
The Influence of Reaction Conditions on the Properties of Graphene Oxide
 , , , , , and
, , , , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Graphene Oxide (GO)
2.3. Characterization
3. Results and Discussion
3.1. Determination of Reaction Yield
3.2. XPS Analysis of GO
3.2.1. The Influence of the Graphite Particle Size
3.2.2. The Influence of the Ratio of Reactants
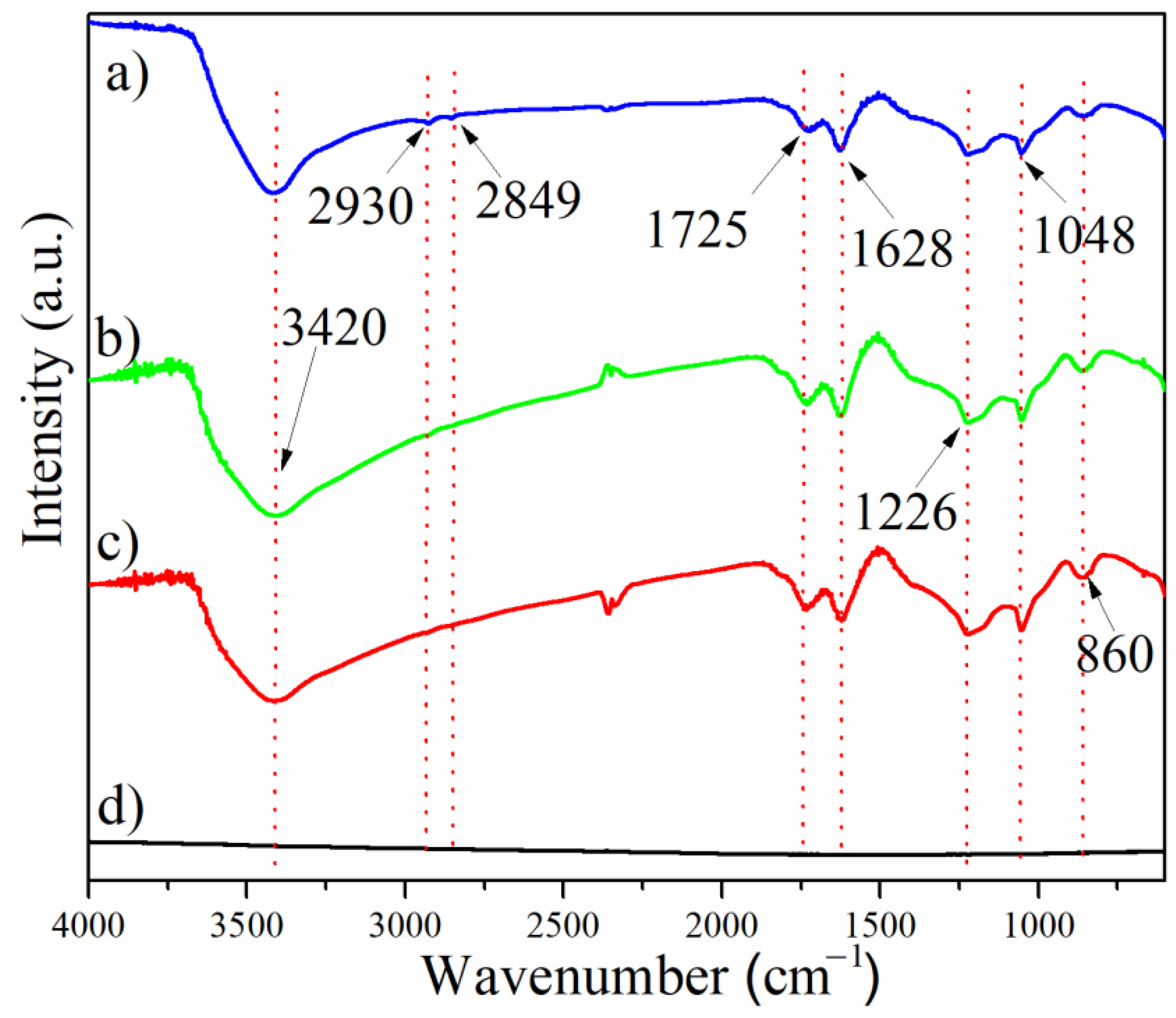
3.3. Characterization of GO Using UV-VIS, FTIR, and Raman Spectroscopy
3.4. Hydrophilicity of GO
3.5. Particle Size Determination
- Graphite-to-oxidant ratio: Adding more oxidant should result in more oxidized GO. The graphite flakes should be more damaged and, therefore, smaller. However, this effect was only observed for the smallest particles (GO45).
