Redox-Active Selenium Compounds—From Toxicity and Cell Death to Cancer Treatment

Abstract
:1. Introduction
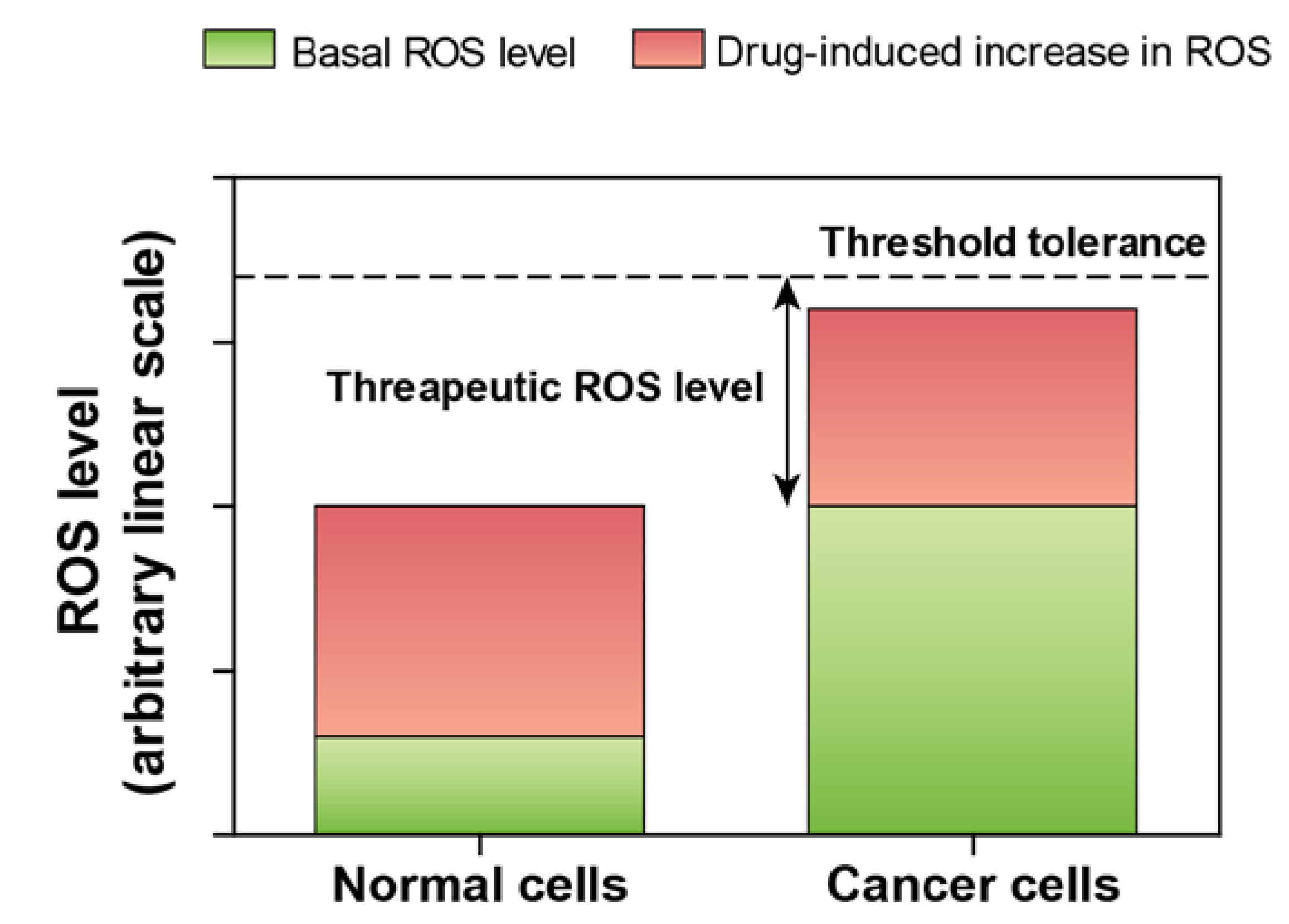
2. Selenium—An Antioxidant with Strong Pro-Oxidant Properties

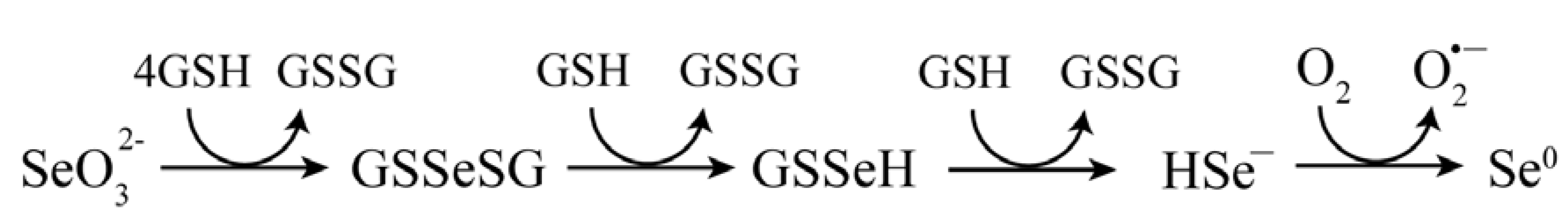
3. Acute and Chronic Selenium Toxicity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
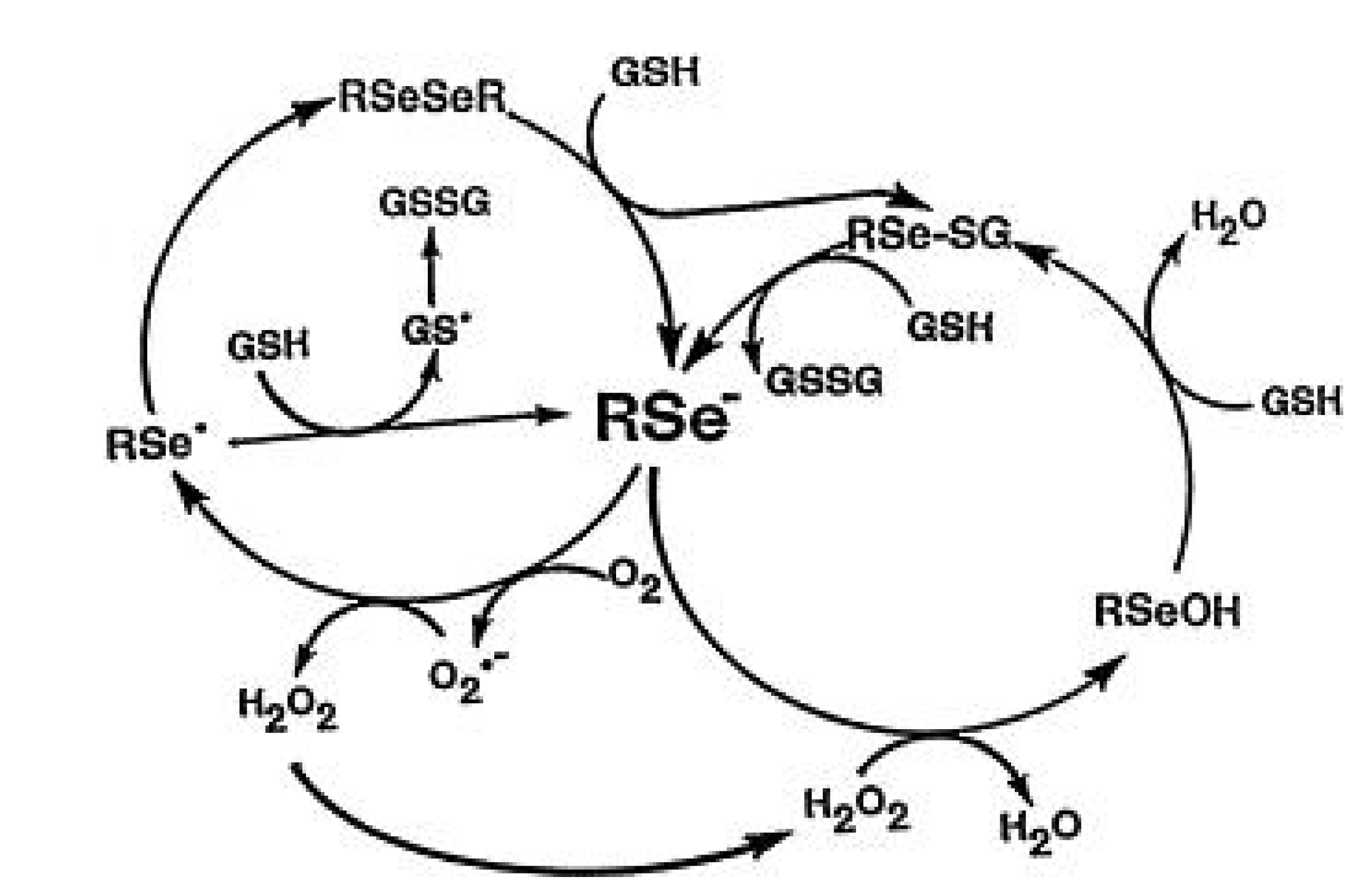
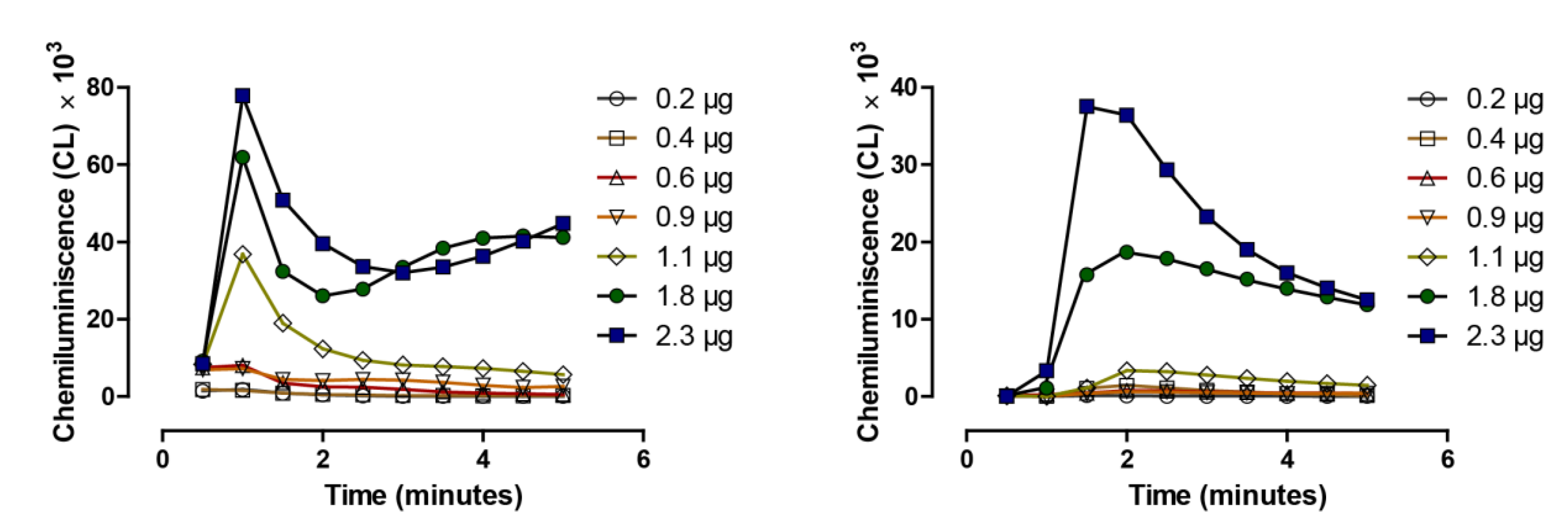
4. Redox Active Selenium Compounds and Detection of Superoxide


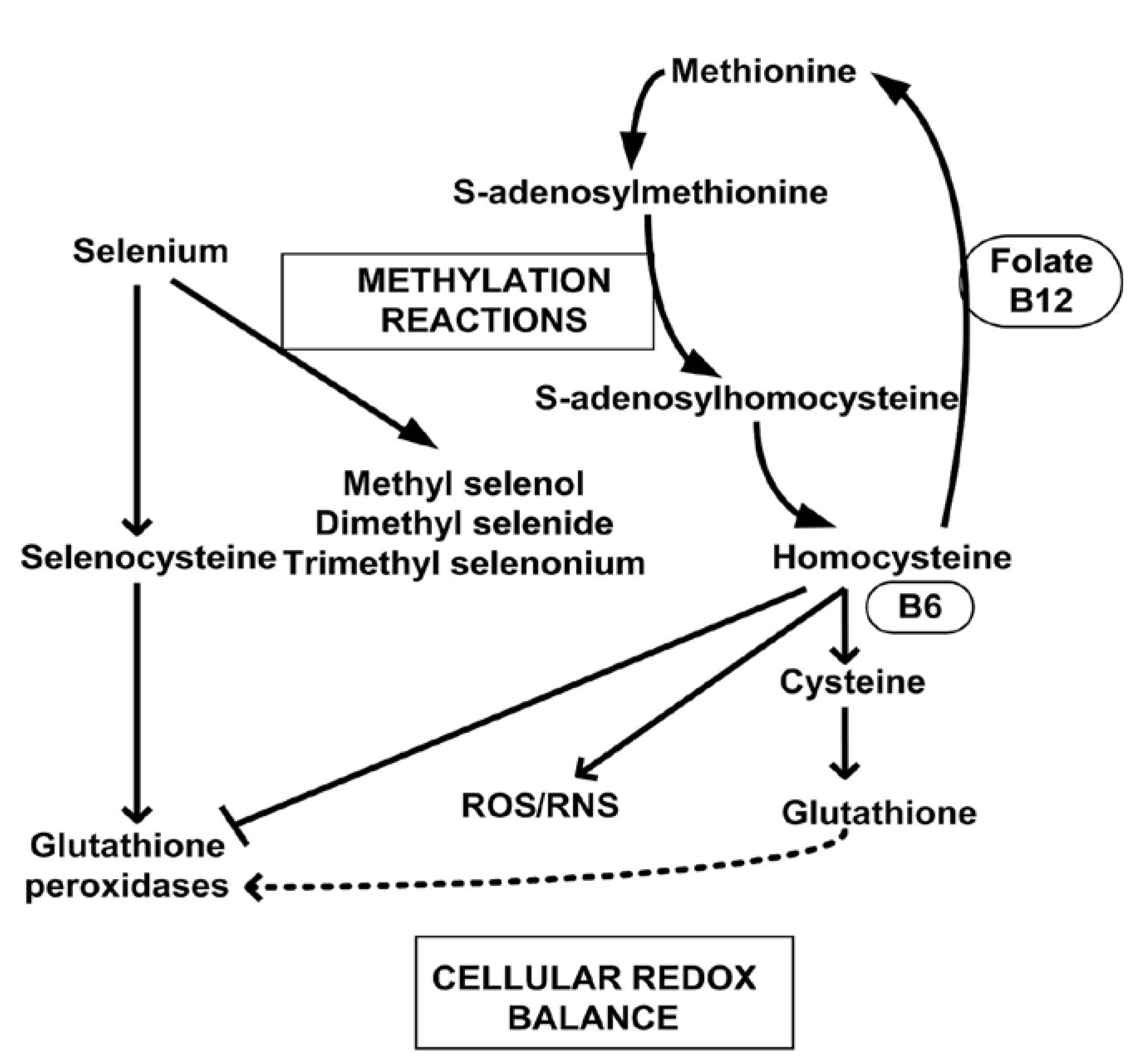
5. Methylation Reactions and Methylated Selenium Species

6. Potent Antitumor Effects of Redox-Active Selenium Compounds
7. Se-Methylselenocysteine
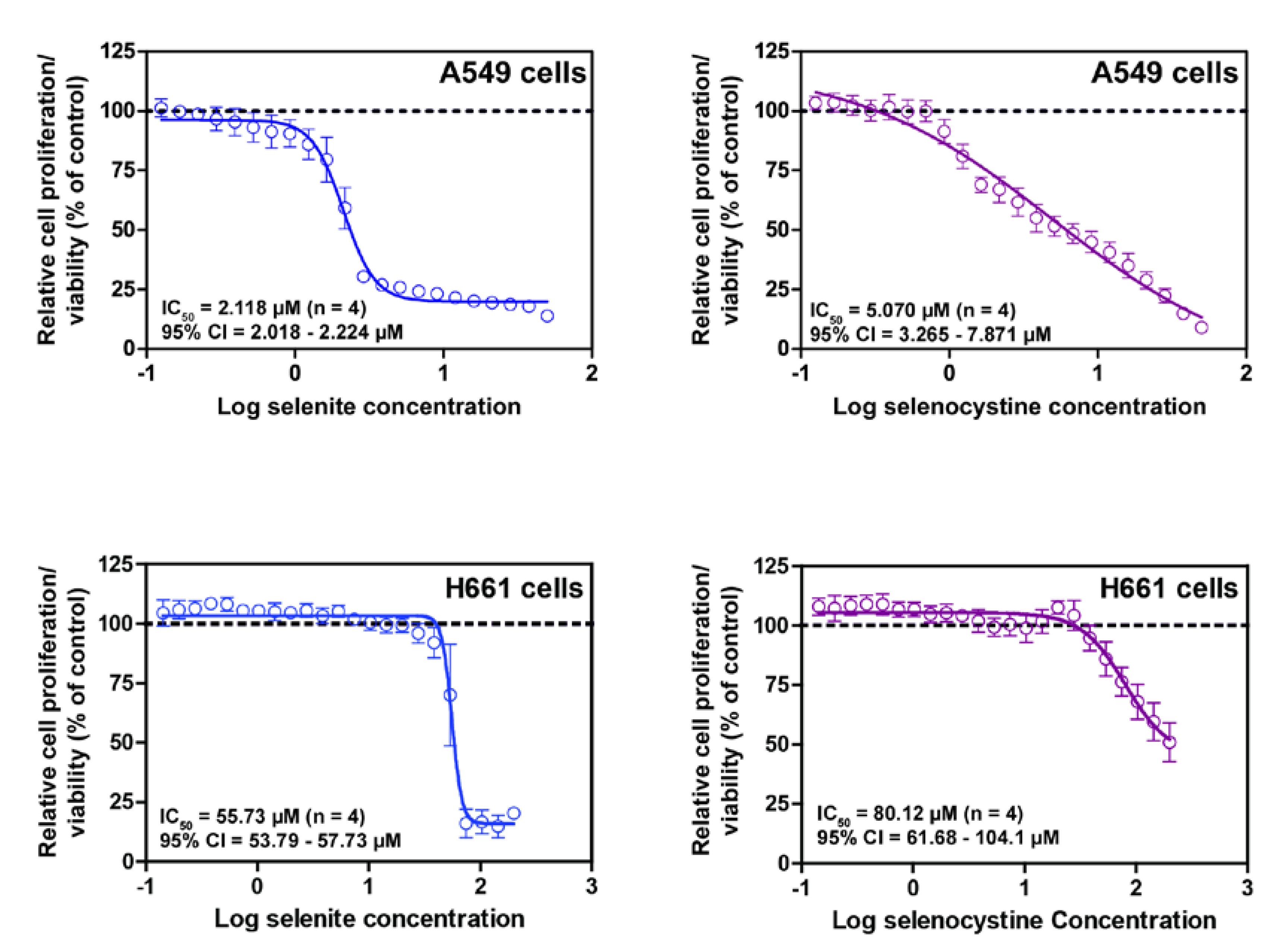
8. Selenocystine

9. Selenium in the Treatment of Cancer—A New Era in Oncology
10. Novel Selenium Compounds and Future Perspectives for Therapeutic Selenium Drugs
11. Summary and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Björnstedt, M.; Kumar, S.; Holmgren, A. Selenodiglutathione is a highly efficient oxidant of reduced thioredoxin and a substrate for mammalian thioredoxin reductase. J. Biol. Chem. 1992, 267, 8030–8034. [Google Scholar] [PubMed]
- Kumar, S.; Björnstedt, M.; Holmgren, A. Selenite is a substrate for calf thymus thioredoxin reductase and thioredoxin and elicits a large non-stoichiometric oxidation of NADPH in the presence of oxygen. Eur. J. Biochem. 1992, 207, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Wallenberg, M.; Olm, E.; Hebert, C.; Björnstedt, M.; Fernandes, A.P. Selenium compounds are substrates for glutaredoxins: A novel pathway for selenium metabolism and a potential mechanism for selenium-mediated cytotoxicity. Biochem. J. 2010, 429, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Selenius, M.; Rundlof, A.K.; Olm, E.; Fernandes, A.P.; Björnstedt, M. Selenium and the selenoprotein thioredoxin reductase in the prevention, treatment and diagnostics of cancer. Antioxid. Redox Signal. 2010, 12, 867–880. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Biochemistry of oxidative stress. Biochem. Soc. Trans. 2007, 35, 1147–1150. [Google Scholar] [CrossRef] [PubMed]
- Labunskyy, V.M.; Hatfield, D.L.; Gladyshev, V.N. Selenoproteins: Molecular pathways and physiological roles. Physiol. Rev. 2014, 94, 739–777. [Google Scholar] [CrossRef] [PubMed]
- Moghadaszadeh, B.; Beggs, A.H. Selenoproteins and their impact on human health through diverse physiological pathways. Physiology (Bethesda) 2006, 21, 307–315. [Google Scholar] [CrossRef]
- Schwarz, K.; Foltz, C.M. Selenium as an integral part of factor 3 against dietary necrotic liver degeneration. J. Am. Chem. Soc. 1957, 79, 3292–3293. [Google Scholar] [CrossRef]
