1. Introduction
Molecular epidemiology of hepatitis C virus (HCV) infection is exceptionally complex due to the highly diverse viral genome, which is classified into >90 genotypes and subtypes [
1]. HCV subtypes such as 1a, 1b, and 3a are considered to be epidemic and are widely distributed worldwide. They are characterized by lower variability and are considered to have spread across the world in the 20th century through unsafe medical procedures, transfusion, blood products, and intravenous drug use [
2,
3,
4,
5,
6]. On the other hand, endemic subtypes are restricted to certain geographic areas such as the Middle East, South and East Asia, and West and Central Africa, have higher genetic variability, and are considered to be much older [
5,
6,
7,
8]. Furthermore, the existence of a quasispecies, which is described as distributions of genetically non-identical but related genomes subjected to a continuous process of competition and selection in a given host, is another factor contributing to the extensive HCV genetic diversity [
9,
10].
Croatia is a South-Eastern European country with a relatively low prevalence of HCV infection. It is estimated that 35,000–45,000 persons, or 0.9% of the Croatian population, are infected with chronic hepatitis C [
11]. HCV genotype distribution in Croatia varies according to different population groups and regions. Genotype 1 (GT1) is the most prevalent among the general population (57–80%), followed by genotype 3 (GT3) (13–48%), while the prevalence of genotype 2 (GT2) (1–2%) and 4 (GT4) (4–7%) is low [
11,
12]. HCV transmission via blood transfusion and unsafe medical procedures was significantly reduced after the introduction of blood donation screening for HCV in the 1990s [
11,
13]. In Croatia, blood transfusion and haemodialysis before 1992 were strongly associated with an infection with HCV GT1, especially subtype 1b (GT1b) [
11,
14]. Today, intravenous drug use represents the major route of HCV transmission in developed countries, with a prevalence of 2–80% among intravenous drug users (IDU) [
15,
16,
17]. Estimated population size of IDU in Croatia is around 15,000, and the most common HCV subtypes in this population are subtype 3a (GT3a) (61%) and subtype 1a (GT1a) (24%) [
11,
18].
The discovery of the new direct acting antivirals (DAA) has revolutionized the treatment of HCV patients and their efficacy has prompted the World Health Organization (WHO) to launch a strategy which calls for the elimination of viral hepatitis as a public health threat by 2030 [
19]. Major changes have been implemented in HCV treatment strategies in Croatia in the last several years. Treatment eligibility was expanded to include patients aged 18–84 years, regardless of fibrosis stage, and the annual treated number of patients tripled from 2015 to 2018 [
20]. However, some quasispecies variants bear polymorphisms in drug-targeted genes, which may negatively impact antiviral treatment and confer resistance to direct acting antivirals in 5–10% of the patients [
21,
22,
23]. The use of direct acting antivirals with high barrier to resistance and a combination of several DAA classes can be beneficial in ensuring a successful achievement of sustained virological response (SVR). A combination of highly efficient pangenotypic DAA has reduced the need for baseline resistance testing [
23,
24,
25,
26]. However, current guidelines suggest testing for NS5A resistance associated substitutions (RAS) in the treatment of naïve patients with HCV GT3 and liver cirrhosis before treatment with sofosbuvir/velpatasvir since Y93H mutation was shown to reduce SVR to 84–88% in these patients [
26,
27]. Furthermore, testing for NS5A RAS is advised before treatment with grazoprevir/elbasvir in patients infected with HCV GT1a [
26,
28]. Literature data on baseline DAA resistance prevalence varies across different studies, partly due to inconsistent classification of observed substitutions as RAS. Our group previously conducted a preliminary analysis of baseline resistance to NS3 protease inhibitors and NS5A inhibitors in patients infected with GT1a and GT1, respectively. High prevalence of NS3 RAS (46.3%), especially Q80K RAS (42.6%), was found in GT1a-infected patients, while clinically relevant NS5A RAS were shown to be more common in GT1b (24.2%) compared with GT1a (7.8%) [
29,
30]. However, epidemic history and resistance prevalence to all DAA classes for the most common HCV genotypes and subtypes circulating in Croatia remain unknown. The aim of this study was to conduct a comprehensive analysis of the molecular, virological, clinical, and epidemiological characteristics of the HCV epidemic in Croatia in a four-year period.