- Acid-to-graphite ratio: Acid enables oxidation and acts as a solvent with a diluting effect. Increasing the ratio from 1:60 to 1:80 reduces the concentration of reactants, which generally reduces the reaction rate. This should result in less oxidation (at a constant reaction time) and a larger particle size. Additionally, the viscosity of the reactant mixture is reduced by the addition of more acid, which reduces the mechanical breaking of the particles.
- Graphite particle size: The smaller the particle size, the higher the proportion of end-C atoms, which are more easily accessible and more reactive than inner C atoms. The oxidation of larger graphite particles is, therefore, slower, which leads to a larger particle size. The viscosity of the reaction mixture, after the reaction has started and GO has formed, increases with increasing graphite particle size. Increased viscosity reduces the mobility of reactants, which could reduce the reaction rate. Therefore, larger GO particles are expected to be obtained by using larger graphite particles. However, the reaction mixture must be mixed to ensure the homogeneous oxidation of all graphite particles. Mechanical mixing of very viscous slurry inevitably leads to the breakage of the particle and, thus, a size reduction. The extent of the breakage depends on the viscosity, which changes with the degree of oxidation and GO concentration.
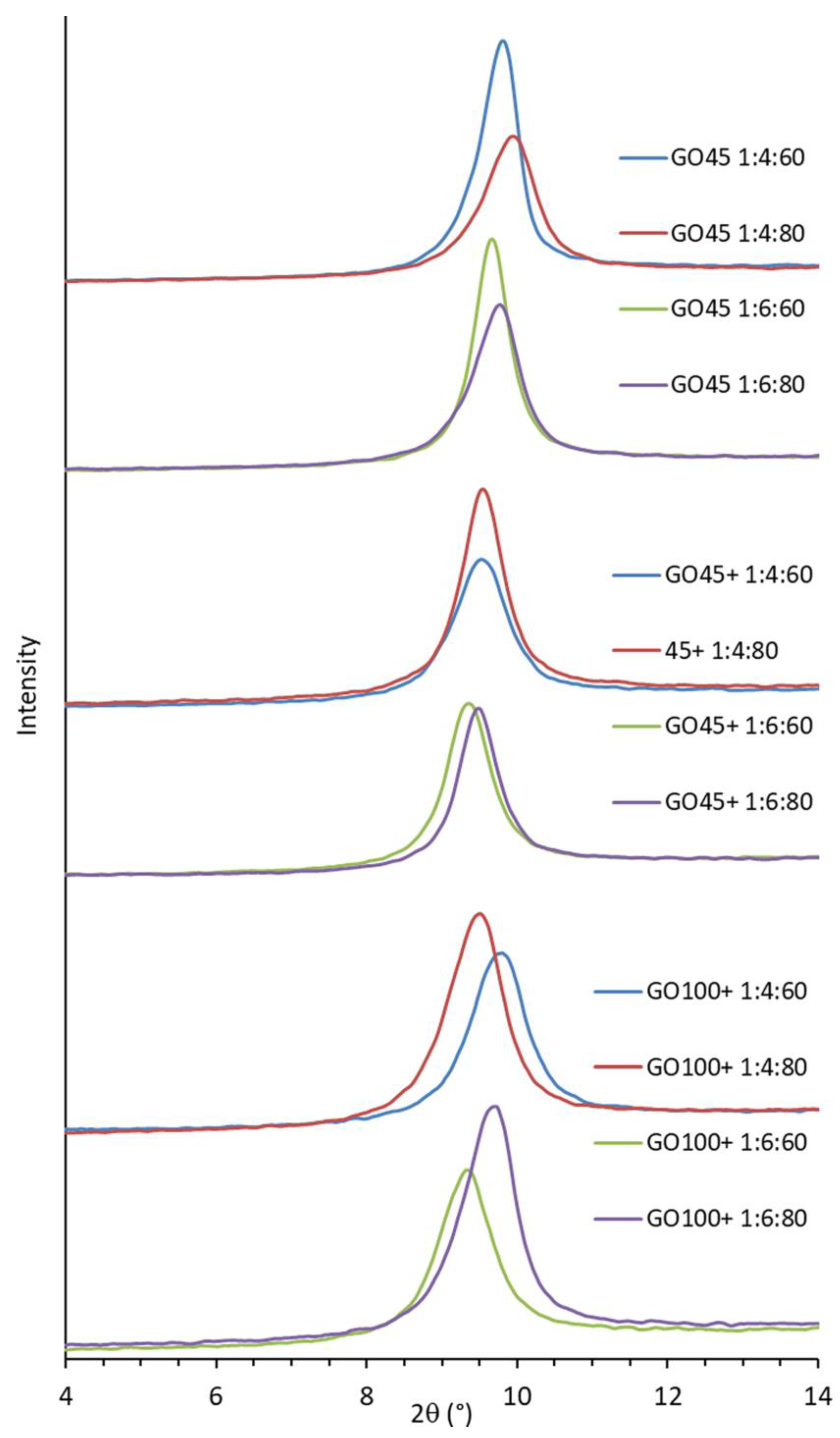
3.6. XRD Analysis of GO
3.7. Thermogravimetric Analysis of GO
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Idumah, C.I.; Hassan, A. Emerging Trends in Graphene Carbon Based Polymer Nanocomposites and Applications. Rev. Chem. Eng. 2016, 32, 223–264. [Google Scholar] [CrossRef]
- Zhu, Y.; Murali, S.; Cai, W.; Li, X.; Suk, J.W.; Potts, J.R.; Ruoff, R.S. Graphene and Graphene Oxide: Synthesis, Properties, and Applications. Adv. Mater. 2010, 22, 3906–3924. [Google Scholar] [CrossRef]
- Kosynkin, D.V.; Higginbotham, A.L.; Sinitskii, A.; Lomeda, J.R.; Dimiev, A.; Price, B.K.; Tour, J.M. Longitudinal Unzipping of Carbon Nanotubes to Form Graphene Nanoribbons. Nature 2009, 458, 872–876. [Google Scholar] [CrossRef]
- Su, C.Y.; Lu, A.Y.; Xu, Y.; Chen, F.R.; Khlobystov, A.N.; Li, L.J. High-Quality Thin Graphene Films from Fast Electrochemical Exfoliation. ACS Nano 2011, 5, 2332–2339. [Google Scholar] [CrossRef] [PubMed]
- Hawal, T.T.; Patil, M.S.; Swamy, S.; Kulkarni, R.M. A Review on Synthesis, Functionalization, Processing and Applications of Graphene Based High Performance Polymer Nanocomposites. Curr. Nanosci. 2021, 18, 167–181. [Google Scholar] [CrossRef]