- Wallenberg, M.; Misra, S.; Wasik, A.M.; Marzano, C.; Björnstedt, M.; Gandin, V.; Fernandes, A.P. Selenium induces a multi-targeted cell death process in addition to ROS formation. J. Cell. Mol. Med. 2014, 18, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Weekley, C.M.; Harris, H.H. Which form is that? The importance of selenium speciation and metabolism in the prevention and treatment of disease. Chem. Soc. Rev. 2013, 42, 8870–8894. [Google Scholar] [CrossRef] [PubMed]
- Guimaraes, M.J.; Peterson, D.; Vicari, A.; Cocks, B.G.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; Ferrick, D.A.; Kastelein, R.A.; Bazan, J.F.; et al. Identification of a novel seld homolog from eukaryotes, bacteria, and archaea: Is there an autoregulatory mechanism in selenocysteine metabolism? Proc. Natl. Acad. Sci. USA 1996, 93, 15086–15091. [Google Scholar] [CrossRef] [PubMed]
- Forstrom, J.W.; Zakowski, J.J.; Tappel, A.L. Identification of the catalytic site of rat liver glutathione peroxidase as selenocysteine. Biochemistry 1978, 17, 2639–2644. [Google Scholar] [CrossRef] [PubMed]
- Spallholz, J.E. On the nature of selenium toxicity and carcinostatic activity. Free Radic. Biol. Med. 1994, 17, 45–64. [Google Scholar] [CrossRef] [PubMed]
- Jukes, T.H. Selenium, an “essential poison”. J. Appl. Biochem. 1983, 5, 233–234. [Google Scholar] [PubMed]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ros-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Wondrak, G.T. Redox-directed cancer therapeutics: Molecular mechanisms and opportunities. Antioxid. Redox Signal. 2009, 11, 3013–3069. [Google Scholar] [CrossRef] [PubMed]
- Kong, Q.; Beel, J.A.; Lillehei, K.O. A threshold concept for cancer therapy. Med. Hypotheses 2000, 55, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Abdullaev, F.I.; MacVicar, C.; Frenkel, G.D. Inhibition by selenium of DNA and RNA synthesis in normal and malignant human cells in vitro. Cancer Lett. 1992, 65, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Caffrey, P.B.; Frenkel, G.D. Selenite cytotoxicity in drug resistant and nonresistant human ovarian tumor cells. Cancer Res. 1992, 52, 4812–4816. [Google Scholar] [PubMed]
- Polo, M.; Marsden, W.; Corbino, J. The travels of marco polo the venetian; J.M. DENT SONS, Ltd.: London, UK, 1908; p. 439. [Google Scholar]
- Anundi, I.; Hogberg, J.; Stahl, A. Involvement of glutathione reductase in selenite metabolism and toxicity, studied in isolated rat hepatocytes. Arch. Toxicol. 1982, 50, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Frenkel, G.D.; Falvey, D. Evidence for the involvement of sulfhydryl compounds in the inhibition of cellular DNA synthesis by selenite. Mol. Pharmacol. 1988, 34, 573–577. [Google Scholar] [PubMed]
