1. Introduction
Cancers are the leading cause of death globally and their treatment mainly involves surgery, chemotherapy, and radiotherapy [
1]. Induction of apoptosis in cancer cells via anticancer drugs is thought to be effective for anticancer therapy [
2]. However, the apoptosis pathway is abnormally regulated in human cancer cells, which may cause these cells to evade radiation and chemotherapy. Cancer cells escape death via resisting apoptosis, which upregulates antiapoptotic signals (e.g., Bcl-2, Akt, and Mcl-1) and downregulates proapoptotic signals (e.g., Bax, Bak, and Bad), thereby initiating and implicating faulty apoptosis [
3,
4]. The concurrent pharmacological strategy targeting autophagy and apoptosis signaling pathways might greatly improve the efficacy of anticancer therapy [
5].
Autophagy is a phenomenon involving the degradation of cellular components and organelles in an autophagosome (double-membrane vesicle), which is transferred to and degraded in the lysosome [
6]. Signaling pathways that are activated by chemotherapeutics and metabolic stress can stimulate autophagy [
7]. After the induction of autophagy, cytoplasmic degradation of constituents in autolysosomes eventually leads to the formation of free nucleotides, amino acids, and fatty acids, which are recycled by the cell for nutrient reuse and energy regeneration. Autophagy can promote cell survival; however, it may also promote cell death [
2,
8], affording dual effects against cancer.
Cell death can occur through three distinct routes of cellular catabolism, namely apoptosis, autophagy, and necrosis. Among these three types of programmed cell death, autophagy-mediated cell death is morphologically defined as cell death without chromatin condensation but is accompanied by the formation of distinctive autophagosomes [
9]. Although apoptosis and autophagy are considered as two different types of cell death, there is a link between them as they share common components that are regulated by common factors [
2]. For instance, various molecular interactions between apoptosis and autophagy have been investigated in apoptosis-specific protein Beclin-1, the Bcl-2 protein family, Ca
2+/calmodulin-dependent protein kinases, phosphatase and tensin homolog (PTEN), protein kinase B (Akt/PKB), and mammalian target of rapamycin (mTOR) [
5,
10]. Therefore, in particular situations, many signaling pathways regulate autophagy, which can also induce apoptosis to cause cell death.
Reactive oxygen species (ROS) have been involved in the signal pathways of cellular apoptosis and autophagy [
11]. It has been reported that high levels of glycation end products increase cellular apoptosis and autophagy via the ROS-mediated extracellular signal-regulated protein kinases (ERK) signaling pathway [
12]. Several anticancer drugs eventually trigger autophagy-mediated cell death via various molecules and signaling pathways [
5]. For instance, tanshinone IIA exerts its autophagy-inducing effect against oral squamous cell carcinoma by inducing the Beclin-1/Atg7/Atg12-Atg5 pathway and suppressing the PI3K/Akt/mTOR survival pathway [
13]. A dietary phenolic compound, fisetin, increases autophagy- and apoptosis-mediated cell death via activation of ERK1/2 and attenuation of Akt/NF-κB/mTOR signaling pathways [
14].
Ginseng (
Panax ginseng Meyer) has been utilized in East Asian traditional medicine for a long time. Red ginseng, which is tinted reddish-brown, is prepared by steaming and then drying [
15]. Korean red ginseng (KRG) is effective against several disorders, including cancer [
15]. KRG extract has been shown in in vivo and in vitro studies to exert anticancer effects in different types of cancer. Oral administration of KRG extract significantly decreases the incidence of lung adenoma and liver cancer [
16]. It prevents gastric cancer caused by
Helicobacter pylori infection by inhibiting H
2S-induced angiogenesis [
17]. It was also shown to inhibit carcinogen-induced mouse skin papilloma and decrease the number of tumors in each mouse [
18]. In addition, in colon cancer, KRG extract inhibits hypoxia-induced epithelial-to-mesenchymal transition and invasion via the NF-κB and ERK1/2 pathways [
19]. However, the effects and mechanisms of action of KRG extract on human colon cancer are not known in detail. In this study, mechanisms of KRG extract-induced cell death were investigated in terms of autophagy- and apoptosis-mediated cell death in human HCT-116 and SNU-1033 colon cancer cells.