- Jiříčková, A.; Jankovský, O.; Sofer, Z.; Sedmidubský, D. Synthesis and Applications of Graphene Oxide. Materials 2022, 15, 920. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.B.; Parvaz, M.; Khan, Z.H. Graphene Oxide: Synthesis and Characterization. In Advanced Structured Materials; Springer: Berlin/Heidelberg, Germany, 2017; Volume 83, pp. 1–28. [Google Scholar]
- Khine, Y.Y.; Wen, X.; Jin, X.; Foller, T.; Joshi, R. Functional Groups in Graphene Oxide. Phys. Chem. Chem. Phys. 2022, 24, 26337–26355. [Google Scholar] [CrossRef]
- Huskić, M.; Bolka, S.; Vesel, A.; Mozetič, M.; Anžlovar, A.; Vizintin, A.; Žagar, E. One-Step Surface Modification of Graphene Oxide and Influence of Its Particle Size on the Properties of Graphene Oxide/Epoxy Resin Nanocomposites. Eur. Polym. J. 2018, 101, 211–217. [Google Scholar] [CrossRef]
- Dimiev, A.; Kosynkin, D.V.; Alemany, L.B.; Chaguine, P.; Tour, J.M. Pristine Graphite Oxide. J. Am. Chem. Soc. 2012, 134, 2815–2822. [Google Scholar] [CrossRef]
- Chen, J.; Yao, B.; Li, C.; Shi, G. An Improved Hummers Method for Eco-Friendly Synthesis of Graphene Oxide. Carbon 2013, 64, 225–229. [Google Scholar] [CrossRef]
- Marcano, D.C.; Kosynkin, D.V.; Berlin, J.M.; Sinitskii, A.; Sun, Z.; Slesarev, A.; Alemany, L.B.; Lu, W.; Tour, J.M. Improved Synthesis of Graphene Oxide. ACS Nano 2010, 4, 4806–4814. [Google Scholar] [CrossRef]
- Casallas Caicedo, F.M.; Vera López, E.; Agarwal, A.; Drozd, V.; Durygin, A.; Franco Hernandez, A.; Wang, C. Synthesis of Graphene Oxide from Graphite by Ball Milling. Diam. Relat. Mater. 2020, 109, 108064. [Google Scholar] [CrossRef]
- Sun, H.; Xu, G.; Lian, W.; Kastiukas, G.; Zhang, J.; Zhang, X.; Liu, W.; Xing, F.; Ren, J. Electrochemical Synthesis and Property Characterisation of Graphene Oxide Using Water as Electrolyte. Chem. Phys. Lett. 2022, 786, 139206. [Google Scholar] [CrossRef]
- Rhazouani, A.; Gamrani, H.; El Achaby, M.; Aziz, K.; Gebrati, L.; Uddin, M.S.; Aziz, F. Synthesis and Toxicity of Graphene Oxide Nanoparticles: A Literature Review of in Vitro and in Vivo Studies. Biomed Res. Int. 2021, 2021, 5518999. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Ullah, S.; Rao, N.; Singh, M.; Patra, I.; Darko, D.A.; Issac, C.P.J.; Esmaeilzadeh-Salestani, K.; Kanaoujiya, R.; Vijayan, V. Synthesis of Three-Dimensional Reduced-Graphene Oxide from Graphene Oxide. J. Nanomater. 2022, 2022, 8731429. [Google Scholar] [CrossRef]
