 7, 1715. [Google Scholar] [CrossRef] [Green Version]
- Jové, M.; Collado, R.; Quiles, J.L.; Sol, J.; Ruiz-sanjuan, M.; Fernandez, M.; De, C.; Pamplona, R. A plasma metabolomic signature discloses human breast cancer. Oncotarget 2017, 8, 19522–19533. [Google Scholar] [CrossRef] [Green Version]
- Flote, V.G.; Vettukattil, R.; Bathen, T.F.; Egeland, T.; McTiernan, A.; Frydenberg, H.; Husøy, A.; Finstad, S.E.; Lømo, J.; Garred, Ø.; et al. Lipoprotein subfractions by nuclear magnetic resonance are associated with tumor characteristics in breast cancer. Lipids Health Dis. 2016, 15, 56. [Google Scholar] [CrossRef] [Green Version]
- Richard, V.; Conotte, R.; Mayne, D.; Colet, J.-M. Does the 1H-NMR plasma metabolome reflect the host-tumor interactions in human breast cancer? Oncotarget 2017, 8, 49915–49930. [Google Scholar] [CrossRef]
- Fuss, T.L.; Cheng, L.L. Evaluation of Cancer Metabolomics Using ex vivo High Resolution Magic Angle Spinning (HRMAS) Magnetic Resonance Spectroscopy (MRS). Metabolites 2016, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Haukaas, T.H.; Euceda, L.R.; Giskeødegård, G.F.; Bathen, T.F. Metabolic portraits of breast cancer by HR MAS MR spectroscopy of intact tissue samples. Metabolites 2017, 7, 18. [Google Scholar] [CrossRef] [Green Version]
- El Ansari, R.; McIntyre, A.; Craze, M.L.; Ellis, I.O.; Rakha, E.A.; Green, A.R. Altered glutamine metabolism in breast cancer; subtype dependencies and alternative adaptations. Histopathology 2017, 72, 183–190. [Google Scholar] [CrossRef]
- Viswanathan, V.S.; Ryan, M.J.; Dhruv, H.D.; Gill, S.; Eichhoff, O.M.; Seashore-Ludlow, B.; Kaffenberger, S.D.; Eaton, J.K.; Shimada, K.; Aguirre, A.J.; et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017, 547, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Mathow, D.; Chessa, F.; Rabionet, M.; Kaden, S.; Jennemann, R.; Sandhoff, R.; Gröne, H.; Feuerborn, A. Zeb1 affects epithelial cell adhesion by diverting glycosphingolipid metabolism. EMBO Rep. 2015, 16, 321–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceppi, P.; Peter, M.E. MicroRNAs regulate both epithelial-to-mesenchymal transition and cancer stem cells. Oncogene 2014, 33, 269–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sciacovelli, M.; Gonçalves, E.; Johnson, T.I.; Zecchini, V.R.; Da Costa, A.S.H.; Gaude, E.; Drubbel, A.V.; Theobald, S.J.; da Costa, A.S.H.; Gaude, E.; et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 2016, 537, 544–547. [Google Scholar] [CrossRef]
- Siddiqui, A.; Gollavilli, P.N.; Schwab, A.; Vazakidou, M.E.; Ersan, P.G.; Ramakrishnan, M.; Pluim, D.; Coggins, S.; Saatci, O.; Annaratone, L.; et al. Thymidylate synthase maintains the de-differentiated state of triple negative breast cancers. Cell Death Differ. 2019, 26, 2223–2236. [Google Scholar] [CrossRef] [Green Version]
- Schwab, A.; Siddiqui, A.; Vazakidou, M.E.; Napoli, F.; Böttcher, M.; Menchicchi, B.; Raza, U.; Saatci, Ö.; Krebs, A.M.; Ferrazzi, F.; et al. Polyol Pathway Links Glucose Metabolism to the Aggressiveness of Cancer Cells. Cancer Res. 2018, 78, 1604–1618. [Google Scholar] [CrossRef] [Green Version]
- Colvin, H.; Nishida, N.; Konno, M.; Haraguchi, N.; Takahashi, H.; Nishimura, J.; Hata, T.; Kawamoto, K.; Asai, A.; Tsunekuni, K.; et al. Oncometabolite D-2-Hydroxyglurate Directly Induces Epithelial-Mesenchymal Transition and is Associated with Distant Metastasis in Colorectal Cancer. Sci. Rep. 2016, 6, 36289. [Google Scholar] [CrossRef] [Green Version]