2. Materials and Methods
2.1. Reagents
Ginsenosides (Rb1, Rc, Rg2, and Rg3), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenylte-trazolium bromide (MTT; cat. M5655), z-VAD-FMK (cat. V116), bafilomycin A1 (cat. B1793), N-acetyl-l-cysteine (NAC; cat. A9165), and propidium iodide (PI; cat. P4170) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Primary antibodies against Atg5 (cat. sc-515347), Bcl-2 (cat. sc-7382), caspase-9 (cat. sc-133109), caspase-3 (cat. sc-7148), poly (ADP-ribose) polymerase 1 (PARP1; cat. sc-8007), and actin (cat. sc-376421) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The LC3 (cat. 2775), Beclin-1 (cat. 3738), and phospho-Bcl-2 (cat. 2827) antibodies were purchased from Cell Signaling Technology (Beverly, MA, USA). Dihydrorhodamine (DHR) 123 (cat. D23806) and acridine orange (cat. A3568) were obtained from Molecular Probes (Eugene, OR, USA) and Invitrogen (Carlsbad, CA, USA), respectively.
2.2. KRG Preparation
KRG extract from the roots of 6-year-old
P. ginseng was prepared by the Korea Ginseng Corporation (Seoul, Korea) as described previously [
20]. Briefly, P. ginseng roots were steamed at 90–100 °C for 3 h, and then dried at 50–80 °C. Next, the KRG extract was isolated by circulating water at 85–90 °C three times for 8 h. The ginsenosides in the KRG extract were compared with ginsenoside standards using ultra-performance liquid chromatography. The KRG extract contained Rb1 (4.62 mg/g), Rb2 (1.83 mg/g), Rc (2.41 mg/g), Rd (0.89 mg/g), Re (0.93 mg/g), Rf (1.21 mg/g), Rg1 (0.71 mg/g), Rg2 (3.21 mg/g), and Rg3 (3.05 mg/g) as the major ginsenosides. The KRG extract powder was dissolved in dimethyl sulfoxide (DMSO; stock solution 100 mg/mL).
2.3. Cell Culture
The human colon cancer cell lines HCT-116, SNU-1033, Caco-2, and HT-29 were purchased from the Korean Cell Line Bank (Seoul, Korea), and normal human colon FHC cells were obtained from the American Type Culture Collection (Rockville, MD, USA). HCT-116, SNU-1033, and HT-29 cells were maintained in RPMI 1640 medium (Gibco, Life Technologies, Grand Island, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS) (Gibco), and Caco-2 cells were cultured in minimum essential medium (Gibco) supplemented with 10% FBS. FHC cells were cultured in a 1:1 mixture of Ham’s F12 and Dulbecco’s modified Eagle’s medium (Gibco) containing 10% FBS, HEPES (25 mM; Gibco), cholera toxin (10 ng/mL; Sigma-Aldrich), insulin (5 μg/mL; Sigma-Aldrich), transferrin (5 μg/mL; Sigma-Aldrich), and hydrocortisone (100 ng/mL; Sigma-Aldrich).
2.4. Cell Viability
Cytotoxicity of KRG or ginsenosides (Rb1 Rc, Rg2, and Rg3) against various colon cancer cells and normal colon cells was evaluated using the MTT assay, as previously described [
21]. Cells were seeded (1.5 × 10
5 cells/well) in 96-well plates and incubated with the KRG extract or ginsenosides for 48 h at various concentrations. For inhibitor or NAC treatment, cells were seeded (1.5 × 10
5 cells/well) and after pre-treatment with the inhibitors (z-VAD-FMK; 5 µM or bafilomycin A1; 1 µM) or NAC (1 mM) for 1 h, they were treated with KRG for 48 h. After incubation with MTT solution for 4 h, the formazan crystals formed were dissolved in DMSO. The absorbance of the formazan product was read via a scanning multi-well spectrophotometer at 540 nm [
21].
