Genome and Phylogenetic Analysis of Infectious Hematopoietic Necrosis Virus Strain SNU1 Isolated in Korea


 , ,
, ,
Abstract
:1. Introduction
2. Material and Methods
2.1. Sample Collection, Examination, and Extraction of Viral RNA
2.2. Virus Isolation and Complete Sequencing
2.3. Phylogenetic Analysis
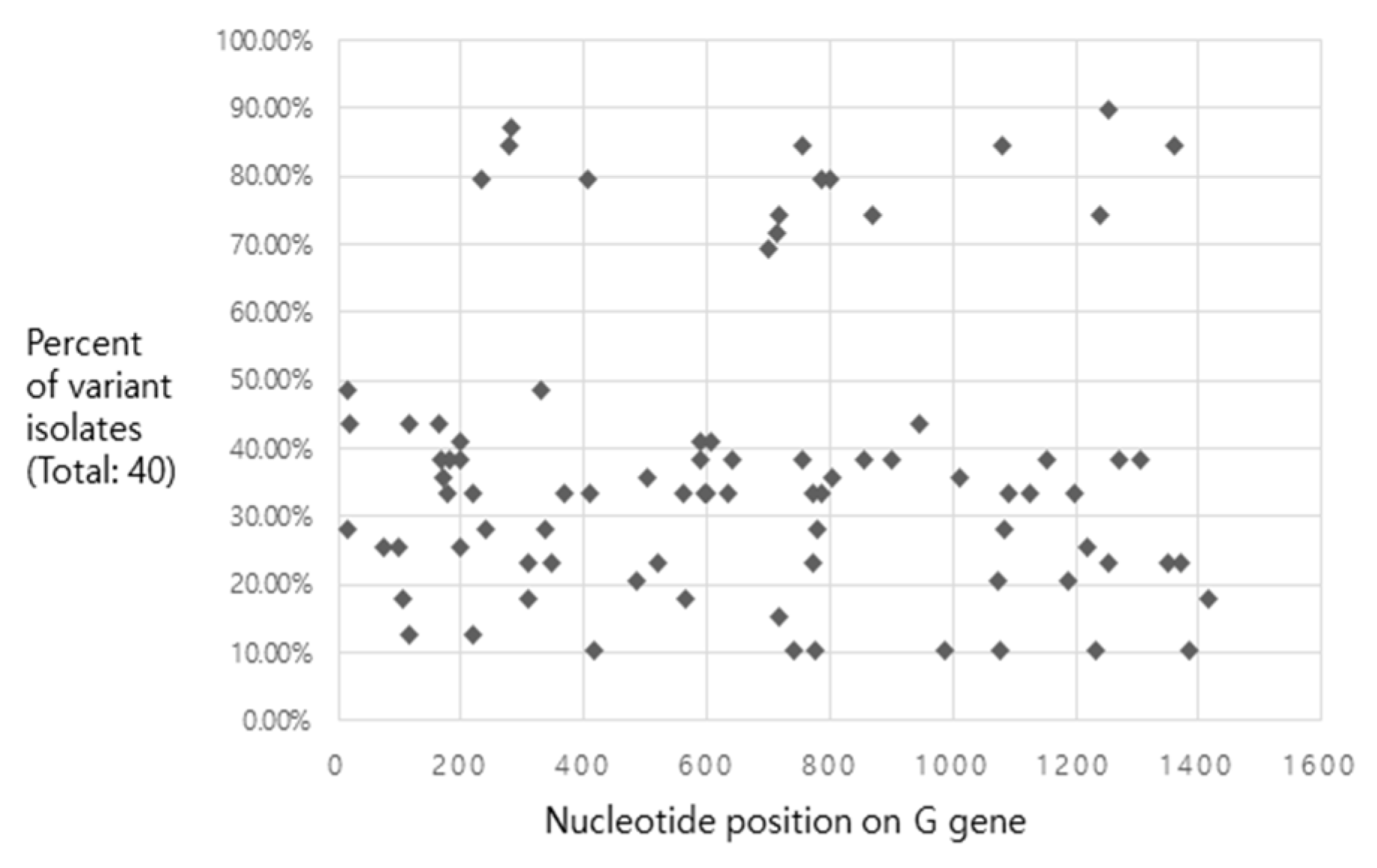
2.4. Analysis of Single Nucleotide Polymorphisms
3. Results and Discussion
3.1. Sample Examination and Virus Isolation
3.2. Complete Sequence of the IHNV Strain SNU1
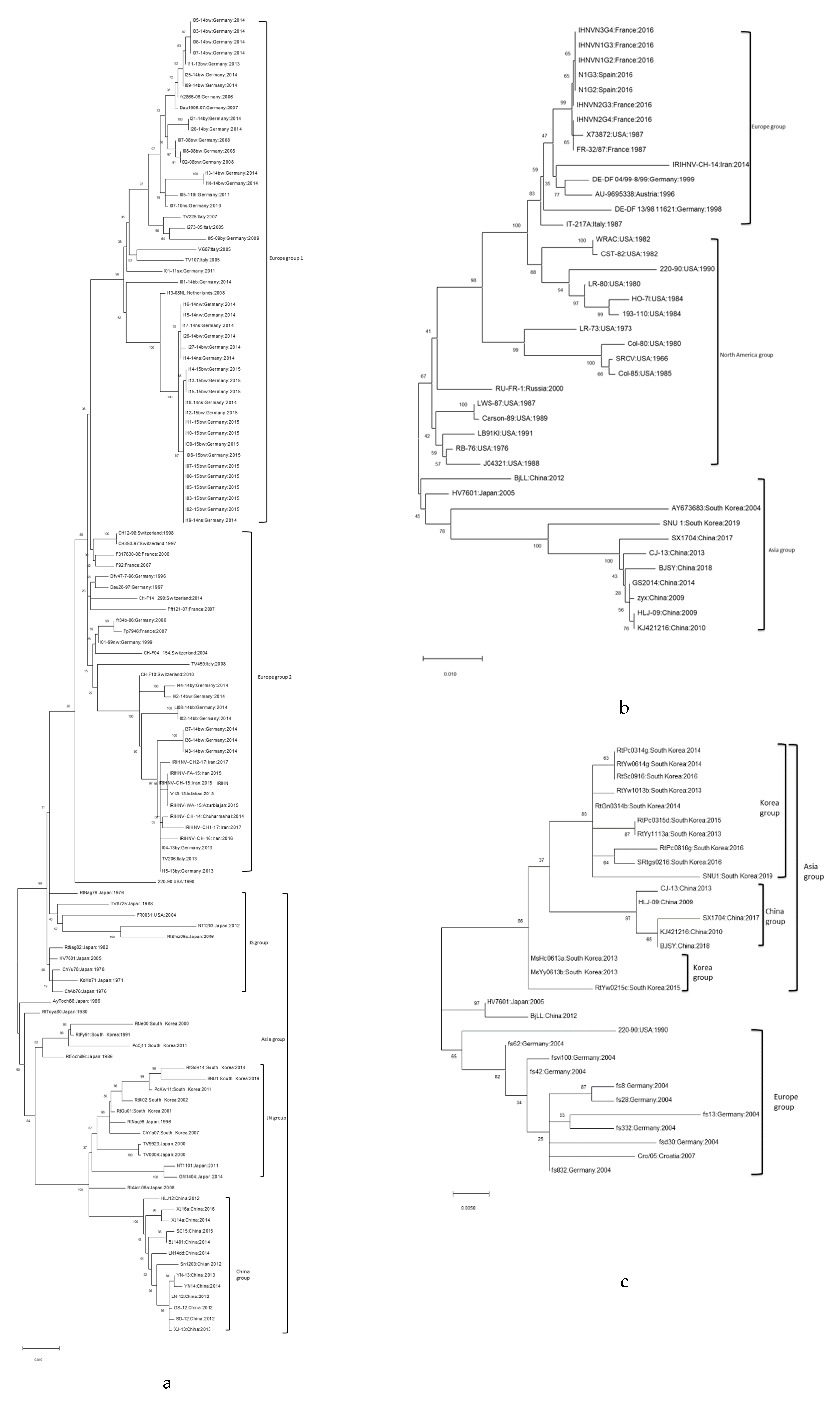
3.3. Phylogenetic Analysis of IHNV Strain SNU1
3.4. Features of IHNV Korean Isolates
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Dixon, P.; Paley, R.; Alegria-Moran, R.; Oidtmann, B. Epidemiological characteristics of infectious hematopoietic necrosis virus (IHNV): A review. Vet. Res. 2016, 47, 63. [Google Scholar] [CrossRef] [PubMed]
- Boudinot, P.; Blanco, M.; de Kinkelin, P.; Benmansour, A. Combined DNA immunization with the glycoprotein gene of viral hemorrhagic septicemia virus and infectious hematopoietic necrosis virus induces double-specific protective immunity and nonspecific response in rainbow trout. Virology 1998, 249, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Wedemeyer, G.A.; Nelson, N.C.; Smith, C.A. Survival of the salmonid viruses infectious hematopoietic necrosis (IHNV) and infectious pancreatic necrosis (IPNV) in ozonated, chlorinated, and untreated waters. J. Fish. Res. Board Can. 1978, 35, 875–879. [Google Scholar] [CrossRef]
- Kurath, G.; Garver, K.A.; Troyer, R.M.; Emmenegger, E.J.; Einer-Jensen, K.; Anderson, E.D. Phylogeography of infectious haematopoietic necrosis virus in North America. J. Gen. Virol. 2003, 84, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.S.; Oh, M.J.; Nishizawa, T.; Park, J.W.; Kurath, G.; Yoshimizu, M. Genotyping of Korean isolates of infectious hematopoietic necrosis virus (IHNV) based on the glycoprotein gene. Arch. Virol. 2007, 152, 2119–2124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishizawa, T.; Kinoshita, S.; Kim, W.S.; Higashi, S.; Yoshimizu, M. Nucleotide diversity of Japanese isolates of infectious hematopoietic necrosis virus (IHNV) based on the glycoprotein gene. Dis. Aquat. Organ. 