- Tian, Q.; Yuan, P.; Quan, C.; Li, M.; Xiao, J.; Zhang, L.; Lu, H.; Ma, T.; Zou, L.; Wang, F.; et al. Phosphorylation of BCKDK of BCAA catabolism at Y246 by Src promotes metastasis of colorectal cancer. Oncogene 2020, 39, 3980–3996. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, R.R.; Leavis, P.C. A Peptide Derived from the Human Leptin Molecule Is a Potent Inhibitor of the Leptin Receptor Function in Rabbit Endometrial Cells. Endocrine 2003, 21, 185–196. [Google Scholar] [CrossRef]
- Liu, Y.; Su, P.; Zhao, W.; Li, X.; Yang, X.; Fan, J.; Yang, H.; Yan, C.; Mao, L.; Ding, Y.; et al. ZNF213 negatively controls triple negative breast cancer progression via Hippo/YAP signaling. Cancer Sci. 2021, 112, 2714–2727. [Google Scholar] [CrossRef]
- Cheng, G.; Zielonka, J.; Dranka, B.P.; McAllister, D.; Mackinnon, A.C.; Joseph, J.; Kalyanaraman, B. Mitochondria-targeted drugs synergize with 2-deoxyglucose to trigger breast cancer cell death. Cancer Res. 2012, 72, 2634–2644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, L.; Feng, X.; Huang, M.; Zhang, K.; Liu, Q. Sonodynamic Therapy Combined to 2-Deoxyglucose Potentiate Cell Metastasis Inhibition of Breast Cancer. Ultrasound Med. Biol. 2019, 45, 2984–2992. [Google Scholar] [CrossRef] [PubMed]
- Bernert, B.; Porsch, H.; Heldin, P. Hyaluronan Synthase 2 (HAS2) Promotes Breast Cancer Cell Invasion by Suppression of Tissue Metalloproteinase Inhibitor 1 (TIMP-1). J. Biol. Chem. 2011, 286, 42349–42359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kast, R.E.; Skuli, N.; Cos, S.; Karpel-Massler, G.; Shiozawa, Y.; Goshen, R.; Halatsch, M.-E. The ABC7 regimen: A new approach to metastatic breast cancer using seven common drugs to inhibit epithelial-to-mesenchymal transition and augment capecitabine efficacy. Breast Cancer: Targets Ther. 2017, 9, 495–514. [Google Scholar] [CrossRef] [Green Version]
- Campbell, E.J.; Dachs, G.U.; Morrin, H.R.; Davey, V.C.; Robinson, B.A.; Vissers, M.C.M. Activation of the hypoxia pathway in breast cancer tissue and patient survival are inversely associated with tumor ascorbate levels. BMC Cancer 2019, 19, 307. [Google Scholar] [CrossRef]
- Ghanem, A.; Melzer, A.M.; Zaal, E.; Neises, L.; Baltissen, D.; Matar, O.; Glennemeier-Marke, H.; Almouhanna, F.; Theobald, J.; Abu el Maaty, M.A.; et al. Ascorbate kills breast cancer cells by rewiring metabolism via redox imbalance and energy crisis. Free Radic. Biol. Med. 2020, 163, 196–209. [Google Scholar] [CrossRef]
- Kirane, A.; Toombs, J.E.; Larsen, J.E.; Ostapoff, K.T.; Meshaw, K.R.; Zaknoen, S.; Brekken, R.A.; Burrows, F.J. Epithelial–mesenchymal transition increases tumor sensitivity to COX-2 inhibition by apricoxib. Carcinogenesis 2012, 33, 1639–1646. [Google Scholar] [CrossRef] [Green Version]
- Mandal, P.K.; Freiter, E.M.; Bagsby, A.L.; Robertson, F.M.; McMurray, J.S. Efficient synthesis of apricoxib, CS-706, a selective cyclooxygenase-2 inhibitor, and evaluation of inhibition of prostaglandin E2 production in inflammatory breast cancer cells. Bioorganic Med. Chem. Lett. 2011, 21, 6071–6073. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Yang, B.; Xiang, T.; Peng, W.; Qiu, Z.; Wan, J.; Zhang, L.; Li, H.; Li, H.; Ren, G. Diallyl disulfide inhibits growth and metastatic potential of human triple-negative breast cancer cells through inactivation of the β-catenin signaling pathway. Mol. Nutr. Food Res. 2015, 59, 1063–1075. [Google Scholar] [CrossRef]