- Gutiérrez-Cruz, A.; Ruiz-Hernández, A.R.; Vega-Clemente, J.F.; Luna-Gazcón, D.G.; Campos-Delgado, J. A Review of Top-down and Bottom-up Synthesis Methods for the Production of Graphene, Graphene Oxide and Reduced Graphene Oxide. J. Mater. Sci. 2022, 57, 14543–14578. [Google Scholar] [CrossRef]
- Bera, M.; Chandravati; Gupta, P.; Maji, P.K. Facile One-Pot Synthesis of Graphene Oxide by Sonication Assisted Mechanochemical Approach and Its Surface Chemistry. J. Nanosci. Nanotechnol. 2017, 18, 902–912. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Bolados, H.; Contreras-Cid, A.; Yazdani-Pedram, M.; Acosta-Villavicencio, G.; Flores, M.; Fuentealba, P.; Neira-Carrillo, A.; Verdejo, R.; López-Manchado, M.A. Synthesis of Fluorinated Graphene Oxide by Using an Easy One-Pot Deoxyfluorination Reaction. J. Colloid Interface Sci. 2018, 524, 219–226. [Google Scholar] [CrossRef]
- Zhou, X.; Liu, Z. A Scalable, Solution-Phase Processing Route to Graphene Oxide and Graphene Ultralarge Sheets. Chem. Commun. 2010, 46, 2611–2613. [Google Scholar] [CrossRef]
- Chen, J.; Chi, F.; Huang, L.; Zhang, M.; Yao, B.; Li, Y.; Li, C.; Shi, G. Synthesis of Graphene Oxide Sheets with Controlled Sizes from Sieved Graphite Flakes. Carbon 2016, 110, 34–40. [Google Scholar] [CrossRef]
- Jia, J.; Kan, C.M.; Lin, X.; Shen, X.; Kim, J.K. Effects of Processing and Material Parameters on Synthesis of Monolayer Ultralarge Graphene Oxide Sheets. Carbon 2014, 77, 244–254. [Google Scholar] [CrossRef]
- Rourke, J.P.; Pandey, P.A.; Moore, J.J.; Bates, M.; Kinloch, I.A.; Young, R.J.; Wilson, N.R. The Real Graphene Oxide Revealed: Stripping the Oxidative Debris from the Graphene-like Sheets. Angew. Chem. Int. Ed. 2011, 50, 3173–3177. [Google Scholar] [CrossRef]
- Daud, N.A.; Chieng, B.W.; Ibrahim, N.A.; Talib, Z.A.; Muhamad, E.N.; Abidin, Z.Z. Functionalizing Graphene Oxide with Alkylamine by Gamma-Ray Irradiation Method. Nanomaterials 2017, 7, 135. [Google Scholar] [CrossRef]
- Xu, G.; Malmstrom, J.; Edmonds, N.; Broderick, N.; Travas-Sejdic, J.; Jin, J. Investigation of the Reduction of Graphene Oxide by Lithium Triethylborohydride. J. Nanomater. 2016, 2016, 4021059. [Google Scholar] [CrossRef]
- Hussein, A.; Sarkar, S.; Lee, K.; Kim, B. Cryogenic Fracture Behavior of Epoxy Reinforced by a Novel Graphene Oxide/Poly(p-Phenylenediamine) Hybrid. Compos. B Eng. 2017, 129, 133–142. [Google Scholar] [CrossRef]
- Eigler, S.; Dotzer, C.; Hof, F.; Bauer, W.; Hirsch, A. Sulfur Species in Graphene Oxide. Chem. Eur. J. 2013, 19, 9490–9496. [Google Scholar] [CrossRef]
