
Evaluation of the Efficiency of Photoelectrochemical Activity Enhancement for the Nanostructured LaFeO3 Photocathode by Surface Passivation and Co-Catalyst Deposition

 , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Synthesis
2.1.1. LaFeO3 Films
2.1.2. Pt Nanoparticles
2.1.3. TiO2 Layers
2.2. Characterization
2.3. Photoelectrochemical Measurements
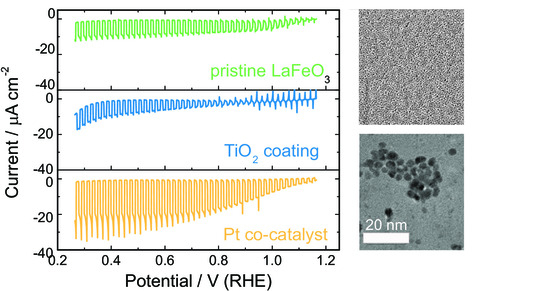
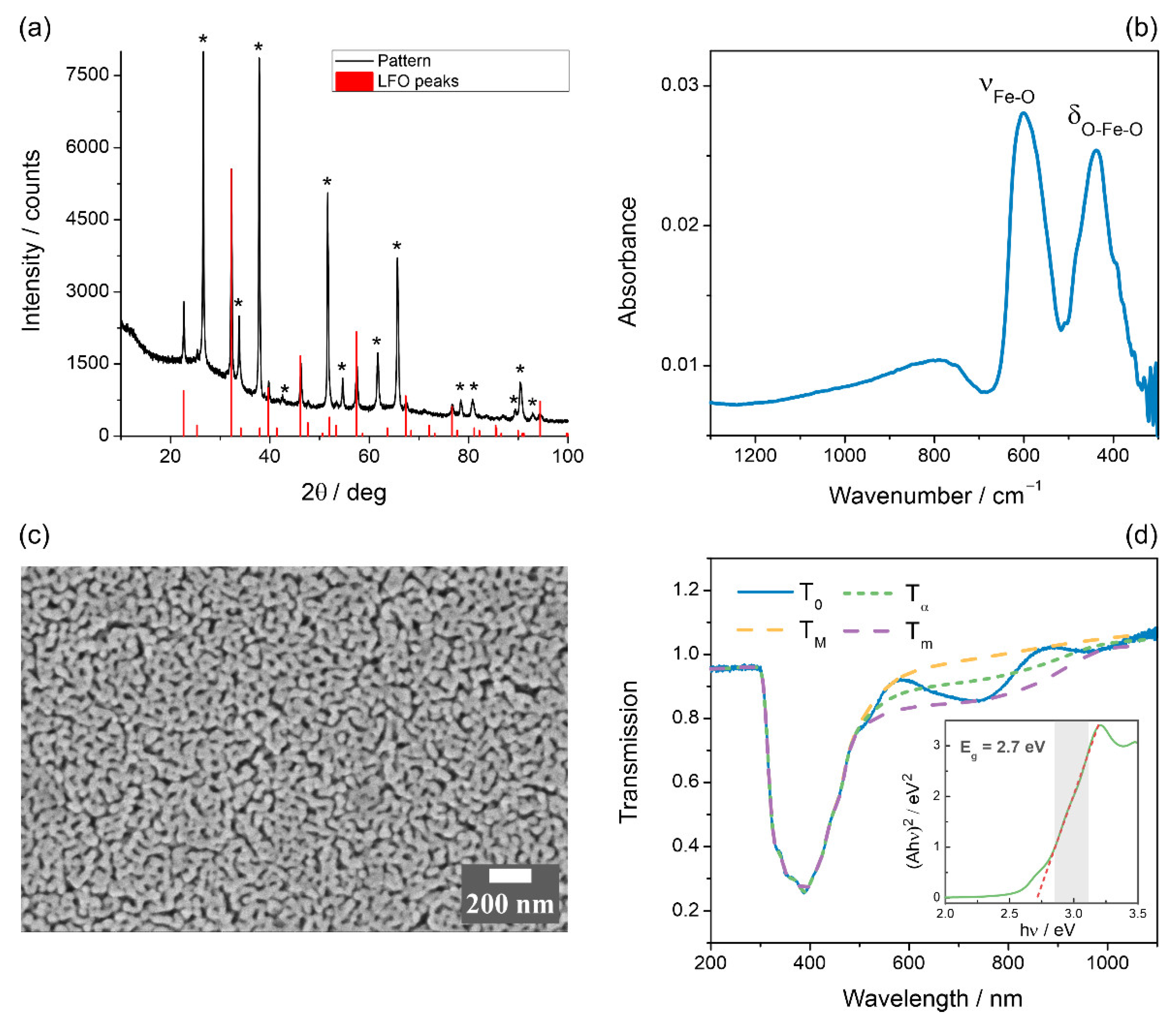
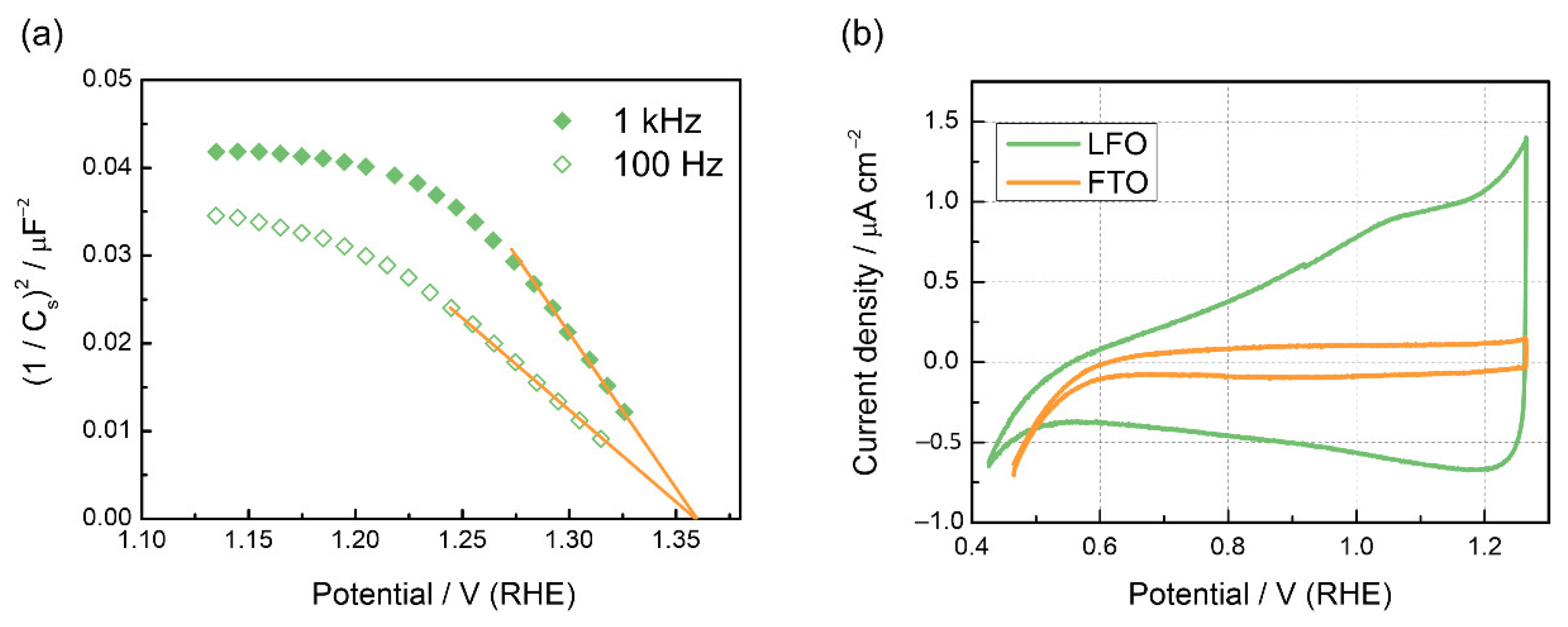
3. Results