- Yip, N.C.; Fombon, I.S.; Liu, P.; Brown, S.; Kannappan, V.; Armesilla, A.; Xu, B.; Cassidy, J.; Darling, J.L.; Wang, W. Disulfiram modulated ROS–MAPK and NFκB pathways and targeted breast cancer cells with cancer stem cell-like properties. Br. J. Cancer 2011, 104, 1564–1574. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Kumar, I.S.; Brown, S.; Kannappan, V.; Tawari, P.E.; Tang, J.Z.; Jiang, W.; Armesilla, A.; Darling, J.L.; Wang, W. Disulfiram targets cancer stem-like cells and reverses resistance and cross-resistance in acquired paclitaxel-resistant triple-negative breast cancer cells. Br. J. Cancer 2013, 109, 1876–1885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.-X.; Zheng, D.-W.; Rong, L.; Zhu, J.-Y.; Hong, S.; Li, C.; Xu, Z.-S.; Cheng, S.-X.; Zhang, X.-Z. Targeting epithelial-mesenchymal transition: Metal organic network nano-complexes for preventing tumor metastasis. Biomaterials 2017, 139, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Ozawa, Y.; Kimura, T.; Sato, Y.; Kuznetsov, G.; Xu, S.; Uesugi, M.; Agoulnik, S.; Taylor, N.P.; Funahashi, Y.; et al. Eribulin mesilate suppresses experimental metastasis of breast cancer cells by reversing phenotype from epithelial–mesenchymal transition (EMT) to mesenchymal–epithelial transition (MET) states. Br. J. Cancer 2014, 110, 1497–1505. [Google Scholar] [CrossRef] [PubMed]
- Orun, O.; Tiber, P.M.; Sevinç, S.K. Apoptotic Effects of Etodolac in Breast Cancer Cell Cultures. In Nonsteroidal Anti-Inflammatory Drugs; IntechOpen: London, UK, 2017; pp. 133–151. [Google Scholar]
- Na, Y.-R.; Yoon, Y.-N.; Son, D.-I.; Seok, S.-H. Cyclooxygenase-2 Inhibition Blocks M2 Macrophage Differentiation and Suppresses Metastasis in Murine Breast Cancer Model. PLoS ONE 2013, 8, e63451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, A.J.; Sullivan, F.J.; Giles, F.J.; Glynn, S.A. The yin and yang of nitric oxide in cancer progression. Carcinogenesis 2013, 34, 503–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granados-Principal, S.; Liu, Y.; Guevara, M.L.; Blanco, E.; Choi, D.S.; Qian, W.; Patel, T.; Rodriguez, A.A.; Cusimano, J.; Weiss, H.L.; et al. Inhibition of iNOS as a novel effective targeted therapy against triple-negative breast cancer. Breast Cancer Res. 2015, 17, 25. [Google Scholar] [CrossRef]
- Dávila-González, D.; Choi, D.S.; Rosato, R.R.; Granados-Principal, S.M.; Kuhn, J.G.; Li, W.F.; Qian, W.; Chen, W.; Kozielski, A.L.; Wong, H.; et al. Pharmacological inhibition of NOS activates ASK1/JNK pathway augmenting docetaxel-mediated apoptosis in triple-negative breast cancer. Clin. Cancer Res. 2018, 24, 1152–1162. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Mao, C.; Liu, S.; Xiao, D.; Shi, Y.; Tao, Y. Proline dehydrogenase in cancer: Apoptosis, autophagy, nutrient dependency and cancer therapy. Amino Acids 2021, 53, 1891–1902. [Google Scholar] [CrossRef]
- Liu, Y.; Mao, C.; Wang, M.; Liu, N.; Ouyang, L.; Liu, S.; Tang, H.; Cao, Y.; Liu, S.; Wang, X.; et al. Cancer progression is mediated by proline catabolism in non-small cell lung cancer. Oncogene 2020, 39, 2358–2376. [Google Scholar] [CrossRef]