2.5. Acridine Orange Staining
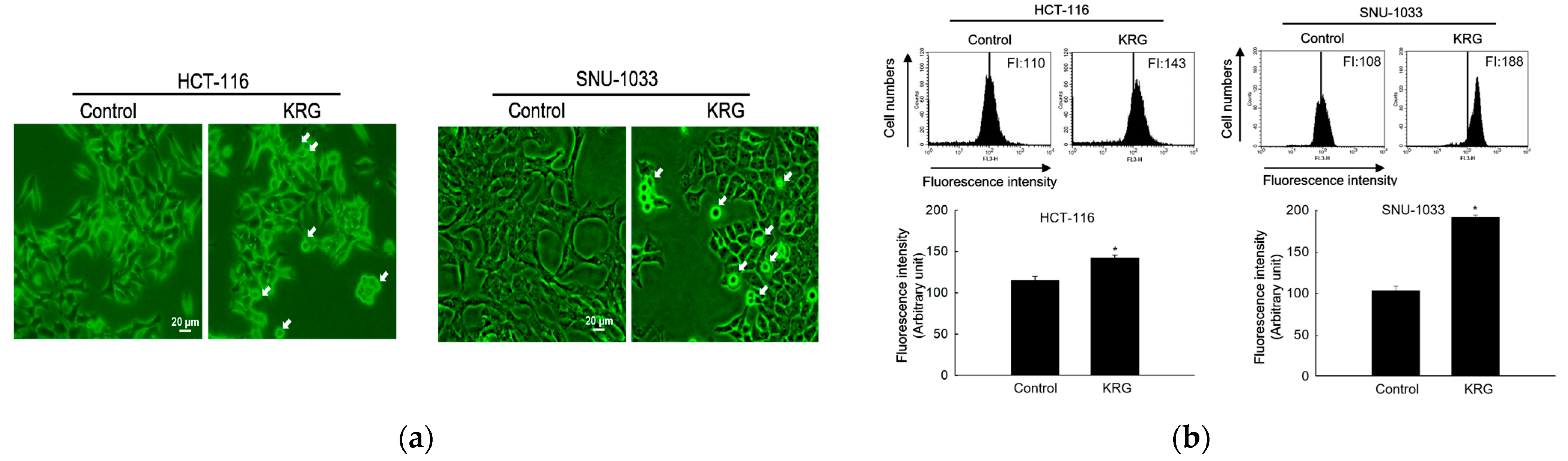
For detection of acidic intracellular compartments, acridine orange staining was performed. Acridine orange in acidic vesicles (autolysosomes) emits bright red fluorescence; it is protonated and forms aggregates, whereas acridine orange in the nucleus emits bright green fluorescence [
22,
23]. For confocal image analysis, colon cancer cells were seeded (0.8 × 10
5 cells/mL) in chamber slides and incubated at 37 °C for 16 h. The cells were treated with KRG (at IC
50 value for each cell line) for 48 h. The cells were stained with 10 μg/mL acridine orange at 37 °C for 30 min and observed under a laser scanning confocal microscope (Carl Zeiss, Jena, Germany). For FACS analysis, colon cancer cells were seeded (0.8 × 10
5 cells/mL) in 6-well plates and incubated at 37 °C for 16 h. The cells were treated with KRG (at IC
50 value for each cell line) for 48 h. Thereafter, the cells were stained with 10 μg/mL acridine orange and red fluorescence was detected using a FACSCalibur™ flow cytometer (Becton Dickinson, Mountain View, CA, USA), separately. Autophagic lysosomes fluoresced orange or red within cytoplasmic vesicles, whereas nuclei fluoresced green.
2.6. LC3 Transfection and Measurement of LC3-Positive Puncta
LC3 is recruited to autophagosomes, forming punctate structures [
24]. The cells were transfected with a green fluorescent protein (GFP)-tagged LC3-expressing plasmid (CBA401; Cell Biolabs, San Diego, CA, USA) using Lipofectamine™ 2000 reagent (Invitrogen) according to the manufacturer’s instructions. The suitable cell confluency in a 60 mm culture dish was 80–90% at the time of transfection. GFP-LC3-expressing plasmid (5 μg/mL) was used for transfection of cells in a culture dish. Lipofectamine™ 2000 reagent and plasmid were freshly diluted to equal volumes with Opti-MEM
® (Gibco 31985070; Invitrogen) medium prior to use. The plasmid/Lipofectamine mixtures were incubated for 15 min at room temperature; the growth medium was replaced by Opti-MEM
® medium and then the mixtures (1000 μL for 60 mm culture dish) were added. After incubation at 37 °C for 4 h, the growth medium was changed, and the cells were incubated with the KRG extract for 48 h. The fluorescence of GFP-LC3 was then detected using a laser scanning confocal microscope (Carl Zeiss).