3. Discussion
In this study, we combined molecular, virological, clinical, and epidemiological data from 300 HCV patients receiving clinical care at the University Hospital for Infectious Diseases Zagreb (UHID) and the Reference Centre for Viral Hepatitis in Croatia from 2016 to 2019, in order to characterize Croatian HCV epidemic. Previous studies of HCV epidemiology in Croatia were mainly focused only on the prevalence and distribution of various HCV genotypes and subtypes, while this study broadens this knowledge by including transmission and phylodynamic analyses. The most common HCV genotypes in the Croatian general population were found to be GT1 and GT3, with no major changes in molecular epidemiology in the last 20 years [
12,
31]. In this study, we observed higher median age of GT1b-infected patients (61 years) compared with GT1a (43 years) and GT3a (44 years), which is similar to data from a previous national study and could be attributed to predominantly iatrogenic mode of HCV transmission in GT1b which was significantly reduced in the last 30 years [
11,
31]. However, median ages of all patients included in this study were overall at least 10 years higher which suggests aging of the patient population and could be attributed to long persistence of HCV infection and the relatively recent availability of the effective direct antiviral therapy. The results of previous European and national studies are concordant with our finding that GT1a and GT3a were more frequently associated with IDU [
3,
11,
17,
32].
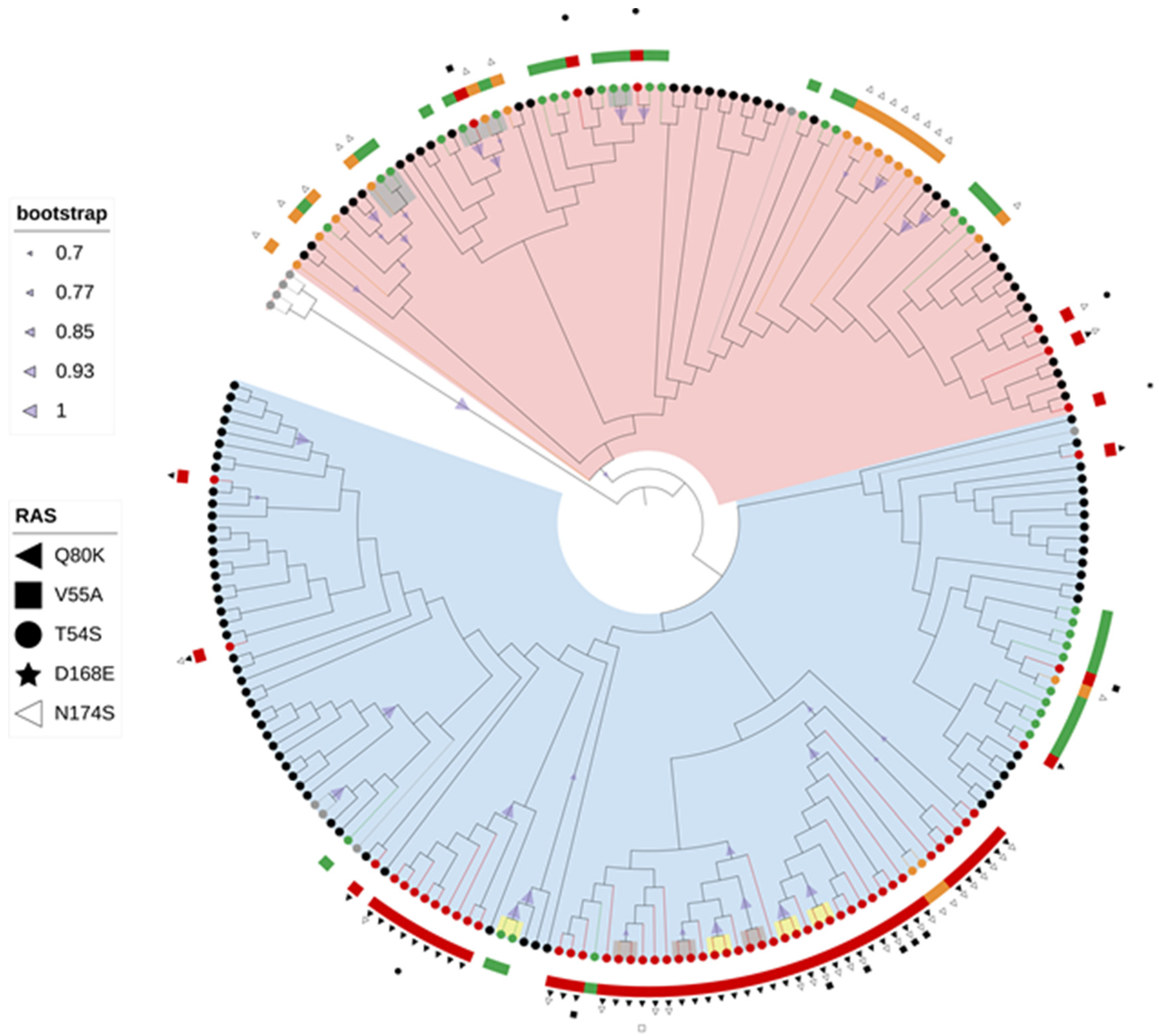
We conducted the first comprehensive analysis of baseline RAS across HCV genome of the most common genotypes circulating in Croatia. The prevalence of RAS varied greatly according to HCV genotypes. The highest prevalence of RAS was observed in the NS3 region (33.0%), especially among GT1a-infected patients (68.8%), with the most commonly detected RAS being Q80K (46.8%) and N174S (39.5%). Preliminary study conducted by Grgic et al. (2017) in the NS3 region of 136 GT1a-infected patients showed similar prevalence of Q80K and N174S RAS [