- Lin, D.; Kuang, G.; Wan, J.; Zhang, X.; Li, H.; Gong, X.; Li, H. Luteolin suppresses the metastasis of triple-negative breast cancer by reversing epithelial-to-mesenchymal transition via downregulation of β-catenin expression. Oncol. Rep. 2017, 37, 895–902. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Huang, J.; Yang, B.; Xiang, T.; Yin, X.; Peng, W.; Cheng, W.; Wan, J.; Luo, F.; Li, H.; et al. Mangiferin exerts antitumor activity in breast cancer cells by regulating matrix metalloproteinases, epithelial to mesenchymal transition, and β-catenin signaling pathway. Toxicol. Appl. Pharmacol. 2013, 272, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Faria, J.; Negalha, G.; Azevedo, A.; Martel, F. Metformin and Breast Cancer: Molecular Targets. J. Mammary Gland Biol. Neoplasia 2019, 24, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Alimova, I.N.; Liu, B.; Fan, Z.; Edgerton, S.M.; Dillon, T.; Lind, S.E.; Thor, A.D. Metformin inhibits breast cancer cell growth, colony formation and induces cell cycle arrest in vitro. Cell Cycle 2009, 8, 909–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schacke, M.; Kumar, J.; Colwell, N.; Hermanson, K.; Folle, G.A.; Nechaev, S.; Dhasarathy, A.; Lafon-Hughes, L. PARP-1/2 inhibitor olaparib prevents or partially reverts EMT induced by TGF-β in NMuMG cells. Int. J. Mol. Sci. 2019, 20, 518. [Google Scholar] [CrossRef] [Green Version]
- Mishra, R.R.; Belder, N.; Ansari, S.A.; Kayhan, M.; Bal, H.; Raza, U.; Ersan, P.G.; Tokat, Ü.M.; Eyüpoğlu, E.; Saatci, Ö.; et al. Reactivation of cAMP Pathway by PDE4D Inhibition Represents a Novel Druggable Axis for Overcoming Tamoxifen Resistance in ER-positive Breast Cancer. Clin. Cancer Res. 2018, 24, 1987–2001. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Uppal, H.; Demaria, M.; Desprez, P.Y.; Campisi, J.; Kapahi, P. Simvastatin suppresses breast cancer cell proliferation induced by senescent cells. Sci. Rep. 2015, 5, 17895. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Seah, S.; Loh, X.; Chan, C.-W.; Hartman, M.; Goh, B.-C.; Lee, S.-C. Simvastatin-induced breast cancer cell death and deactivation of PI3K/Akt and MAPK/ERK signalling are reversed by metabolic products of the mevalonate pathway. Oncotarget 2016, 7, 2532–2544. [Google Scholar] [CrossRef] [Green Version]
- Ghosh-Choudhury, N.; Mandal, C.C.; Ghosh-Choudhury, N.; Choudhury, G.G. Simvastatin induces derepression of PTEN expression via NFκB to inhibit breast cancer cell growth. Cell. Signal. 2010, 22, 749–758. [Google Scholar] [CrossRef] [Green Version]
- Lustberg, M.B.; Pant, S.; Ruppert, A.S.; Shen, T.; Wei, Y.; Chen, L.; Brenner, L.; Shiels, D.; Jensen, R.R.; Berger, M.; et al. Phase I/II trial of non-cytotoxic suramin in combination with weekly paclitaxel in metastatic breast cancer treated with prior taxanes. Cancer Chemother. Pharmacol. 2012, 70, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Gilboa-Geffen, A.; Hamar, P.; Le, M.T.; Wheeler, L.A.; Trifonova, R.; Petrocca, F.; Wittrup, A.; Lieberman, J. Gene Knockdown by EpCAM Aptamer-siRNA Chimeras Suppresses Epithelial Breast Cancers and Their Tumor-Initiating Cells. Mol. Cancer Ther. 2015, 14, 2279–2291. [Google Scholar] [CrossRef] [Green Version]
- Camorani, S.; Crescenzi, E.; Gramanzini, M.; Fedele, M.; Zannetti, A.; Cerchia, L. Aptamer-mediated impairment of EGFR-integrin _v_3 complex inhibits vasculogenic mimicry and growth of triple-negative breast cancers. Sci. Rep. 2017, 7, 46659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Reinhard, F.; Pan, E.; Soutschek, J.; Bhat, B.; Marcusson, E.G.; Teruya-Feldstein, J.; Bell, G.W.; Weinberg, R.A. Therapeutic silencing of miR-10b inhibits metastasis in a mouse mammary tumor model. Nat. Biotechnol. 