- Nagyte, V.; Kelly, D.J.; Felten, A.; Picardi, G.; Shin, Y.Y.; Alieva, A.; Worsley, R.E.; Parvez, K.; Dehm, S.; Krupke, R.; et al. Raman Fingerprints of Graphene Produced by Anodic Electrochemical Exfoliation. Nano Lett. 2020, 20, 3411–3419. [Google Scholar] [CrossRef]
- Eckmann, A.; Felten, A.; Verzhbitskiy, I.; Davey, R.; Casiraghi, C. Raman Study on Defective Graphene: Effect of the Excitation Energy, Type, and Amount of Defects. Phys. Rev. B Condens. Matter Mater. Phys. 2013, 88, 035426. [Google Scholar] [CrossRef]
- Eckmann, A.; Felten, A.; Mishchenko, A.; Britnell, L.; Krupke, R.; Novoselov, K.S.; Casiraghi, C. Probing the Nature of Defects in Graphene by Raman Spectroscopy. Nano Lett. 2012, 12, 3925–3930. [Google Scholar] [CrossRef]
- Casiraghi, C.; Hartschuh, A.; Qian, H.; Pliscanec, S.; Georgia, C.; Fasoli, A.; Novoselov, K.S.; Basko, D.M.; Ferrari, A.C. Raman Spectroscopy of Graphene Edges. Nano Lett. 2009, 9, 1433–1441. [Google Scholar] [CrossRef]
- Li, Z.; Wang, Y.; Kozbial, A.; Shenoy, G.; Zhou, F.; McGinley, R.; Ireland, P.; Morganstein, B.; Kunkel, A.; Surwade, S.P.; et al. Effect of Airborne Contaminants on the Wettability of Supported Graphene and Graphite. Nat. Mater. 2013, 12, 925–931. [Google Scholar] [CrossRef]
- Lee, H.C.; Liu, W.W.; Chai, S.P.; Mohamed, A.R.; Aziz, A.; Khe, C.S.; Hidayah, N.M.S.; Hashim, U. Review of the Synthesis, Transfer, Characterization and Growth Mechanisms of Single and Multilayer Graphene. RSC Adv. 2017, 7, 15644–15693. [Google Scholar] [CrossRef]
- Lotya, M.; Rakovich, A.; Donegan, J.F.; Coleman, J.N. Measuring the Lateral Size of Liquid-Exfoliated Nanosheets with Dynamic Light Scattering. Nanotechnology 2013, 24, 265703. [Google Scholar] [CrossRef]
- Chen, X.; Jiang, W.; Hu, B.; Liang, Z.; Zhang, Y.; Kang, J.; Cao, Y.; Xiang, M. Effects of Graphene Oxide Size on Curing Kinetics of Epoxy Resin. RSC Adv. 2021, 11, 29215–29226. [Google Scholar] [CrossRef]
- Schniepp, H.C.; Li, J.L.; McAllister, M.J.; Sai, H.; Herrera-Alonson, M.; Adamson, D.H.; Prud’homme, R.K.; Car, R.; Seville, D.A.; Aksay, I.A. Functionalized Single Graphene Sheets Derived from Splitting Graphite Oxide. J. Phys. Chem. B 2006, 110, 8535–8539. [Google Scholar] [CrossRef]
- Szabó, T.; Berkesi, O.; Dékány, I. DRIFT Study of Deuterium-Exchanged Graphite Oxide. Carbon 2005, 43, 3186–3189. [Google Scholar] [CrossRef]
- Lakhe, P.; Kulhanek, D.L.; Sun, W.; Zhang, B.; Green, M.J.; Mannan, M.S. Calorimetry of Explosive Thermal Decomposition of Graphite Oxide. J. Hazard. Mater. 2019, 366, 275–281. [Google Scholar] [CrossRef]