29]. The prevalence of NS3 RAS was shown to be much lower in GT1b (5.0%) and GT3a (0.9%), especially when the analysis was limited to clinically relevant RAS. Literature data from other studies in Southern and Eastern Europe is limited. One Italian study showed RAS prevalence of 20.4% in the NS3 region, with the majority of NS3 RAS observed in GT1a (45.2%) and GT1b (10.8%). Similarly, to our study, most frequently observed RAS was Q80K in GT1a (17.0%) [
33]. Q80K RAS was common in other European studies. Jimenez-Sousa et al. found prevalence of Q80K in the range from 7.3% to 22.2% in 2568 patients with chronic hepatitis C in 115 hospitals in Spain [
34]. Beloukas et al. (2015) studied the occurrence of Q80K in 238 treatment-naïve HCV-1a carriers in England and found 14.9–27.1% prevalence of Q80K RAS [
35]. It should be noted that Q80K mutation is considered clinically relevant in respect to simeprevir, which is first generation protease inhibitor. Today, most of the DAA combinations include third generation protease inhibitors such as grazoprevir, glecaprevir, and voxilaprevir, which show higher genetic resistance barrier and better pangenotypic activity [
26,
36,
37]. However, Q80K RAS is still relevant in the context of new protease inhibitors since it causes reduced susceptibility to voxilaprevir [
22,
38]. In our study, Y56F RAS, which causes reduced susceptibility to grazoprevir, was common in GT1b (22.5%), followed by T54S (2.5%), V55A (1.3%), and N174F (1.3%). In Italy, several studies found very low prevalence of Y56F in GT1b (0–0.2%), while the most common RAS was T54S (1.9–4%) [
33,
39].