2006, 71, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Park, M.A.; Sohn, S.G.; Lee, S.D.; Chun, S.K.; Park, J.W.; Fryer, J.L.; Hah, Y.C. Infectious haematopoietic necrosis virus from salmonids cultured in Korea. J. Fish. Dis. 1993, 16, 471–478. [Google Scholar] [CrossRef]
- Troyer, R.M.; Kurath, G. Molecular epidemiology of infectious hematopoietic necrosis virus reveals complex virus traffic and evolution within southern Idaho aquaculture. Dis. Aquat. Organ. 2003, 55, 175–185. [Google Scholar] [CrossRef]
- Enzmann, P.J.; Castric, J.; Bovo, G.; Thiery, R.; Fichtner, D.; Schütze, H.; Wahli, T. Evolution of infectious hematopoietic necrosis virus (IHNV), a fish rhabdovirus, in Europe over 20 years: Implications for control. Dis. Aquat. Organ. 2010, 89, 9–15. [Google Scholar] [CrossRef]
- Enzmann, P.J.; Kurath, G.; Fichtner, D.; Bergmann, S.M. Infectious hematopoietic necrosis virus: Monophyletic origin of European isolates from North American genogroup M. Dis. Aquat. Organ. 2005, 66, 187–195. [Google Scholar] [CrossRef]
- Xu, L.; Zhao, J.; Liu, M.; Kurath, G.; Breyta, R.B.; Ren, G.; Lu, T. Phylogeography and evolution of infectious hematopoietic necrosis virus in China. Mol. Phylogenet. Evol. 2019, 131, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Jia, P.; Breyta, R.B.; Li, Q.; Qian, X.; Wu, B.; Zheng, W.; Jin, N. Insight into infectious hematopoietic necrosis virus (IHNV) in Chinese rainbow trout aquaculture from virus isolated from 7 provinces in 2010–2014. Aquaculture 2018, 496, 239–246. [Google Scholar] [CrossRef]
- Jia, P.; Zheng, X.C.; Shi, X.J.; Kan, S.F.; Wang, J.J.; He, J.Q.; Liu, H. Determination of the complete genome sequence of infectious hematopoietic necrosis virus (IHNV) Ch20101008 and viral molecular evolution in China. Infect. Genet. Evol. 2014, 27, 418–431. [Google Scholar] [CrossRef] [PubMed]
- Alonso, M.; Rodrıguez, S.; Prieto, S.I.P. Nested PCR improves detection of infectious hematopoietic necrosis virus in cells coinfected with infectious pancreatic necrosis virus. J. Virol. Methods 1999, 81, 1–9. [Google Scholar] [CrossRef]