2010, 28, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Li, W.; Liu, C.; Li, W.; Yu, H.; Lei, B.; Ren, Y.; Li, Z.; Pang, D.; Qian, C. MiR-23a promotes TGF-1-induced EMT and tumor metastasis in breast cancer cells by directly targeting CDH1 and activating Wnt/β-catenin signaling. Oncotarget 2017, 8, 69538–69550. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, K.; Lowry, M.C.; Corcoran, C.; Martinez, V.G.; Daly, M.; Rani, S.; Gallagher, W.M.; Radomski, M.W.; MacLeod, R.A.; O’Driscoll, L. miR-134 in extracellular vesicles reduces triple-negative breast cancer aggression and increases drug sensitivity. Oncotarget 2015, 6, 32774–32789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knezevic, J.; Pfefferle, A.D.; Petrovic, I.; Greene, S.B.; Perou, C.M.; Rosen, J.M. Expression of miR-200c in claudin-low breast cancer alters stem cell functionality, enhances chemosensitivity and reduces metastatic potential. Oncogene 2015, 34, 5997–6006. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Wei, L.; Huang, Y.; Wu, Y.; Su, M.; Pang, X.; Wang, N.; Ji, F.; Zhong, C.; Chen, T.; et al. miR520c blocks EMT progression of human breast cancer cells by repressing STAT3. Oncol. Rep. 2017, 37, 1537–1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabbah, M.; Emami, S.; Redeuilh, G.; Julien, S.; Prévost, G.; Zimber, A.; Ouelaa, R.; Bracke, M.; De Wever, O.; Gespach, C. Molecular signature and therapeutic perspective of the epithelial-to-mesenchymal transitions in epithelial cancers. Drug Resist. Updates 2008, 11, 123–151. [Google Scholar] [CrossRef] [PubMed]
- Sethi, N.; Kang, Y. Notch signaling: Mediator and therapeutic target of bone metastasis. BoneKEy Rep. 2012, 1, 3. [Google Scholar] [CrossRef] [Green Version]
- Hyman, D.M.; Taylor, B.S.; Baselga, J. Implementing Genome-Driven Oncology. Cell 2017, 168, 584–599. [Google Scholar] [CrossRef] [Green Version]
- Ramos, P.; Bentires-Alj, M. Mechanism-based cancer therapy: Resistance to therapy, therapy for resistance. Oncogene 2014, 34, 3617–3626. [Google Scholar] [CrossRef]
- Dang, C.V.; Reddy, E.P.; Shokat, K.M.; Soucek, L. Drugging the ‘undruggable’ cancer targets. Nat. Rev. Cancer 2017, 17, 502–508. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Molecular Subtype | Normal Breast Like | Luminal A | Luminal B | HER2+ | Basal Like | Claudin Low | |
|---|---|---|---|---|---|---|---|
| Hormonal | ER | ± | + | ± | − | − | − |
| PR | ± | + | ± | − | − | − | |
| HER2 | − | − | + | + | − | − | |
| Proliferation Rate | Low | Low | High | High | High | High | |
| Frequency of incidence (%) | 5–10 | 50–60 | 10–20 | 15–20 | 10–20 | 12–14 | |
| Prognosis | Intermediate | Good | Intermediate | Poor | Poor | Poor | |
| P53 mutation | Low | Low | Intermediate | High | High | High | |
| Site of metastasis | Unclear | Bone | Liver, Bone | Lung, Brain | Lung | Unclear | |
| Features/Name of Marker | Phenotype (State of Tissues/Cells) | |
|---|---|---|
| Epithelial | Mesenchymal | |
| Shape | Elongated | Rounded |
| Motility | Sessile | Motile |
| Adherence | Adherent to neighbours | Non-adherent to neighbours |
| Proliferation | Higher proliferation | Lower proliferation |
| Invasion | Non-invasive | Invasive |
| Micro tentacles | Absent | Present |
| EMT markers | ||
| Expression | Epithelial state | Mesenchymal state |
| N-cadherin | Decreased | Increased |
| E-cadherin | Increased | Decreased |
| Vimentin | Decreased | Increased |
| Occludin | Increased | Decreased |