- Ganther, H.E. Selenotrisulfides. Formation by the reaction of thiols with selenious acid. Biochemistry 1968, 7, 2898–2905. [Google Scholar] [CrossRef] [PubMed]
- Ganther, H.E. Reduction of the selenotrisulfide derivative of glutathione to a persulfide analog by glutathione reductase. Biochemistry 1971, 10, 4089–4098. [Google Scholar] [CrossRef] [PubMed]
- Seko, Y.; Imura, N. Active oxygen generation as a possible mechanism of selenium toxicity. Biomed. Environ. Sci. 1997, 10, 333–339. [Google Scholar] [PubMed]
- Chaudiere, J.; Courtin, O.; Leclaire, J. Glutathione oxidase activity of selenocystamine: A mechanistic study. Arch. Biochem. Biophys. 1992, 296, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.; Boylan, L.M.; Wu, C.K.; Spallholz, J.E. Oxidation of glutathione and superoxide generation by inorganic and organic selenium compounds. Biofactors 2007, 31, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, K. Biological potency of organic selenium compounds. I. Aliphatic monoseleno- and diseleno-dicarboxylic acids. J. Biol. Chem. 1969, 244, 2103–2110. [Google Scholar] [PubMed]
- Spallholz, J.E.; Shriver, B.J.; Reid, T.W. Dimethyldiselenide and methylseleninic acid generate superoxide in an in vitro chemiluminescence assay in the presence of glutathione: Implications for the anticarcinogenic activity of l-selenomethionine and l-se-methylselenocysteine. Nutr. Cancer 2001, 40, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Seko, Y.; Saito, Y.; Kitahara, J.; Imura, N. Active oxygen generation by the reaction of selenite with reduced glutathione in vitro. In Selenium in Biology and Medicine; Wendel, A., Ed.; Springer Berlin Heidelberg: Tübingen, Germany, 1989; pp. 70–73. [Google Scholar]
- Feigl, F.; West, P.W. Test for selenium based on a catalytic effect. Anal. Chem. 1947, 19, 351–353. [Google Scholar] [CrossRef]
- Tsen, C.C.; Tappel, A.L. Catalytic oxidation of glutathione and other sulfhydryl compounds by selenite. J. Biol. Chem. 1958, 233, 1230–1232. [Google Scholar] [PubMed]
- Rhead, W.J.; Schrauzer, G.N. The selenium catalyzed reduction of methylene blue by thiols. Bioinorg. Chem. 1974, 3, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Spallholz, J.E.; Whittam, J.H. Seleniurn toxicity interpreted from biological, catalytic, chemilurninescent and scanning electron microscopic data. In Proceedings of the Fifth International Symposium on Selenium in Biology and Medicine, Nashville, TN, USA, 20–23 July 1992.
- Levander, O.A.; Morris, V.C.; Higgs, D.J. Selenium as a catalyst for the reduction of cytochrome C by glutathione. Biochemistry 1973, 12, 4591–4595. [Google Scholar] [CrossRef] [PubMed]
- McCord, J.M.; Fridovich, I. The utility of superoxide dismutase in studying free radical reactions. I. Radicals generated by the interaction of sulfite, dimethyl sulfoxide, and oxygen. J. Biol. Chem. 1969, 244, 6056–6063. [Google Scholar] [PubMed]