2.7. Western Blot Analysis
The expression levels of proteins associated with cell apoptosis and autophagy were detected using western blot analysis. Cells were seeded (1.5 × 105 cells/well) in 100 mm culture dish and incubated with the KRG extract for 48 h. Total protein was extracted from the cells using the RIPA lysis buffer (Thermo Fisher, Life Technologies, Grand Island, Carlsbad, CA, USA), containing protease inhibitors (Thermo Fisher), according to the manufacturer’s instructions. Extracted proteins were subjected to SDS-PAGE, and the separated proteins were then transferred from the gel onto nitrocellulose membranes. After blocking with 10% BSA for 1 h, the membranes were incubated with primary antibodies against Atg5, Beclin-1, phospho-Bcl-2, Bcl-2, caspase-9, caspase-3, PARP1, LC3, and actin, and then horseradish peroxidase-conjugated immunoglobulin G secondary antibodies (Pierce, Rockford, IL, USA). Incubation with primary and secondary antibodies was carried out for 2 h at room temperature. Protein bands were detected using the Amersham ECL western blotting Detection Reagent (GE Healthcare Life Sciences, Buckinghamshire, UK).
2.8. Detection of Sub-G1 Hypodiploid Cells
To determine the apoptotic effect of KRG, flow cytometry was performed using propidium iodide (PI) staining of cells. The cells were seeded (0.8 × 105 cells/mL) in 6-well plates and incubated at 37 °C for 16 h. The cells were treated with KRG (at IC50 value for each cell line) for 48 h. These cells were fixed with 70% ethanol for 30 min at 4 °C and treated with PI and RNase A for 30 min at 37 °C. The PI-stained cells were evaluated with a FACSCalibur™ flow cytometer (Becton Dickinson) and the sub-G1 hypodiploid cells were analyzed via histogram data assessed using the CellQuest™ and ModFit software.
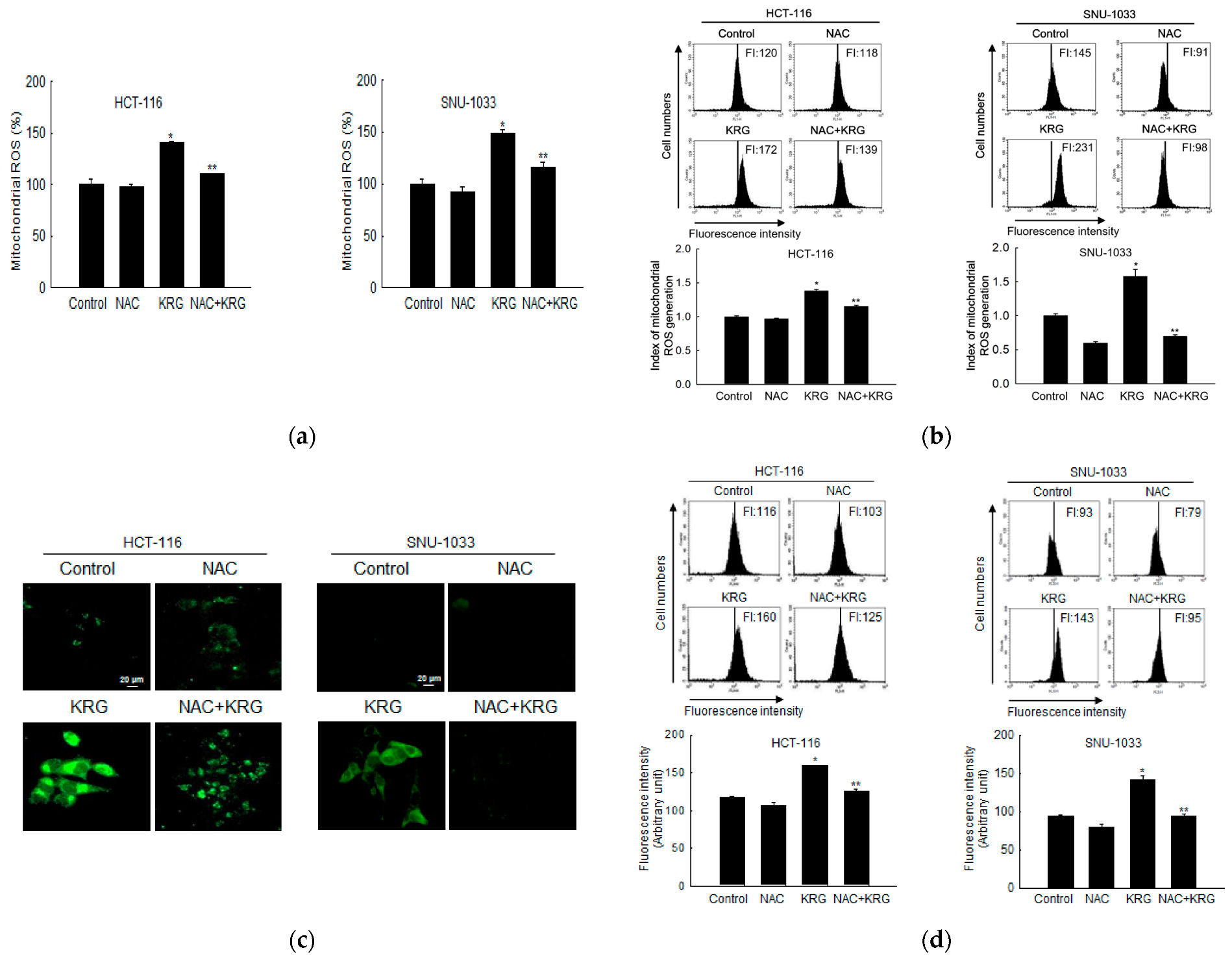
2.9. Mitochondrial ROS Measurement
To determine the ROS levels in mitochondria, the cells were seeded (0.8 × 105 cells/mL) in 6-well plates and after treatment with NAC (1 mM) for 1 h, they were treated with KRG for 48 h. After the cells were reacted with 20 μM DHR 123 for 30 min at 37 °C, images of the stained cells were observed using a laser scanning confocal microscope (Carl Zeiss). The DHR 123-stained cells were also analyzed using a flow cytometer and a fluorospectrometer.
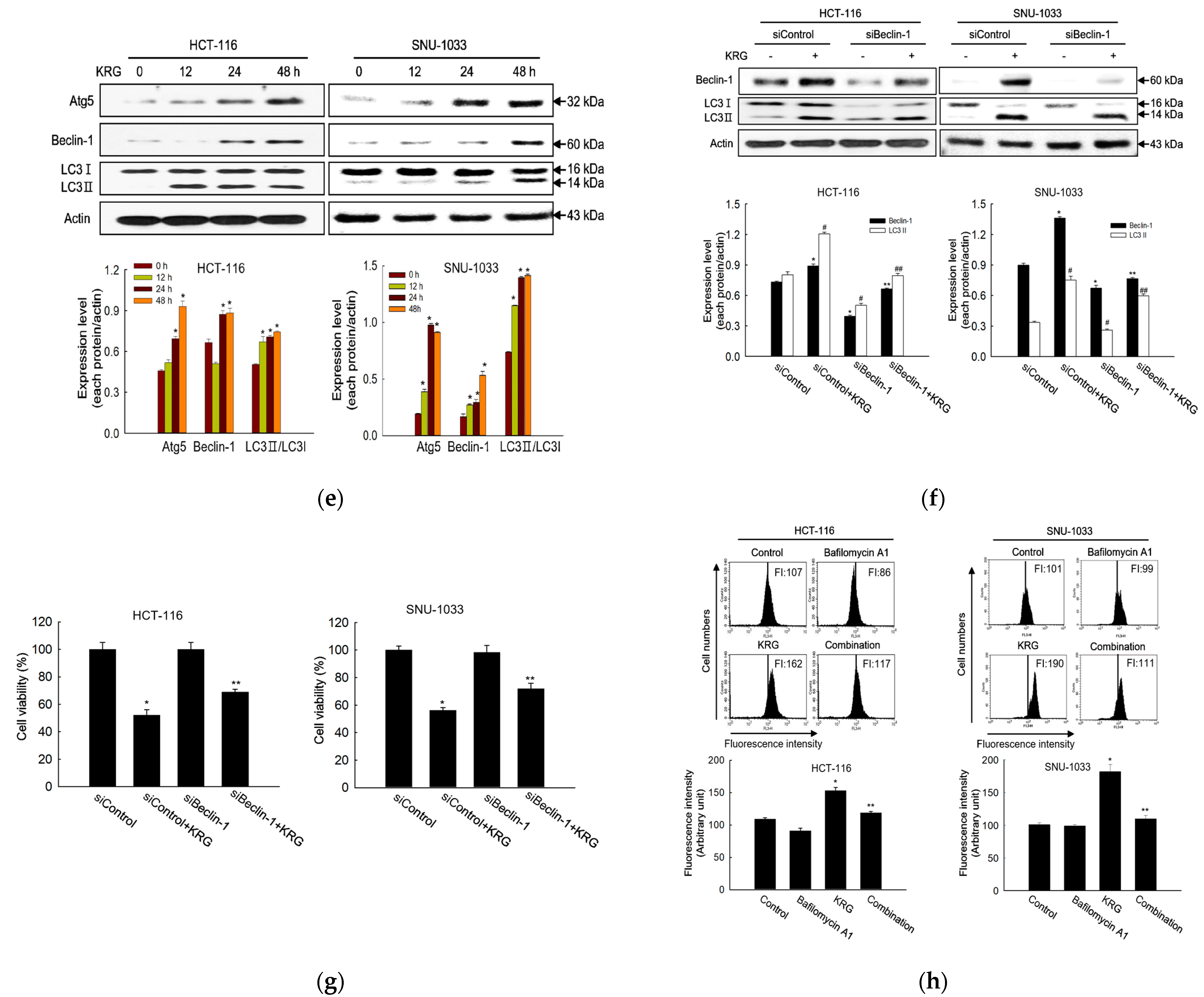
2.10. Transfection with Short-Interfering RNA (siRNA)
To determine the role of Beclin-1 in KRG-treated colon cancer cells, we performed transient knockdown of Beclin-1 using siRNA transfection for various studies. The transfection was performed in 1000 μL of