| Fibronectin | Decreased | Increased |
| ZO-1 | Increased | Decreased |
| MMP-9 | Decreased | Increased |
| MMP-2 | Decreased | Increased |
| S. No. | Metabolite | Metabolite Inhibitors/Metabolic Inhibitor of EMT | Mechanism | Ref. |
|---|---|---|---|---|
| Glucose Metabolism | ||||
| 1. | Pyruvate kinase M2 (PKM2) | PKM2 is the limiting step of glycolysis and is strongly related to the EMT process. | [90] | |
| Fructose 1,6-bisphosphatase (FBP1) | Inhibits PKM2 activation to block glycolysis; boosts mitochondrial complex I activity; enhances OXPHOS; ultimately inhibit EMT. | |||
| Leptin inhibitors | Leptin promotes EMT via high PKM2 expression and activation of PI3K/AKT signalling cascade. Thus, Leptin receptor inhibition required blocking EMT-induced chemoresistance. | [91] | ||
| LPR-2 inhibits chemotherapeutics resistance on ER- breast cancer cells. | [149] | |||
| Anti-ObR and PI3K/AKT signalling pathway inhibitor LY294002 significantly abolished leptin-induced PKM2 expression and EMT program. | [91] | |||
| Silencing YAP | PKM2 phosphorylation on tyrosine 105 promotes BCSCs via activation of downstream signals of self-associated protein (YAP); enhances nuclear translocation of YAP. Therefore, silencing YAP impairs BCSCs mediated oncogenic kinases and hence inhibit EMT-induced chemoresistance. | [92,93,150] | ||
| 2. | Pyruvate dehydrogenase kinase 1 (PDK1) | Enzyme prevents pyruvate dehydrogenase complex to initiate TCA cycle; enhance glycolytic metabolism to initiate liver metastases. PDK1 inhibition is necessary for blocking EMT program and prevents liver and lung specific metastasis. | [94] | |
| Silencing long non-coding RNA- H19 | H19 required for glycolytic activity and BCSC characteristics; highly associated with PDK1 expression. H19 silencing abolishes PDK1 expression under hypoxia, glycolysis, and self-renewal circumstances. | [95] | ||
| Aspirin (acetylsalicylic acid) | Inhibits both H19 and PDK1; significantly reduce of BCSC characteristics and block EMT program. | [96] | ||
| 3. | Phosphoglucose isomerase (PGI) | Catalyzes interconversion of G-6-P and F-6-P; overexpression enhances EMT via increasing NF-kB activity to regulate ZEB transcription. | [97] | |
| MicroRNA-200 | Inhibit ZEB expression and reverses other targets involved in PGI-induced EMT program. | [98] | ||
| 4. | NADH and NADPH | Significant sources of reducing equivalent ROS detoxification; serve as contributors in decreasing intracellular ROS; NAD (P) H level act as a link between ROS and EMT process. Overexpression of NQO1 promotes PKLR in breast cancer. NQO1 interacts with PKLR to promote glycolysis while preserving NAD (P) H homeostasis. | [103] | |
| Silencing NQO1 | Silencing NQO1 to significantly rise intracellular ROS; which hinders the EMT process. | [104] | ||
| 5. | Matrix metalloproteinase-3 (MMP-3) | MMP-3 inhibitors | MMP-3 overexpression serves as signal from the breast cancer microenvironment to mediate the ROS in breast cancer cells which further promotes snail and EMT expression. | [107] |
| Lipid Metabolism | ||||
| 6. | Sterol regulatory element-binding transcription protein 1 (SREBP1) | Main transcriptional promoter of lipogenesis; de novo lipogenesis; inhibit E-cadherin (epithelial marker) expression in breast cancer by forming a co-repressor structure with snail and histone deacetylase. | [110] | |
| miR-18a-5p | Inhibit SREBP1 to block EMT program and breast cancer lung metastasis. | |||