Results of this research showed higher prevalence of baseline NS5A RAS in GT1 (18.0%) compared with GT3a (6.3%). Furthermore, even though the overall prevalence of RAS was similar among different subtypes of GT1, a large difference was observed in the prevalence of clinically relevant RAS with 20.0% of GT1b-infected patients and 4.6% of GT1a-infected patients having resistance conferring mutations. Such discrepancy was first observed in our preliminary study where Y93H was the only RAS detected in GT1b-infected patients (24.2%) [
30]. Current analysis revealed other RAS in GT1b-infected patients with the prevalence >5% (R30Q, L31M), while the prevalence of Y93H was lower (11.3%), even though it still represents the most common NS5A RAS. In this study, Y93H RAS was not detected in GT1a, while its prevalence in GT3a-infected patients was very low (1.8%). NS5A represents the most important genomic region for resistance testing since it contains the largest number of possible mutation sites, while many of the most commonly prescribed DAA combinations include NS5A inhibitor [
38,
40]. There is no recent data on the prevalence of resistance to NS5A inhibitors in south eastern Europe. An Italian observational study which included 1032 treatment naive patients from 23 clinical centres found NS5A RAS prevalence of 6.8% in GT1a, 10.3% in GT1b, and 8.5% in GT3a when the analysis was limited to clinically relevant RAS [
33]. Other Italian studies found similar prevalence of NS5A RAS in GT3a (11.5%), GT1a (4.9%), and GT1b (23.0%) [
41,
42], while national studies across Europe found NS5A RAS prevalence of 2–18.9% in GT1a, 13.0–43.3% in GT1b, and 3–23.5% in GT3a [
40,
43,
44,
45,
46]. It should be noted that such high variation in RAS prevalence among a single genotype could at least partly be attributed to the lack of standardisation in definition of RAS.
For the first time, this study analysed the prevalence of resistance associated substitutions to nucleoside NS5B inhibitors in Croatia. High genetic barrier to resistance and pangenetic activity of NS5B inhibitors are possible due to the highly conserved NS5B active site [
23]. The only NI NS5B RAS found in the current study was L159F in GT1b (31.3%). Literature data on the prevalence of NS5B resistance in national studies are scarce. Several studies in southern Europe found similar prevalence of NI NS5B RAS in GT1b (14.8–21.1%) [
33,
40]. No NI NS5B RAS were observed in GT1a or GT3a patients [
33,
41].
Since every DAA class induces specific mutation profile characteristic for various HCV genotypes and subtypes, the use of combination therapy with agents targeting various viral proteins has been successful in obtaining high rates of SVR [
23,
24,
25,
26]. Only a few studies analysed the presence of RAS to all three DAA classes for every patient, mainly due to the lack of sequences spanning across all relevant genome regions. Our data show that multiclass resistance is relatively uncommon in Croatia (8.3%), with the most frequently observed RAS in both the NS3 and NS5A region (5.0%). The prevalence of combination RAS involving the NS5B region was low (<2%), especially NS3 + NS5A + NS5B RAS (0.7%). Global analysis of DAA RAS using published GenBank data similarly found low prevalence of multiple RAS in different regions of the same sequence (1.2–3.5%), with the exception of NS3 + NS5A RAS (15.6%) [
47]. In a study conducted as a part of the AVIATOR, a phase 2 clinical trial, none of the patients had baseline RAS in all three targets [
48]. Among European national studies, authors of a Portuguese study found 4.9% prevalence of NS5A + NS5B RAS, while an Italian study found 7.3% prevalence of multiclass RAS, with RAS simultaneously observed in the NS3 + NS5A region (2.7%), NS3 + NS5B region (1.9%) and NS5A + NS5B region (1.6%) [
40]. The effect of the presence of multiple resistance substitutions is not extensively studied, however, it should be considered when administering DAA combinations which involve NS3 and NS5A inhibitors [
33].