Opti-MEM® medium with 2 μL of Lipofectamine® RNAiMAX (Thermo Fisher 13778100, Waltham, MA, USA), and 10–50 nM siRNA per 60 mm culture dish. After 30 min, the mixture was added to the cells cultured at 37 °C in a CO2 incubator for 24 h. The siRNAs used for transfection were mismatched siRNA control (Control siRNA-A; sc-37007, sequence not disclosed by the manufacturer; Santa Cruz Biotechnology) and siRNA against Beclin-1 (siBeclin-1; Bioneer Inc., Daejeon, Korea; sense, 5′-ACUUUGCUGUAACCCUGUA (dTdT)-3′; antisense, 5′-UACAGGGUUACA GCAAAGU (dTdT)-3′).
2.11. Hoechst 33342 Staining
To measure the formation of apoptotic bodies, the cells were seeded (0.8 × 105 cells/mL) in 6-well plates and incubated at 37 °C for 16 h. The cells were then treated with KRG or ginsenosides for 48 h. The treated cells were stained with Hoechst 33342 dye (Sigma-Aldrich) for 10 min at 37 °C, and images were captured using a fluorescence microscope equipped with a CoolSNAP-Pro color digital camera (Media Cybernetics, Rockville, MD, USA). Apoptotic cells were regarded as cells with Hoechst 33342-stained bright and fragmented nuclei. The numbers of cells in both viable and apoptotic single cells were measured. The index of apoptotic cells was determined as the apoptotic single cells/total cells (viable and apoptotic single cells).