- Goswami, D. Cytotoxic Effects of Selenium Conjugated Transferrins on Leukemia Cell Lines. Ph.D Thesis, Texas Tech University, Lubbock, TX, USA, 2014. [Google Scholar]
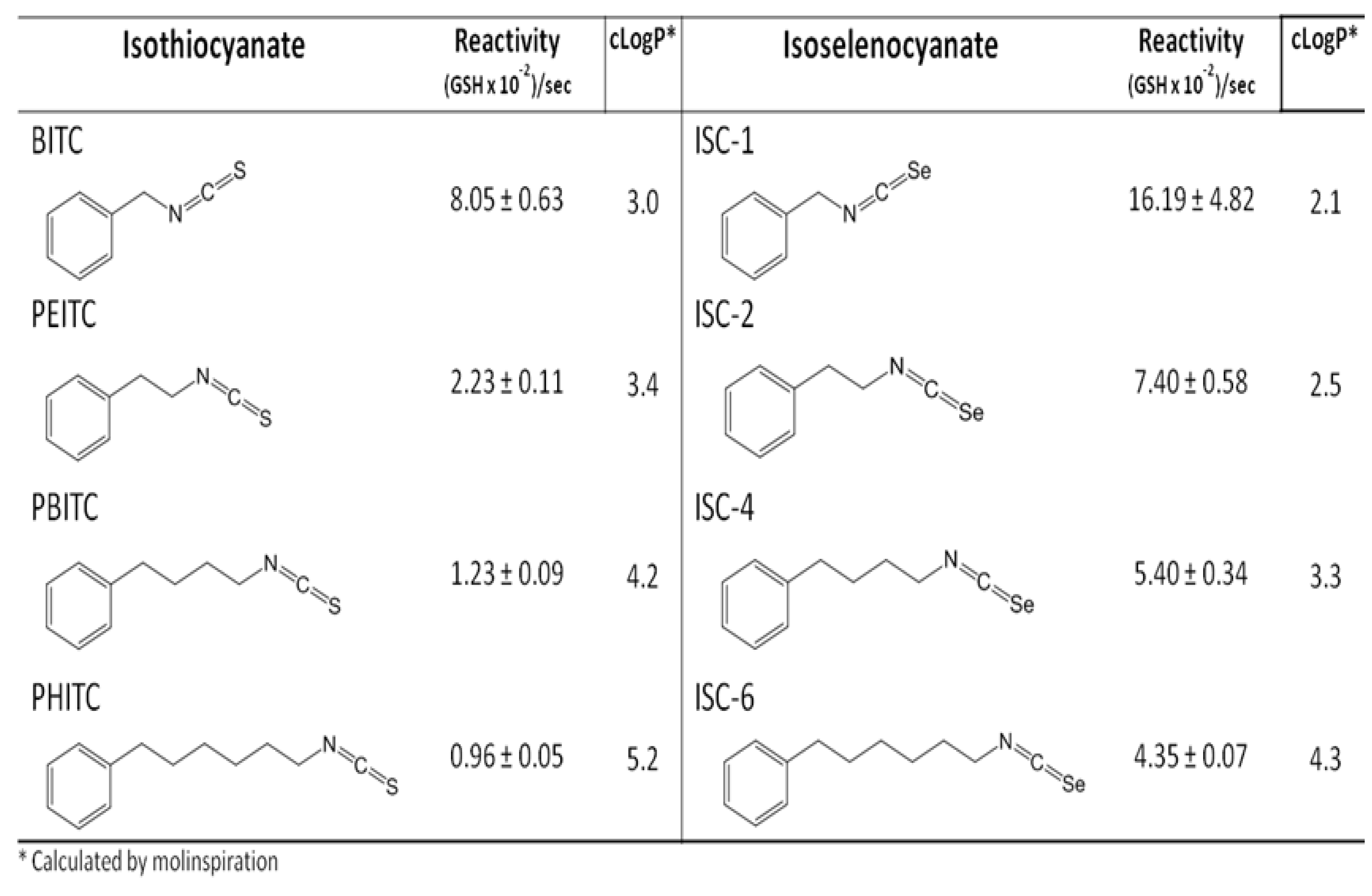
- Crampsie, M.A.; Pandey, M.K.; Desai, D.; Spallholz, J.; Amin, S.; Sharma, A.K. Phenylalkyl isoselenocyanates vs phenylalkyl isothiocyanates: Thiol reactivity and its implications. Chemico-Biol. Interact. 2012, 200, 28–37. [Google Scholar] [CrossRef]
- Lenzi, M.; Fimognari, C.; Hrelia, P. Sulforaphane as a promising molecule for fighting cancer. Cancer Treat. Res. 2014, 159, 207–223. [Google Scholar] [PubMed]
- Lee, Y.J.; Lee, S.H. Sulforaphane induces antioxidative and antiproliferative responses by generating reactive oxygen species in human bronchial epithelial beas-2b cells. J. Korean Med. Sci. 2011, 26, 1474–1482. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zeng, L.; Jiang, W.; Fu, Y.; Zheng, W.; Chen, T. Rational design of cancer-targeted selenium nanoparticles to antagonize multidrug resistance in cancer cells. Nanomedicine 2015. [Google Scholar] [CrossRef]
- Misra, H.P. Generation of superoxide free radical during the autoxidation of thiols. J. Biol. Chem. 1974, 249, 2151–2155. [Google Scholar] [PubMed]
- Gutteridge, J.M.; Beard, A.P.; Quinlan, G.J. Superoxide-dependent lipid peroxidation. Problems with the use of catalase as a specific probe for fenton-derived hydroxyl radicals. Biochem. Biophys. Res. Commun. 1983, 117, 901–907. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.L.; Spallholz, J.E. In vitro hemolysis of rat erythrocytes by selenium compounds. Biochem. Pharmacol. 1983, 32, 957–961. [Google Scholar] [CrossRef] [PubMed]
- Tran, P.L.; Hammond, A.A.; Mosley, T.; Cortez, J.; Gray, T.; Colmer-Hamood, J.A.; Shashtri, M.; Spallholz, J.E.; Hamood, A.N.; Reid, T.W. Organoselenium coating on cellulose inhibits the formation of biofilms by pseudomonas aeruginosa and staphylococcus aureus. Appl. Environ. Microbiol. 2009, 75, 3586–3592. [Google Scholar] [CrossRef] [PubMed]
- Ganther, H.E. Enzymic synthesis of dimethyl selenide from sodium selenite in mouse liver extracts. Biochemistry 1966, 5, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Loeschner, K.; Hadrup, N.; Hansen, M.; Pereira, S.A.; Gammelgaard, B.; Moller, L.H.; Mortensen, A.; Lam, H.R.; Larsen, E.H. Absorption, distribution, metabolism and excretion of selenium following oral administration of elemental selenium nanoparticles or selenite in rats. Metallomics 2014, 6, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Ip, C.; Thompson, H.J.; Zhu, Z.; Ganther, H.E. In vitro and in vivo studies of methylseleninic acid: Evidence that a monomethylated selenium metabolite is critical for cancer chemoprevention. Cancer Res. 2000, 60, 2882–2886. [Google Scholar] [PubMed]