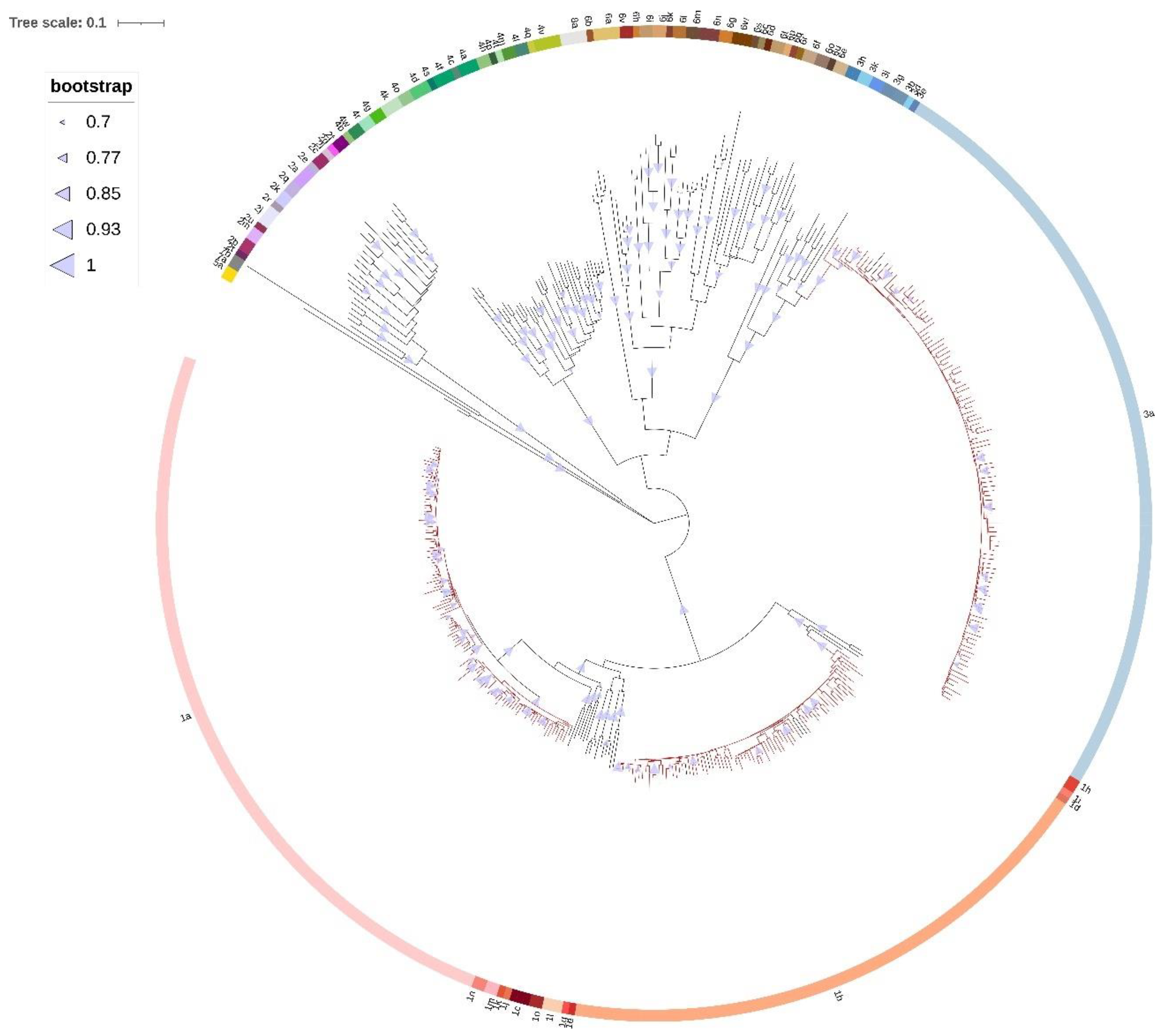
High molecular diversity of HCV genome due to continuous process of genetic variation enables the detection of evolutionary and epidemiological patterns in a relatively short time span [
9,
32,
49,
50,
51]. Phylogenetic analyses of NS5A, NS5B, and NS3 sequences showed consistent separation of GT1a sequences into two distinct clades: clade I (62.4%) and clade II (37.6%). Proportion of clade I sequences seems to be slightly higher compared to previous preliminary national studies based on single gene phylogenies [
29,
30]. The prevalence of NS3 RAS was shown to be associated with clade, with NS3 RAS observed more often in clade I (79.4%) compared with clade II (51.2%) (
p = 0.003). This phenomenon was even more pronounced when taking into consideration only clinically relevant RAS (75.0% clade I vs. 14.6% clade II,
p < 0.001), especially Q80K mutation (73.5% clade I vs. 2.4% clade II,
p < 0.001). This divergence of GT1a sequences in two distinct clades and preferential association of Q80K RAS in NS3 with clade I is in accordance with previous studies [
34,
48,
52,
53,
54]. Picket et al. showed the existence of many clade-informative sites within the NS3, NS5A, and NS5B non-structural coding regions using full-genome sequencing data [
52]. Krishnan et al. found that the majority GT1a sequences with Q80K RAS classified into clade I (98%), with overall prevalence of Q80K in clade I of 66.4% [
48]. Overall prevalence of Q80K was 46.8% in the United States and 13.5% in the European Union [
48]. Jimenes Souisa et al. also confirmed a higher prevalence of Q80K RAS in patients infected with clade I GT1a (41.5%) compared to clade II (1.6%) (
p <0.001) [
34]. De Luca et al. (2015) observed a significant difference in clade prevalence according to geographic origin, with higher prevalence of clade I among non-European sequences, represented mostly by sequences from the United States, compared with European sequences (75.7% vs. 49.3%;
p < 0.001) Q80K RAS was detected exclusively in clade I sequences with prevalence of 51.6% [
54]. Several studies suggest the origin of the Q80K polymorphism in the United States in clade I in the middle of the 20th century [