- Lopez-Lastra, M.; Gonzalez, M.; Jashes, M.; Sandino, A.M. A detection method for infectious pancreatic necrosis virus (IPNV) based on reverse transcription (RT)-polymerase chain reaction (PCR). J. Fish. Dis. 1994, 17, 269–282. [Google Scholar] [CrossRef]
- Chico, V.; Gomez, N.; Estepa, A.; Perez, L. Rapid detection and quantitation of viral hemorrhagic septicemia virus in experimentally challenged rainbow trout by real-time RT-PCR. J. Virol. Methods 2006, 132, 154–159. [Google Scholar] [CrossRef]
- Marshall, S.; Heath, S.; Henríquez, V.; Orrego, C. Minimally Invasive Detection ofPiscirickettsia salmonis in Cultivated Salmonids via the PCR. Appl. Environ. Microbiol. 1998, 64, 3066–3069. [Google Scholar]
- Metselaar, M.; Thompson, K.D.; Gratacap, R.M.L.; Kik, M.J.; LaPatra, S.E.; Lloyd, S.J.; Adams, A. Association of red-mark syndrome with a Rickettsia-like organism and its connection with strawberry disease in the USA. J. Fish. Dis. 2010, 33, 849–858. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. In Nucleic Acids Symposium Series; Information Retrieval Ltd.: London, UK, 1999; Volume 41, pp. 95–98. [Google Scholar]
- Amend, D.F. Control of infectious hematopoietic necrosis virus disease by elevating the water temperature. J. Fish. Res. Board Can. 1970, 27, 265–270. [Google Scholar] [CrossRef]
- Schönherz, A.A.; Lorenzen, N.; Guldbrandtsen, B.; Buitenhuis, B.; Einer-Jensen, K. Ultra-deep sequencing of VHSV isolates contributes to understanding the role of viral quasispecies. Vet. Res. 2016, 47, 10. [Google Scholar] [CrossRef]
- Panzarin, V.; Holmes, E.C.; Abbadi, M.; Zamperin, G.; Quartesan, R.; Milani, A.; Toffan, A. Low evolutionary rate of infectious pancreatic necrosis virus (IPNV) in Italy is associated with reduced virulence in trout. Virus Evol. 2018, 4, vey019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbadi, M.; Fusaro, A.; Ceolin, C.; Casarotto, C.; Quartesan, R.; Dalla Pozza, M.; Panzarin, V. Molecular evolution and phylogeography of co-circulating IHNV and VHSV in Italy. Front. Microbiol. 2016, 7, 1306. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhao, L.L.; Li, Y.J.; Tang, L.J.; Qiao, X.Y.; Jiang, Y.P.; Liu, M. Analysis of the genome sequence of infectious hematopoietic necrosis virus HLJ-09 in China. Virus Genes 2016, 52, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Yong, C.Y.; Ong, H.K.; Tang, H.C.; Yeap, S.K.; Omar, A.R.; Ho, K.L.; Tan, W.S. Infectious hematopoietic necrosis virus: Advances in diagnosis and vaccine development. PeerJ 2019, 7, e7151. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| IHNV Primer | Sequences (5’-3’) | Position | Annealing Temp, Time |
|---|---|---|---|
| IHNV 1FF | GTATAAGAAAAGTAACTTGACT | 1 | 50 °C, 30 s |
| IHNV 1RR | CAATCCCTTGGCTGGTTGC | 670 | |
| IHNV 2FF | CAAGCTCGAGGTCTTGCAA | 610 | 55 °C, 30 s |
| IHNV 2RR | CCCTCTCCGGTTGAGCCAT | 1220 | |
| IHNV 3FF | GAGATCGCTCGTCTCCTTGT | 1160 | 50 °C, 30 s |
| IHNV 3RR | GGCCTGGTGGGCCTGTCT | 1760 | |
| IHNV 4FF | GAGAGCTGTCAGGATGCC | 1690 | 54 °C, 30 s |
| IHNV 4RR | CCTCTCCTCGTCTCCGCT | 2330 | |
| IHNV 5FF | CAAACGAGAGCATGTCTATTTTCA | 2240 | 55 °C, 30 s |
| IHNV 5RR | TTTGGCTGTTTGCTCCGCAG | 3060 | |
| IHNV 6FF | CCTGTTTTCATCCAGCCATGT | 2690 | 55 °C, 60 s |
| IHNV 6RR | CCTCAAGACATTCCTCTCTG | 3940 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oh, W.T.; Jun, J.W.; Giri, S.S.; Yun, S.; Kim, H.J.; Kim, S.G.; Kim, S.W.; Han, S.J.; Kwon, J.; Park, S.C. Genome and Phylogenetic Analysis of Infectious Hematopoietic Necrosis Virus Strain SNU1 Isolated in Korea. Pathogens 2019, 8, 200. https://doi.org/10.3390/pathogens8040200
Oh WT, Jun JW, Giri SS, Yun S, Kim HJ, Kim SG, Kim SW, Han SJ, Kwon J, Park SC. Genome and Phylogenetic Analysis of Infectious Hematopoietic Necrosis Virus Strain SNU1 Isolated in Korea. Pathogens. 2019; 8(4):200. https://doi.org/10.3390/pathogens8040200
Chicago/Turabian StyleOh, Woo Taek, Jin Woo Jun, Sib Sankar Giri, Saekil Yun, Hyoun Joong Kim, Sang Guen Kim, Sang Wha Kim, Se Jin Han, Jun Kwon, and Se Chang Park. 2019. "Genome and Phylogenetic Analysis of Infectious Hematopoietic Necrosis Virus Strain SNU1 Isolated in Korea" Pathogens 8, no. 4: 200. https://doi.org/10.3390/pathogens8040200