- Qiu, Y.; Guo, F.; Hurt, R.; Külaots, I. Explosive Thermal Reduction of Graphene Oxide-Based Materials: Mechanism and Safety Implications. Carbon 2014, 72, 215–223. [Google Scholar] [CrossRef]
- Navaee, A.; Salimi, A. Efficient Amine Functionalization of Graphene Oxide through the Bucherer Reaction: An Extraordinary Metal-Free Electrocatalyst for the Oxygen Reduction Reaction. RSC Adv. 2015, 5, 59874–59880. [Google Scholar] [CrossRef]
- Xiang, B.Q.; Hou, Y.G.; Wu, J.X.; Ma, Y.X.; Gan, X.T.; Zhao, J. In-fiber photoelectric device based on graphene-coated tilted fiber grating. Opto Electron. Sci. 2023, 2, 230012. [Google Scholar] [CrossRef]
- Jovanović, S.; Huskić, M.; Kepić, D.; Yasir, M.; Haddadi, K. A review on graphene and graphene composites for application in electromagnetic shielding. Graphene 2D Mater. 2023, 8, 59–80. [Google Scholar] [CrossRef]
- Worku, A.K.; Ayele, D.W. Recent advances of graphene-based materials for emerging technologies. Results Chem. 2023, 5, 100971. [Google Scholar] [CrossRef]
- Kumar, N.; Srivastava, V.C. Simple Synthesis of Large Graphene Oxide Sheets via Electrochemical Method Coupled with Oxidation Process. ACS Omega 2018, 3, 10233–10242. [Google Scholar] [CrossRef]
- Hou, Y.; Lv, S.; Liu, L.; Liu, X. High-quality preparation of graphene oxide via the Hummers' method: Understanding the roles of the intercalator, oxidant, and graphite particle size. Ceram. Int. 2020, 46, 2392–2402. [Google Scholar] [CrossRef]
- Zhang, Q.; Yang, Y.; Fan, H.; Feng, L.; Wen, G.; Qin, L. Synthesis of graphene oxide using boric acid in hummers method. Colloids Surf. A 2022, 652, 129802. [Google Scholar] [CrossRef]
- Costa, M.C.F.; Marangoni, V.S.; Ng, P.R.; Nguyen, H.T.L.; Carvalho, A.; Castro Neto, A.H. Accelerated Synthesis of Graphene Oxide from Graphene. Namomaterials 2021, 11, 551. [Google Scholar] [CrossRef] [PubMed]
- Muniyalakshmi, M.; Sethuraman, K.; Silambarasan, D. Synthesis and characterization of graphene oxide nanosheets. Mater. Today Proc. 2020, 21, 408–410. [Google Scholar] [CrossRef]
- Kotsyubynsky, V.O.; Boychuk, V.M.; Budzulyak, I.M.; Rachiy, B.I.; Hodlevska, M.A.; Kachmar, A.I.; Hodlevsky, M.A. Graphene oxide synthesis using modified Tour method. Adv. Nat. Sci. Nanosci. Nanotechnol. 2021, 12, 035006. [Google Scholar] [CrossRef]
- Díez-Pascual, A.M.; Sainz-Urruela, C.; Vallés, C.; Vera-López, S.; Paz San Andrés, M. Tailorable Synthesis of Highly Oxidized Graphene Oxides via an Environmentally-Friendly Electrochemical Process. Nanomaterials 2020, 10, 239. [Google Scholar] [CrossRef]