- Metes-Kosik, N.; Luptak, I.; Dibello, P.M.; Handy, D.E.; Tang, S.S.; Zhi, H.; Qin, F.; Jacobsen, D.W.; Loscalzo, J.; Joseph, J. Both selenium deficiency and modest selenium supplementation lead to myocardial fibrosis in mice via effects on redox-methylation balance. Mol. Nutr. Food Res. 2012, 56, 1812–1824. [Google Scholar] [CrossRef] [PubMed]
- Stohs, S.J.; Bagchi, D. Oxidative mechanisms in the toxicity of metal ions. Free Radic. Biol. Med. 1995, 18, 321–336. [Google Scholar] [CrossRef] [PubMed]
- Ong, T.P.; Moreno, F.S.; Ross, S.A. Targeting the epigenome with bioactive food components for cancer prevention. J. Nutrigenet. Nutrigenomics 2011, 4, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Hoefig, C.S.; Renko, K.; Kohrle, J.; Birringer, M.; Schomburg, L. Comparison of different selenocompounds with respect to nutritional value vs. Toxicity using liver cells in culture. J. Nutr. Biochem. 2011, 22, 945–955. [Google Scholar] [CrossRef] [PubMed]
- Weekley, C.M.; Aitken, J.B.; Vogt, S.; Finney, L.A.; Paterson, D.J.; de Jonge, M.D.; Howard, D.L.; Musgrave, I.F.; Harris, H.H. Uptake, distribution, and speciation of selenoamino acids by human cancer cells: X-ray absorption and fluorescence methods. Biochemistry 2011, 50, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Turowski, S.G.; San Martin, I.D.; Rajput, A.; Rustum, Y.M.; Hoffman, R.M.; Seshadri, M. Magnetic resonance and fluorescence-protein imaging of the anti-angiogenic and anti-tumor efficacy of selenium in an orthotopic model of human colon cancer. Anticancer Res. 2011, 31, 387–393. [Google Scholar] [PubMed]
- Yin, M.B.; Li, Z.R.; Toth, K.; Cao, S.; Durrani, F.A.; Hapke, G.; Bhattacharya, A.; Azrak, R.G.; Frank, C.; Rustum, Y.M. Potentiation of irinotecan sensitivity by se-methylselenocysteine in an in vivo tumor model is associated with downregulation of cyclooxygenase-2, inducible nitric oxide synthase, and hypoxia-inducible factor 1alpha expression, resulting in reduced angiogenesis. Oncogene 2006, 25, 2509–2519. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A. Methylselenocysteine: A promising antiangiogenic agent for overcoming drug delivery barriers in solid malignancies for therapeutic synergy with anticancer drugs. Expert Opin. Drug Deliv. 2011, 8, 749–763. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Seshadri, M.; Oven, S.D.; Toth, K.; Vaughan, M.M.; Rustum, Y.M. Tumor vascular maturation and improved drug delivery induced by methylselenocysteine leads to therapeutic synergy with anticancer drugs. Clin. Cancer Res. 2008, 14, 3926–3932. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Durrani, F.A.; Toth, K.; Rustum, Y.M. Se-methylselenocysteine offers selective protection against toxicity and potentiates the antitumour activity of anticancer drugs in preclinical animal models. Br. J. Cancer 2014, 110, 1733–1743. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, K.; Watanabe, H.; Yamazaki, S.; Toda, S. Glutathione peroxidase activity of d,l-selenocystine and selenocystamine. Biochem. Biophys. Res. Commun. 1980, 96, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Björnstedt, M.; Kumar, S.; Bjorkhem, L.; Spyrou, G.; Holmgren, A. Selenium and the thioredoxin and glutaredoxin systems. Biomed. Environ. Sci. 1997, 10, 271–279. [Google Scholar] [PubMed]
- Hondal, R.J.; Marino, S.M.; Gladyshev, V.N. Selenocysteine in thiol/disulfide-like exchange reactions. Antioxid. Redox Signal. 2013, 18, 1675–1689. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Wong, Y.S. Selenocystine induces reactive oxygen species-mediated apoptosis in human cancer cells. Biomed. Pharmacother. 2009, 63, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.S.; Kunwar, A.; Ahmad, A.; Kumbhare, L.B.; Jain, V.K.; Priyadarsini, K.I. In vitro radioprotection studies of organoselenium compounds: Differences between mono- and diselenides. Radiat. Environ. Biophys. 2009, 48, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Ošťádalová, I.; Babický, A. Toxic effect of various selenium compounds on the rat in the early postnatal period. Arch. Toxicol. 1980, 45, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, T.; Taniguchi, S.; Mihara, M.; Nakamuro, K.; Sayato, Y. Toxicity and chemical form of selenium in the liver of mice orally administered selenocystine for 90 days. Arch. Toxicol. 1994, 68, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, T.; Mihara, M.; Nakamuro, K.; Sayato, Y. Distribution and chemical form of selenium in mice after administration of selenocystine. Biol. Pharm Bull. 1994, 17, 1215–1219. [Google Scholar] [CrossRef] [PubMed]
- Lane, H.W.; Teer, P.; Dukes, J.; Johnson, J.; White, M.T. The effect of four chemical forms of selenium on mammary tumor incidence in balb/c female mice treated with 7–12-dimethylbenz[a]anthracene. Cancer Lett. 1990, 50, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Whiting, R.F.; Wei, L.; Stich, H.F. Unscheduled DNA synthesis and chromosome aberrations induced by inorganic and organic selenium compounds in the presence of glutathione. Mutat. Res. 1980, 78, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Weisberger, A.S.; Suhrland, L.G. Studies on analogues of l-cysteine and l-cystine. III. The effect of selenium cystine on leukemia. Blood 1956, 11, 19–30. [Google Scholar] [PubMed]