53,
54]. Our study shows high prevalence of clade I and Q80K RAS, which is comparable with data from the United States, suggesting efficient transfer of this polymorphism from the United States to Europe.
Sequence analysis of fast evolving viruses such as HCV was shown to be efficient method for viral transmission tracing [
16,
17,
49,
51]. Phylogenetic analysis demonstrated that 27 (9.0%) of Croatian HCV sequences had a presumed epidemiological link with another sequence and classified into 13 transmission pairs or clusters which were the most common in GT3a, while IDU was shown to be a risk factor statistically associated with clustering. Several studies performed phylogenetic cluster analysis among IDU to infer epidemiologic links and factors associated with clustering. Hackman et al. (2020) found that 46% of 820 community recruited IDU in Baltimore had genetically linked HCV infections with an average cluster size of 2–3 patients [
49]. Clipman et al. (2021) analysed 483 HCV sequences from IDU in India and found transmission cluster prevalence of 28.8% with a median cluster size of three individuals [
16]. Cluster analysis showed that younger age (<35 years) was significantly associated with being in a cluster, similarly to our study [
16,
50]. A study by Parczewski et al. (2018) showed that sequences obtained from patients with F3-F4 liver fibrosis less commonly formed clusters and pairs (22.2% vs. 43.7%,
p < 0.001) and that NS5A RAS were less frequent among clustered sequences (5.2% vs. 11.2%,
p = 0.039) [
55]. However, in our research, fibrosis stage and presence of RAS showed no association with clustering. Palladino et al. (2020) showed similar prevalence of transmission clusters (10.5%) in the HCV GT1a population in Spain based on the NS5A region. However, male-dominated transmission pairs and predomination of clade II viruses were shown to be specific for the Spanish HCV-GT1a population [
51]. In our research, the transmission pairs and clusters comprised exclusively of clade I viruses in GT1a, while the patients’ gender showed no association with clustering.