2.12. Immunoprecipitation
Immunoprecipitation (IP) was performed to detect the interaction of Bcl-2 with Beclin-1. Cell lysates were collected and immunoprecipitated using an anti-Beclin-1 antibody in an immunoprecipitation buffer, and subsequently incubated with an agarose-conjugated secondary antibody. Western blotting was performed using anti-Bcl-2 to detect the association of Beclin-1 and Bcl-2 [
25].
2.13. Statistical Analysis
Results are shown as means ± standard errors of the mean (SEM). The Sigma Stat software v12 (SPSS, Chicago, IL, USA) was used for statistical analyses. The data were examined using a one-way analysis of variance (ANOVA) followed by Tukey’s post-hoc test. A value of p < 0.05 was considered statistically significant.
4. Discussion
Ginseng is a valuable agricultural commodity grown for use in many traditional medicinal therapies. KRG is a type of ginseng preparation that is produced by steaming and drying fresh ginseng [
26]. Ginseng saponin consists of triterpenoid glycosides of dammarane containing glucose, arabinose, xylose, and rhamnose [
27]. Ginsenosides, the important bioactive components in ginseng, possess a common triterpene backbone and exhibit structural diversity. To date, about 200 ginsenosides have been isolated from various strains of ginseng. These include 20(S)-ginsenoside-Rg3, ginsenoside-Rh2, Rs1, Rs2, Rs3, Rs4, and Rg5; notoginsenoside-R4 in the protopanaxadiol group; and 20(R)-ginsenoside-Rh1, ginsenoside-Rh4 and F4 in the protopanaxatriol group [
27]. Moreover, some ginsenosides, such as Rg2, Rg3, and Rh1, exist in different stereoisomeric forms, 20-(S) and 20-(R), depending on the position of the hydroxyl group in C-20. Other ginsenosides, including Rb2, Rb3, Rc, aglycone, Rg1, and F11, contain different saccharide substituents [
28]. Ginsenosides have various pharmaceutical activities [
29]. For example, CK, Rd, Rg2, Rg3, and Rg5 ginsenosides exhibit their anticancer effects by inhibiting differentiation, proliferation, and metastasis and induce autophagy, apoptosis, and cell cycle arrest by altering pathways related to carcinogenesis in breast, brain, lung, liver, and gastric cancer cell lines [
29].
Ginsenoside Rg2 is the major component of KRG. The pharmacological properties of Rg2 have been demonstrated in many in vitro and in vivo studies. Rg2 has a role in inhibiting hepatic glucose production in HepG2 cells; this is achieved by the activation of the AMP-activated protein kinase pathway [
30]. Rg2 has also been found to have a neuroprotective effect on glutamate-induced neurotoxicity, a result of mechanisms related to antiapoptosis and antioxidation [
31]. Moreover, it has been demonstrated that Rg2 mediates anticancer effects by activating cell cycle arrest and signaling pathways related to mitochondrial damage-induced ROS production and apoptosis [
32].
We showed, using our system, that the KRG extract exerts cytotoxicity in human colon cancer cells by triggering both autophagy- and apoptosis-mediated cell death pathways via ROS production. Although autophagy has a dual effect in facilitating cell survival and cell death, it was confirmed that autophagy induced by the KRG extract triggered cell death in HCT-116 and SNU-1033 cells, rather than survival, as observed in our cell viability assays using siBeclin-1-transfected cells and bafilomycin A1, a specific autophagy inhibitor. Despite the distinct characteristics of autophagy- and apoptosis-mediated cell death, many connections between them have been identified. Molecular mediators involved in apoptosis simultaneously trigger autophagy-mediated cell death. For instance, p53, which is a famous transcription factor that regulates apoptosis and cell cycle progression by promoting cyclin-dependent kinase inhibitors (e.g., p16 and p21) and active caspases, may also regulate autophagy-mediated cell death. Atg5 forms a ubiquitin-like conjugation system with Atg12 to enhance the elongation of the autophagosome membrane during vacuole formation [
33]. Following several proapoptotic signals, Atg5 can be cleaved and translocated from the cytosol to the mitochondria in a truncated form to activate caspase-dependent apoptosis by interacting with Bcl-X
L and promoting cytochrome c release [