- Brodin, O.; Eksborg, S.; Wallenberg, M.; Asker-Hagelberg, C.; Huusfeldt Larsen, E.; Mohlkert, D.; Lenneby-Helleday, C.; Jacobsson, H.; Linder, S.; Misra, S.; et al. Pharmacokinetics and toxicity of sodium selenite in the treatment of patients with carcinoma in a phase I clinical trial: The SECAR study. Nutrients 2015. submitted for publication. [Google Scholar]
- Greeder, G.A.; Milner, J.A. Factors influencing the inhibitory effect of selenium on mice inoculated with ehrlich ascites tumor cells. Science 1980, 209, 825–827. [Google Scholar] [CrossRef] [PubMed]
- Watson-Williams, E. A preliminary note on the treatment of inoperable carcinoma with selenium. Br. Med. J. 1919, 2, 463–464. [Google Scholar] [CrossRef] [PubMed]
- Asfour, I.A.; El-Tehewi, M.M.; Ahmed, M.H.; Abdel-Sattar, M.A.; Moustafa, N.N.; Hegab, H.M.; Fathey, O.M. High-dose sodium selenite can induce apoptosis of lymphoma cells in adult patients with non-hodgkin’s lymphoma. Biol. Trace Elem. Res. 2009, 127, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Asfour, I.A.; Fayek, M.; Raouf, S.; Soliman, M.; Hegab, H.M.; El-Desoky, H.; Saleh, R.; Moussa, M.A.R. The impact of high-dose sodium selenite therapy on bcl-2 expression in adult non-hodgkin’s lymphoma patients: Correlation with response and survival. Biol. Trace Elem. Res. 2007, 120, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lippman, S.M.; Klein, E.A.; Goodman, P.J.; Lucia, M.S.; Thompson, I.M.; Ford, L.G.; Parnes, H.L.; Minasian, L.M.; Gaziano, J.M.; Hartline, J.A.; et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: The selenium and vitamin E cancer prevention trial (SELECT). JAMA 2009, 301, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.-J.; Chen, Y.; Zhang, Y.-Q.; Zhou, M.-Z.; Song, X.-M.; Zhang, B.-Z.; Luo, L.; Xu, P.-M.; Zhao, Y.-N.; Zhao, Y.-B.; et al. The protective role of selenium on the toxicity of cisplatin-contained chemotherapy regimen in cancer patients. Biol. Trace Elem. Res. 1997, 56, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Sieja, K.; Talerczyk, M. Selenium as an element in the treatment of ovarian cancer in women receiving chemotherapy. Gynecol. Oncol. 2004, 93, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Olm, E.; Fernandes, A.P.; Hebert, C.; Rundlof, A.K.; Larsen, E.H.; Danielsson, O.; Björnstedt, M. Extracellular thiol-assisted selenium uptake dependent on the x(c)- cystine transporter explains the cancer-specific cytotoxicity of selenite. Proc. Natl. Acad. Sci. USA 2009, 106, 11400–11405. [Google Scholar] [CrossRef] [PubMed]
- Liu, D. Speciation of Arsenic and Selenium in Rabbit Using X-Ray Absorption Spectroscopy. Master’s Thesis, University of Saskatchewan, Saskatoon, Canada, 2011. [Google Scholar]