- Marković, Z.M.; Budimir, M.D.; Kepić, D.P.; Holclajtner-Antunović, I.D.; Marinović-Cincović, M.T.; Dramićanin, M.D.; Spasojević, V.D.; Peruško, D.B.; Špitalský, Z.; Mičušik, M.; et al. Semi-transparent, conductive thin films of electrochemical exfoliated graphene. RSC Adv. 2016, 6, 39275–39283. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Yield (%) | Yield (%) | Yield (%) | |||
|---|---|---|---|---|---|
| GO45 1:4:60 | 210 | GO45+ 1:4:60 | 223 | GO100+ 1:4:60 | 223 |
| GO45 1:4:80 | 217 | GO45+ 1:4:80 | 218 | GO100+ 1:4:80 | 223 |
| GO45 1:6:60 | 226 | GO45+ 1:6:60 | 226 | GO100+ 1:6:60 | 227 |
| GO45 1:6:80 | 228 | GO45+ 1:6:80 | 222 | GO100+ 1:6:80 | 226 |
| C-C/C=C (%) | C-O (%) | C=O (%) | COOH (%) | O/C | |
|---|---|---|---|---|---|
| GO45 1:4:60 | 43.7 | 44.7 | 8.1 | 3.5 | 0.52 |
| GO45 1:4:80 | 38.7 | 47.4 | 9.8 | 4.1 | 0.58 |
| GO45 1:6:60 | 35.5 | 50.2 | 10.4 | 3.8 | 0.62 |
| GO45 1:6:80 | 38.2 | 45.1 | 11.9 | 4.8 | 0.58 |
| GO100+ 1:4:60 | 43.6 | 44.0 | 7.9 | 4.5 | 0.55 |
| GO100+ 1:4:80 | 47.8 | 45.0 | 3.5 | 3.7 | 0.44 |
| GO100+ 1:6:60 | 42.5 | 46.1 | 7.4 | 4.0 | 0.54 |
| GO100+ 1:6:80 | 44.7 | 45.8 | 5.9 | 3.6 | 0.50 |
| Sample | ID/IG Ratio | Sample | ID/IG Ratio |
|---|---|---|---|
| GO45 1:4:60 | 0.92 | GO100+ 1:4:60 | 0.89 |
| GO45 1:4:80 | 0.92 | GO100+ 1:4:80 | 0.92 |
| GO45 1:6:60 | 0.95 | GO100+ 1:6:60 | 0.92 |
| GO45 1:6:80 | 0.92 | GO100+ 1:6:80 | 0.88 |
| Sample | Contact Angle | Sample | Contact Angle |
|---|---|---|---|
| GO45 1:4:60 | 21.0° | GO100+ 1:4:60 | 24.0° |
| GO45 1:4:80 | 22.6° | GO100+ 1:4:80 | 34.3° |
| GO45 1:6:60 | 28.5° | GO100+ 1:6:60 | 25.3° |
| GO45 1:6:80 | 28.3° | GO100+ 1:6:80 | 23.8° |
| Slurry | Dried Samples | |||
|---|---|---|---|---|
| Z-aver. (nm) | PDI | Z-aver. (nm) | PDI | |
| GO45 1:4:60 | 5249 | 0.524 | 7020 | 0.730 |
| GO45 1:4:80 | 3984 | 0.407 | 5062 | 0.569 |
| GO45 1:6:60 | 2637 | 0.416 | 4579 | 0.834 |
| GO45 1:6:80 | 2851 | 0.364 | 4868 | 0.784 |
| GO45+ 1:4:60 | 2800 | 0.493 | 3688 | 0.573 |
| GO45+ 1:4:80 | 2577 | 0.530 | 4161 | 0.535 |
| GO45+ 1:6:60 | 3418 | 0.682 | 4465 | 0.892 |
| GO45+ 1:6:80 | 3701 | 0.528 | 5438 | 0.454 |
| GO100+ 1:4:60 | 3486 | 0.253 | 5420 | 0.406 |
| GO100+ 1:4:80 | 4096 | 0.368 | 7131 | 0.568 |
| GO100+ 1:6:60 | 5151 | 0.639 | 7187 | 0.885 |
| GO100+ 1:6:80 | 4306 | 0.472 | 6141 | 0.508 |
| Sample | 2θ (°) | d-Spacing (nm) | FWHM | Cryst. Size (nm) |
|---|---|---|---|---|
| GO45 1:4:60 | 9.765 | 0.905 | 0.659 | 13.3 |
| GO45 1:4:80 | 9.907 | 0.892 | 0.862 | 9.9 |
| GO45 1:6:60 | 9.661 | 0.915 | 0.613 | 14.4 |
| GO45 1:6:80 | 9.731 | 0.908 | 0.774 | 11.1 |
| GO45+ 1:4:60 | 9.521 | 0.928 | 0.858 | 10.0 |