34]. The autophagic function of Beclin-1 is inhibited by the interaction of Bcl-2 via the Beclin-1–Bcl-2 complex. Bcl-2 acts as a potent antiapoptotic molecule regulating Beclin-1-mediated autophagy and cell death. Moreover, Bcl-2 prevents autophagy-mediated cell death by inhibiting calcium release from the endoplasmic reticulum through Ca
2+/calmodulin-dependent kinase β and AMP-activated protein kinase activation by calcium, leading to inhibition of mTOR signaling that triggers autophagy [
35]. Bcl-2 also negatively regulates autophagy by binding with Beclin-1 through a BH3 domain to interfere with the beclin-1-mediated Type III PI3K complex that plays a role in the formation of autophagic vesicles [
36]. However, phosphorylation of Bcl-2 disrupts its interaction with Beclin-1, promoting autophagy by releasing Beclin-1. Therefore, phosphorylation of Bcl-2 can induce apoptosis [
33]. Furthermore, signals that promote autophagy or BH3 domain-competitive displacement of Beclin-1 from Bcl-2 by other BH3-containing proteins may lead to the release of Beclin-1 from the Beclin-1–Bcl-2 complex [
37]. For example, under starvation stress, c-Jun
N-terminal protein kinase 1 (JNK1) induces phospho-Bcl-2 and triggers Bcl-2 dissociation from Beclin-1, thereby inducing autophagy-mediated cell death [
38].
In the present study, total Bcl-2 levels and Beclin-1–Bcl-2 complex formation decreased upon treatment of colon cancer cells with the KRG extract, both indicating the participation of the Bcl-2 signaling pathway in the KRG extract-induced autophagy-mediated cell death. Moreover, because the KRG extract increased phospho-Bcl-2 levels in a time-dependent manner, it may be assumed that phosphorylated Bcl-2 is linked to apoptosis-mediated cell death, resulting in simultaneous activation of both these processes upon treatment with the KRG extract.
In addition to their molecular connections, apoptosis and autophagy-mediated cell deaths are also regulated by the mitochondria, which function as a switch between them. Briefly, autophagy is induced under conditions of mild mitochondrial stress, whereas apoptosis occurs upon moderate mitochondrial stress caused by the dissociation of proapoptotic factors from the mitochondria [
9]. There is a complicated interaction between ROS and autophagy-mediated cell death pathways, which are critical regulators of autophagy-mediated cell death in response to cellular stress. Mitochondrial ROS modulates autophagy during starvation [
33] and causes autophagy by inducing nerve growth factor deprivation in sympathetic neurons [
34]. It has been reported that ROS modulates the process of autophagy and apoptosis through increasing phospho-Bcl-2 (Ser70) and phospho-Beclin-1 (Thr119) [
39]. Aflatoxin B2-generated mitochondrial ROS in hepatocytes causes PI3K/Akt/mTOR-mediated autophagy [
40]. In our system, staining of cells with DHR 123, which is a dye used for detecting mitochondrial ROS, revealed that the KRG extract increased ROS generation in the mitochondria of colon cancer cells.
Pre-treatment with NAC (an inhibitor of ROS) significantly rescued the mitochondria from ROS-induced damage (i.e., fewer acridine orange-positive cells), which suggested that autophagy-mediated cell death in colon cancer cells was induced by the KRG extract via increased mitochondrial ROS-mediated cell death. Furthermore, ROS, as a cell death stimulus, also activates the intrinsic or mitochondrial apoptotic pathway by disrupting mitochondrial membrane potential and releasing intermembrane proteins, which are involved in the downregulation of Bcl-2, into the cytosol. Following the formation of the apoptosome, cleaved caspase-9 activates caspase-3, an apoptosis executor, leading to apoptosis [
41]. As a result, the increased cleaved caspase-9 and caspase-3 levels showed that the KRG extract exhibited cytotoxicity by activating the intrinsic apoptosis pathway simultaneously with autophagic cell death.
In conclusion, our novel findings indicate that the KRG extract caused cytotoxicity through autophagy- and apoptosis-mediated cell death in human colon cancer cells by inducing mitochondrial ROS generation and by increasing the expression of Atg5, Beclin-1, phospho-Bcl-2, active caspase-9, and caspase-3 (
Figure 6).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