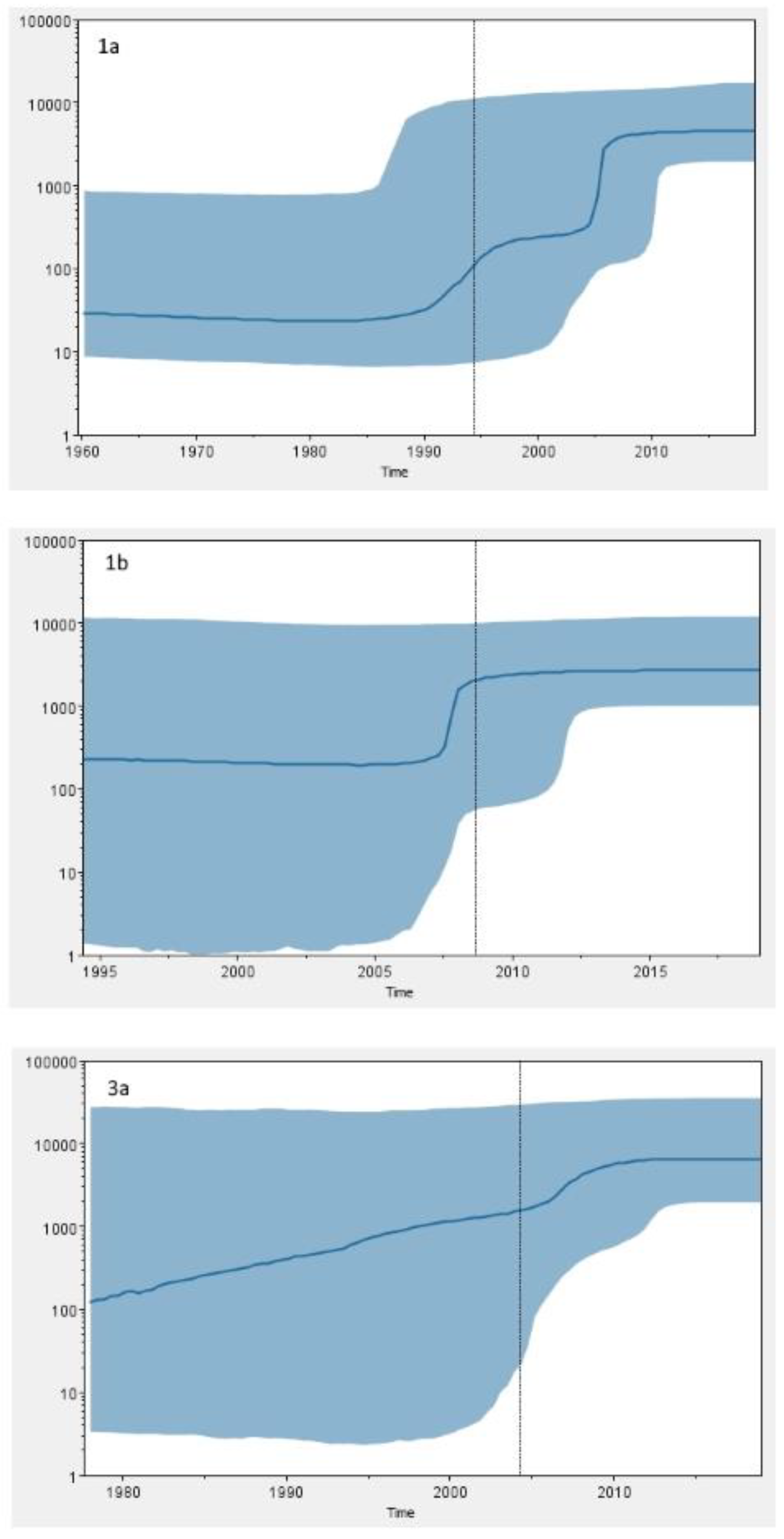
Phylodynamic analysis was performed on concatenated NS3, NS5A, and NS5B sequences of every patient in order to increase the confidence of inferred evolutionary relationships. Several studies have tried to reconstruct the origin and evolutionary history of the most common epidemic HCV subtypes such as GT1a, GT1b, and GT3a [
4,
32,
50,
56,
57,
58,
59]. A study by Margiokinis et al. (2009) supports a massive expansion of the GT1a and GT1b epidemics between 1940 and 1980, with the expansion of HCV GT1b preceding that of GT1a by 15–17 years. Authors concluded that the global epidemic of both subtypes coincides with the vast increase of unsafe medical procedures during and after World War II, while GT1a expansion was at least in part driven by the increase of IDU after 1960 [
4]. Cuypers et al. (2017) found that the first introduction of GT1a in Italy was timed around 1958 (95% HPD, 1949–1964), probably on multiple occasions from the US and Western Europe [
56]. Our study shows that last common ancestor of Gt1a sequences dates around 1960 (95% HPD, 1911–1994), a period of the post-World War II and Cold War–era migrations. Hoshino et al. (2018) found the increase of GT1a population in Okinawa, Japan in two periods—from 1965 to 1980, which could be attributed to the US occupation after World War II and in the beginning of the 21st century which could be associated with an increase in the illicit drug use [
59]. The Bayesian skyline analysis of Croatian GT1a sequences revealed an exponential increase of HCV GT1a infections in the 1990s and its continued growth throughout the first decade of the 2000s, which is in accordance with a huge increase in the number of IDU registered in this period in Croatia [
60]. Unlike many other studies [
4,
32,
50], the most recent common ancestor of Croatian GT1b sequences dates to the 1990s (95% HPD, 1975–2011), which is a period of introduction of HCV antibody screening of all donor blood and subsequent reduction in iatrogenic transmission of HCV characteristic for GT1b. The Bayesian skyline analysis suggests that GT1b population in Croatia was relatively constant in the last 30 years. However, the results have to be taken with caution due to the broad estimation errors (95% HPD limits). Furthermore, the tMRCA for GT1b sequences seems to be too recent to fit epidemiological data. Further research is needed in order to infer epidemiologic history of GT1b in Croatia.
Phylodynamic and phylogeographic analyses of GT3a epidemic history suggest that the most probable origin of this subtype is the Golden Crescent and Indian subcontinent [
32,
57]. Recent Montenegrin studies showed that GT3a was most likely exported from this area, which has a long history of opium production, to Europe in the first half of the 20th century by illicit drug trade, and reached Montenegro in the 1960s or 1970s, followed by exponential increase of effective population size [
32,