| GO45+ 1:4:80 | 9.543 | 0.926 | 0.751 | 11.5 |
| GO45+ 1:6:60 | 9.357 | 0.944 | 0.752 | 11.5 |
| GO45+ 1:6:80 | 9.486 | 0.932 | 0.687 | 12.7 |
| GO100+ 1:4:60 | 9.754 | 0.906 | 0.834 | 10.3 |
| GO100+ 1:4:80 | 9.455 | 0.935 | 0.910 | 9.4 |
| GO100+ 1:6:60 | 9.638 | 0.917 | 0.819 | 10.5 |
| GO100+ 1:6:80 | 9.318 | 0.948 | 0.908 | 9.4 |
| Weight loss | Peak | ||||
|---|---|---|---|---|---|
| 1st st. (%) | 2nd st. (%) | 3rd st. (%) | 4th st. (%) | (°C) | |
| GO45 1:4:60 | 8.8 | 28.9 | 11.7 | 6.2 | 172 |
| GO45 1:4:80 | 8.5 | 30.3 | 12.7 | 4.6 | 162 |
| GO45 1:6:60 | 8.1 | 32.7 | 13.5 | 3.4 | 170 |
| GO45 1:6:80 | 7.9 | 29.8 | 12.8 | 4.0 | 159 |
| GO45+ 1:4:60 | 7.7 | 31.8 | 13.4 | 4.4 | 168 |
| GO45+ 1:4:80 | 9.3 | 30.9 | 13.5 | 6.0 | 173 |
| GO45+ 1:6:60 | 7.7 | 31.3 | 12.0 | 4.7 | 170 |
| GO45+ 1:6:80 | 7.8 | 29.9 | 11.6 | 3.8 | 173 |
| GO100+ 1:4:60 | 7.4 | 30.0 | 13.1 | 6.5 | 163 |
| GO100+ 1:4:80 | 8.0 | 30.4 | 13.6 | 3.0 | 164 |
| GO100+ 1:6:60 | 7.5 | 31.3 | 12.0 | 3.7 | 170 |
| GO100+ 1:6:80 | 7.8 | 31.1 | 14.5 | 4.4 | 166 |
| Method | ID/IG | Sheet Size (μm) | TGA WL 500 °C (%) | XRD (°) | Ref. |
|---|---|---|---|---|---|
| Electrochemical followed by oxidation with KMnO4 | 1.24 | ≈10 | ≈80 | 8.88 | [44] |
| Hummers’ method | 0.66–0.90 | 0.98 | 61.55 | 10.50 | [45] |
| Improved Hummers’ method with boric acid | 0.92–1.04 | / | 46.22 | 10.10 | [46] |
| Modified Hummers’ | 1.0 | ≈10 | / | / | [47] |
| Modified Hummers’ | 0.83 | / | / | 10.44 | [48] |
| Tour method | 1.77 | / | / | 10.44 | [49] |
| Electrochemical synthesis | 0.41–1.71 | / | / | 9–12 | [50] |
| Electrochemical synthesis | 0.04 | Up to 12 | [51] | ||
| Modified Hummers’ | 0.92 | Up to 5.25 | / | 9.91 | This study |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huskić, M.; Kepić, D.; Kleut, D.; Mozetič, M.; Vesel, A.; Anžlovar, A.; Bogdanović, D.B.; Jovanović, S. The Influence of Reaction Conditions on the Properties of Graphene Oxide. Nanomaterials 2024, 14, 281. https://doi.org/10.3390/nano14030281
Huskić M, Kepić D, Kleut D, Mozetič M, Vesel A, Anžlovar A, Bogdanović DB, Jovanović S. The Influence of Reaction Conditions on the Properties of Graphene Oxide. Nanomaterials. 2024; 14(3):281. https://doi.org/10.3390/nano14030281
Chicago/Turabian StyleHuskić, Miroslav, Dejan Kepić, Duška Kleut, Miran Mozetič, Alenka Vesel, Alojz Anžlovar, Danica Bajuk Bogdanović, and Svetlana Jovanović. 2024. "The Influence of Reaction Conditions on the Properties of Graphene Oxide" Nanomaterials 14, no. 3: 281. https://doi.org/10.3390/nano14030281