| 7. | Fatty Acid Synthase (FASN) | A lipogenic enzyme required for EMT expansion in breast tumors. | [111,112] | |
| Cerulenin | Block FASN and slows down the EMT program; also reverses the hyperglycaemia-induced EMT phenotype of breast cancer. | |||
| 8. | Acetyl-CoA carboxylase1 (ACC1) | Involved in protein acetylation and stimulates conversion of acetyl-CoA to malonyl-CoA. ACC1 inhibition raises acetyl-CoA level leading to acetylation of Smad2; Leptin or TGF-β signal activation in obese-breast cancer patients to promote EMT program. Thus, targeting ACC1-dependent EMT axis is a promising platform of research in obese breast cancer patients. | [114] | |
| 9. | Aldo-keto Reductase 1 family B1 (AKR1B1) | Enzyme converts prostaglandin H2 to prostaglandin F2a. Twist promotes NF-kB activation and induce EMT program to improve BCSC-like features. | [115] | |
| Epalrestat | Anti-AKR1B1 drug drastically reduces EMT; drug for TNBC targeting AKR1B1. | |||
| 10. | Lipid transfer protein (Nir2) | Acts as unique EMT controller in breast tumor cells; TGFβ1-induced EMT is slowed when Nir2 is silenced; thus, it is a promising beneficial target. | [116] | |
| Amino Acid Metabolism | ||||
| 11. | Asparagine synthetase | Rate-limiting enzyme in asparagine biosynthesis and utilized as therapeutic target to reduce asparagine bioavailability in the tumor micro-environment; could block EMT program to impair invasiveness and metastasis of breast cancer. | [126,127] | |
| L-asparaginase or Asparagine as dietary intake | Supplementing L-asparaginase or improving the dietary content of asparagine for breast cancer patients prevents the EMT program and reduce metastasis. | |||
| 12. | Cystine | Cystine deprivation induces necrosis in the TNBC phenotype while limited cell death in the luminal subtype of breast cancer. | [128] | |
| MiR-200c | Transfection of MiR-200c in cystine-enriched breast cancer phenotypes reverses mesenchymal features. | |||
| 13. | Glutaminase-2 (GLS2) | Mediates expression of mesenchymal markers, invasion, and metastasis in TNBC; EMT is inversely linked to GLS2 levels. The loss of GLS2 expression during EMT leads to an enhanced glutamine-independent phenotype and decreased mitochondrial activity, while, GLS2 restoration in GLS2-negative breast cancer cells exhibits enhanced consumption of mitochondrial gluta-mine and impairs BCSC-like properties. | [130,131] | |
| 14. | Inositol-1,4,5-trisphosphate receptors (IPR3) | Highly expressed in breast cancer patients with enhanced lactate, lysine, alanine, lipoproteins and low serum pyruvate and glucose levels compared to healthy individuals. | [123] | |
| 15. | Glycine | Glycine biosynthetic pathway is highly upregulated in rapid proliferating breast cancer cells. Thus, its supplementation is not recommended in diet which might worsen breast cancer patient’s condition into tumor metastasis and proliferation. Glycine is a potential biomarker and therapeutic response tracking. | [133] | |
| 16. | Thymidylate synthase (TS) | Nucleotide metabolic enzyme is associated with cell proliferation, de-differentiation, and EMT phenotype of breast cancer de-differentiation, requiring DPYD-dependent pyrimidine catabolism. | [145] | |
| S. No. | Drugs/Formulation | Target | Ref |
|---|---|---|---|
| 1. | 2-deoxyglucose (2-DG) | Inhibit glycolysis | [151,152] |
| 2. | 4-methylumbelliferone | Hyaluronan synthase-2 inhibitor | [153] |
| 3. | Agomelatine | Melatonergic receptors agonist and 5-HT2C antagonist | [154] |