58]. The results of our research show that the last common ancestor of GT3a sequences in Croatia dates to 1978 (95% HPD, 1933–2004), causing an epidemic which exponentially grew until the last decade, suggesting similar temporal pattern. However, continuously high upper limit of 95% HPD prevents any definitive conclusions. The observed epidemic growth coincides with the huge increase in the number of new registered addicts in the early 1990s in Croatia [
60]. The rise in illegal drug use in Croatia, including heroin, is considered to have begun in the 1960s, but became a pronounced social problem during the 1980s [
60]. The first harm reduction programs were implemented in the early 2000s, and their efficiency could be partly observed in HCV GT1a and GT3a dynamics, with the effective population size of both subtypes being in a nonexpanding phase in the last decade.
There are some important limitations to this study. All samples were obtained from patients in the chronic phase of HCV infection; therefore, it is possible that the obtained sequences do not necessarily reflect true composition of viral quasispecies at the time of HCV transmission due to high variability of HCV genome. However, due to the lack of symptoms, HCV infection is rarely diagnosed in the acute phase and such samples are not readily available. Our research was conducted on sequences from three key non-structural regions of the HCV genome in order to cover DAA resistance associated sites, while the most accurate phylogenetic relationships would have been obtained by whole genome sequencing. We used Sanger sequencing which is a standard methodology used in molecular epidemiology studies. It allows detection of variants present in >15% of virus quasispecies which is sufficient for resistance analysis, while deep sequencing could possibly be beneficial for more confident tracing of viral transmission events and HCV genotype diversity, including mixed infections. Due to the lack of the patient follow-up, the effect of specific RAS on achievement of sustained viral response could not be evaluated. Furthermore, the concept of HCV compartmentalization suggests that viruses sampled from the blood at a given time point do not fully represent HCV infection dynamics [
15]. However, the use of additional sites such as liver biopsies is ethically not acceptable since the successful implementation of non-invasive methods for assessing liver fibrosis. Our study covered the time span of four years which is a relatively short period. The accuracy of coalescent analysis could be improved by increasing the time frame of sampling. The presumptive mode of HCV transmission was unknown for 48.7% patients which limited the analysis of risk factors associated with HCV transmission. However, we remain confident that the results of this study provide useful and novel insights for tracing HCV genetic diversity, transmission dynamics, and epidemic history in Croatia.


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}