- Weekley, C.M.; Aitken, J.B.; Vogt, S.; Finney, L.A.; Paterson, D.J.; de Jonge, M.D.; Howard, D.L.; Witting, P.K.; Musgrave, I.F.; Harris, H.H. Metabolism of selenite in human lung cancer cells: X-ray absorption and fluorescence studies. J. Am. Chem. Soc. 2011, 133, 18272–18279. [Google Scholar] [CrossRef] [PubMed]
- Misra, S.; Peak, D.; Niyogi, S. Application of XANES spectroscopy in understanding the metabolism of selenium in isolated rainbow trout hepatocytes: Insights into selenium toxicity. Metallomics 2010, 2, 710–717. [Google Scholar] [CrossRef] [PubMed]
- Wallenberg, M.; Jawad, R.; Arodin, L.; Björnstedt, M.; Karolinska Institute, Stockholm, Sweden. 2015.
- Clark, L.C.; Dalkin, B.; Krongrad, A.; Combs, G.F., Jr.; Turnbull, B.W.; Slate, E.H.; Witherington, R.; Herlong, J.H.; Janosko, E.; Carpenter, D.; et al. Decreased incidence of prostate cancer with selenium supplementation: Results of a double-blind cancer prevention trial. Br. J. Urol. 1998, 81, 730–734. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Sk, U.H.; Zhang, Y.; Ren, X.; Zhang, L.; Huber-Keener, K.J.; Sun, Y.W.; Liao, J.; Amin, S.; Sharma, A.K.; et al. Rational incorporation of selenium into temozolomide elicits superior antitumor activity associated with both apoptotic and autophagic cell death. PLoS ONE 2012, 7, e35104. [Google Scholar] [CrossRef] [PubMed]
- Goswami, D.; Bapat, P.; Shastri, A.; Boylan, L.M.; Harris, E.; Spallholz, J. Seleno-transferrin for treatment of leukemia. In Proceedings of the International Meeting on Selenium in Biology and Medicine, Berlin, Germany, 14–18 September 2013; p. 75.
- Bapat, P.; Goswami, D.; Shastri, A.; Boylan, M.; Zapada, K.; Cobos, E.; Plano, D.; Sharma, A.; Spallholz, J.E. A photographic comparison of seleno-trastuzumab, trastuzumab, and selenite on inhibition of the HER2+ human breast cancer cell line BT-474. In Proceedings of the International Meeting on Selenium in Biology and Medicine, Berlin, Germany, 14–18 September 2013; p. S15.
- Sanmartin, C.; Plano, D.; Sharma, A.K.; Palop, J.A. Selenium compounds, apoptosis and other types of cell death: An overview for cancer therapy. Int. J. Mol. Sci. 2012, 13, 9649–9672. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Misra, S.; Boylan, M.; Selvam, A.; Spallholz, J.E.; Björnstedt, M. Redox-Active Selenium Compounds—From Toxicity and Cell Death to Cancer Treatment. Nutrients 2015, 7, 3536-3556. https://doi.org/10.3390/nu7053536
Misra S, Boylan M, Selvam A, Spallholz JE, Björnstedt M. Redox-Active Selenium Compounds—From Toxicity and Cell Death to Cancer Treatment. Nutrients. 2015; 7(5):3536-3556. https://doi.org/10.3390/nu7053536
Chicago/Turabian StyleMisra, Sougat, Mallory Boylan, Arun Selvam, Julian E. Spallholz, and Mikael Björnstedt. 2015. "Redox-Active Selenium Compounds—From Toxicity and Cell Death to Cancer Treatment" Nutrients 7, no. 5: 3536-3556. https://doi.org/10.3390/nu7053536