| 4. | Ascorbate | Vitamin C | [155,156] |
| 5. | Apricoxib | COX-2 Inhibitor | [157,158] |
| 6. | Diallyl disulfide | Increases expression of epithelial marker E-cadherin and decreased expression of mesenchymal markers such as Vimentin, N-cadherin and Snail. | [159] |
| 7. | Disulfiram | ALDH1 inhibitor | [160,161] |
| 8. | Epigallocatechin gallate/iron nano-complexes (EIN) | Versatile coating material which eliminates EMT-type cancer cells in-vitro, and in-vivo studies. Thus, inhibit EMT program and improves conventional chemotherapy response via preventing drug chemoresistance. | [162] |
| 9. | Erbulin | A microtubule inhibitor induces MET in TNBC cells and inhibit migration and invasiveness to lungs. | [163] |
| 10. | Etodolac | COX-2 Inhibitor | [164] |
| 11. | L-NAME | pan-NOS inhibitors | [165,166] |
| 12. | L-NMMA | pan-NOS inhibitors | [167,168] |
| 13. | L-tetrahydro-2-furoic acid (L-THFA) | proline dehydrogenase inhibitor | [169,170] |
| 14. | Luteolin | Inhibited cell migration and invasion, and reversed EMT program in dose dependent manner | [171] |
| 15. | Mangiferin | Matrix metalloproteinase (MMP)-7 and -9 | [172] |
| 16. | Metformin | AMPK, mTOR inhibitor | [154,173,174] |
| 17. | Olaparib | PARP inhibitor | [175] |
| 18. | Pirfenidone | TGF-β inhibitor | [154] |
| 19. | Propranolol | β-adrenergic receptors antagonist | [154] |
| 20. | Quetiapine | RANK/RANKL inhibitor | [154] |
| 21. | Ribavirin | eiF4E, MNK, IMPDH | [154] |
| 22. | Rifabutin | BCL-6, β-catenin | [154] |
| 23. | Rolipram | PDE4 inhibitor | [176] |
| 24. | Simvastatin | HMG-CoA reductase inhibitor | [177,178,179] |
| 25. | Suramin | Heparinase inhibitor | [180] |
| Aptamer | |||
| 26. | EpCAM | Aptamer targeting EpCAM inhibit CSCs linked to siRNAs against PLK1; causes tumor regression when injected in TNBC xenograft model. | [181] |
| 27. | 39mer EGFR CL4 aptamer | Impairs the integrin- αvβ3 EGFR complex on TNBC cells | [182] |
| MiRNAs | |||
| 28. | miR-10b antagomirs | Inhibit metastasis in a mouse mammary tumor model. | [183] |
| 29. | miR-23a | Its inhibition suppressed the TGF-1-induced EMT, migration, invasion, and metastasis of breast cancer cells | [184] |
| 30. | miR-134 | Delivery of miR-134 by exosomes in TNBC cells caused the reduction of cellular migration and invasion. | [185] |
| 31. | miR200c | Expression significantly enhanced the chemosensitivity and decreased the metastatic potential of a p53(null) claudin-low tumor model | [186] |
| 32. | miR520c | Inhibit breast cancer EMT by targeting STAT3 signaling pathway. | [187] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pal, A.K.; Sharma, P.; Zia, A.; Siwan, D.; Nandave, D.; Nandave, M.; Gautam, R.K. Metabolomics and EMT Markers of Breast Cancer: A Crosstalk and Future Perspective. Pathophysiology 2022, 29, 200-222. https://doi.org/10.3390/pathophysiology29020017
Pal AK, Sharma P, Zia A, Siwan D, Nandave D, Nandave M, Gautam RK. Metabolomics and EMT Markers of Breast Cancer: A Crosstalk and Future Perspective. Pathophysiology. 2022; 29(2):200-222. https://doi.org/10.3390/pathophysiology29020017
Chicago/Turabian StylePal, Ajay Kumar, Prateek Sharma, Alishan Zia, Deepali Siwan, Dipali Nandave, Mukesh Nandave, and Rupesh K. Gautam. 2022. "Metabolomics and EMT Markers of Breast Cancer: A Crosstalk and Future Perspective" Pathophysiology 29, no. 2: 200-222. https://doi.org/10.3390/pathophysiology29020017