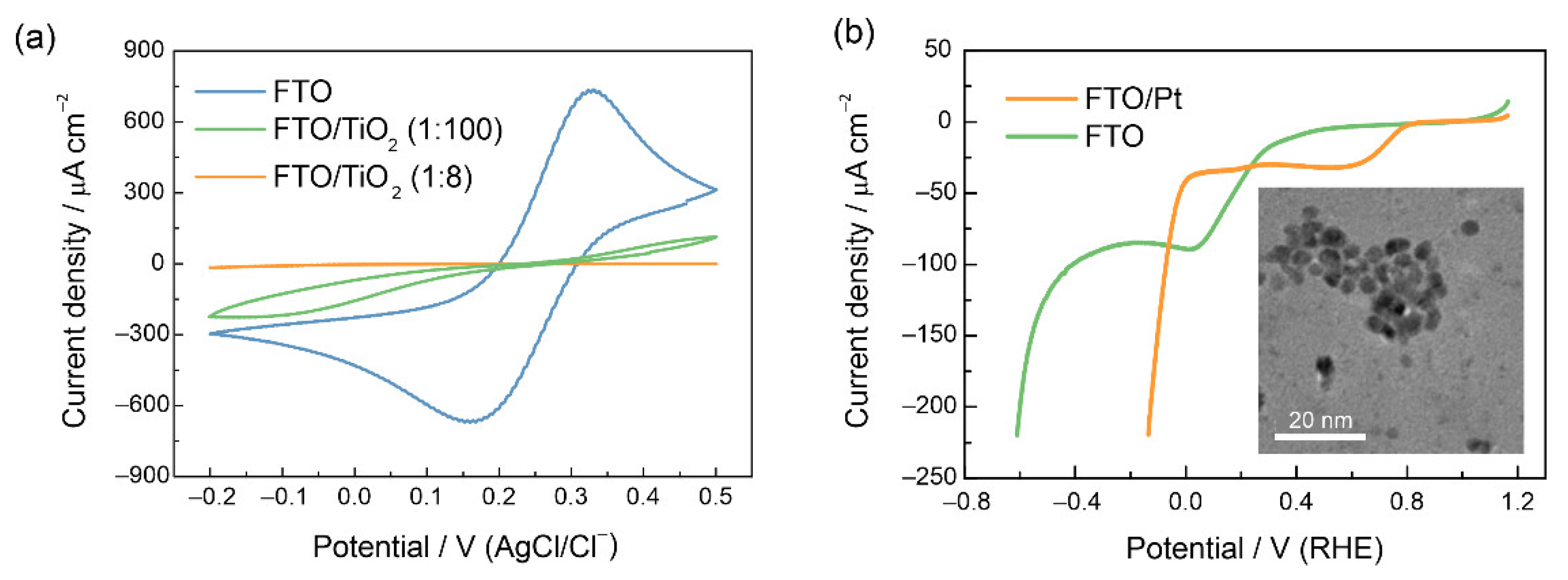
3.1. Characterization of the LFO Films
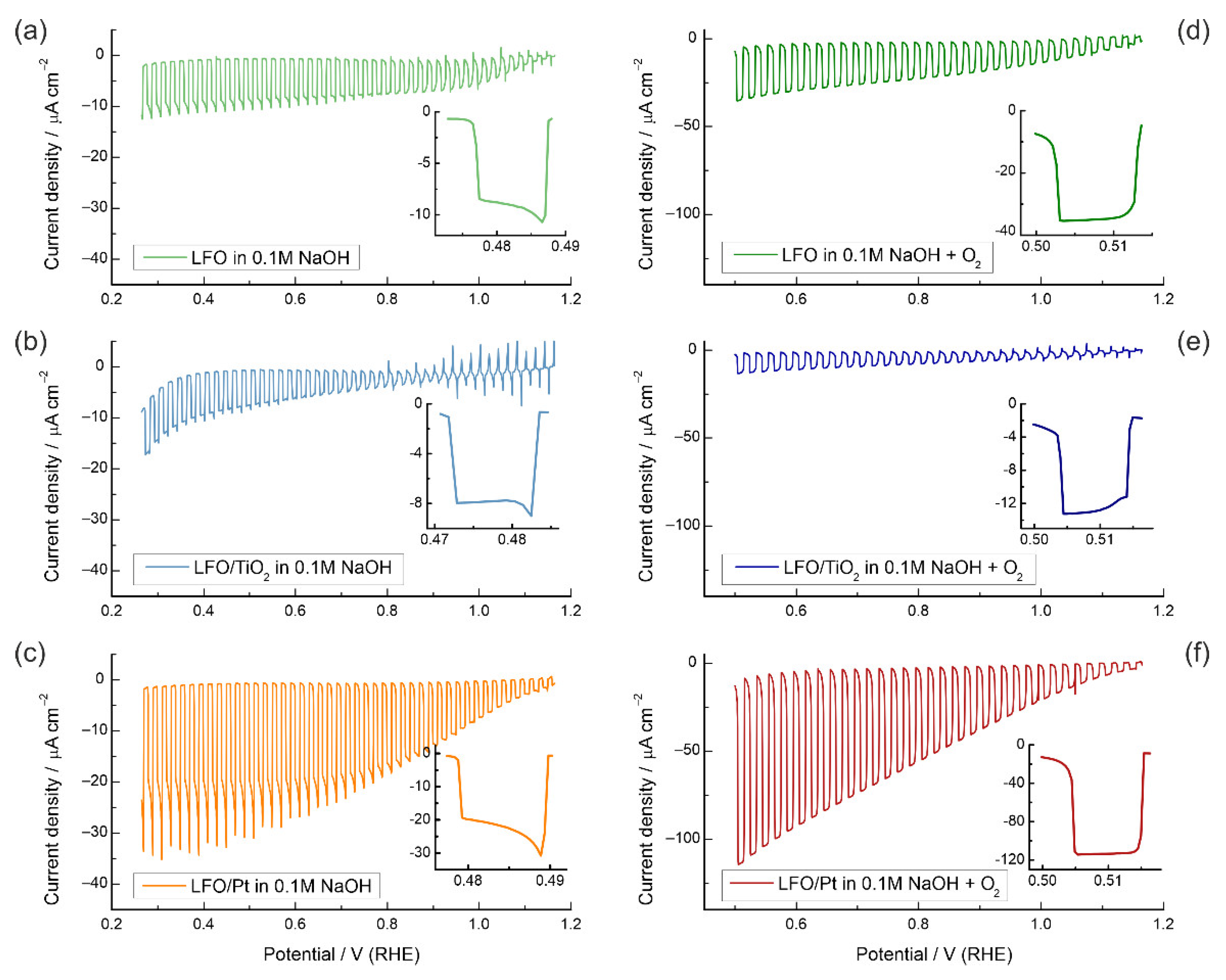
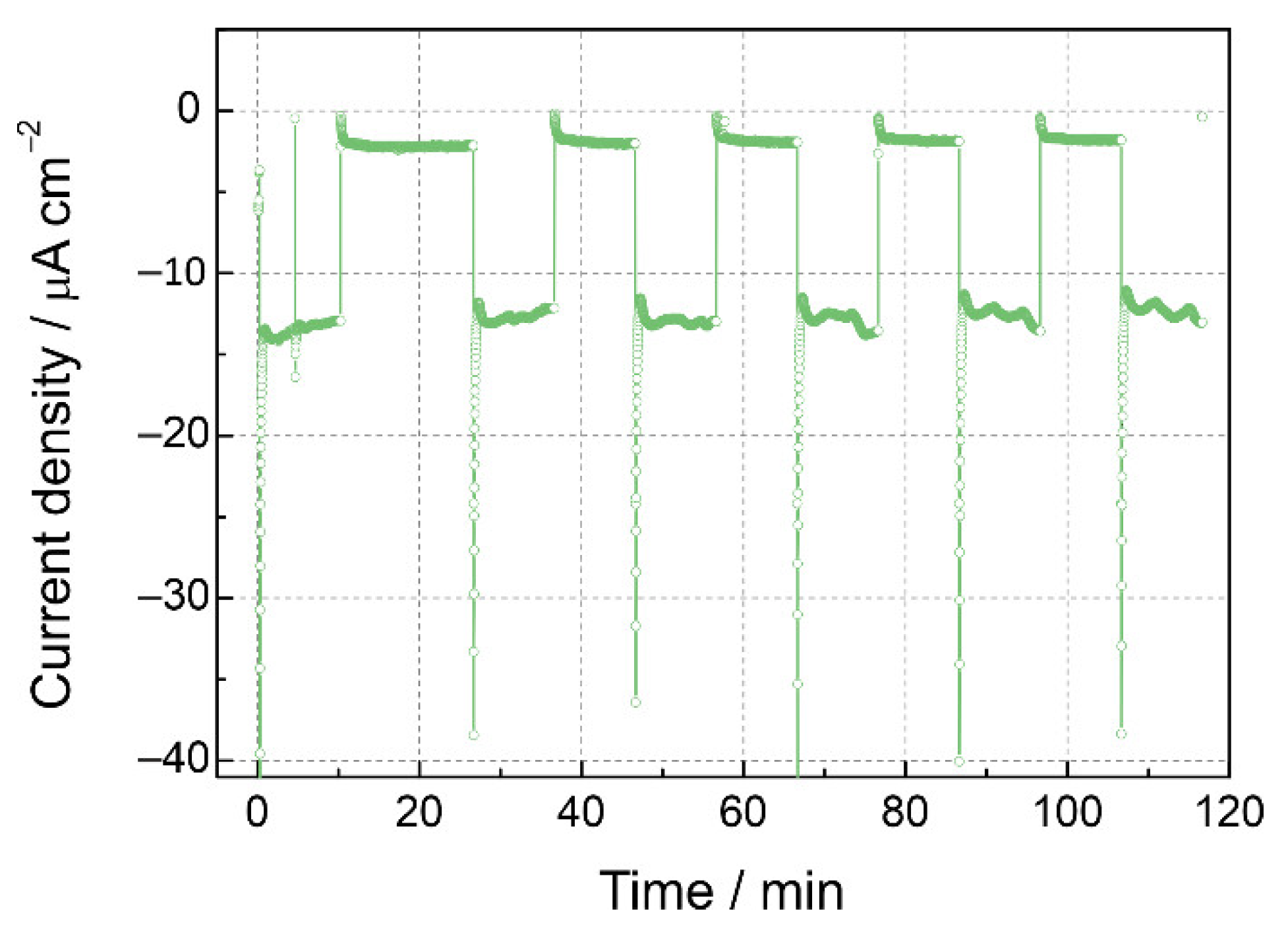
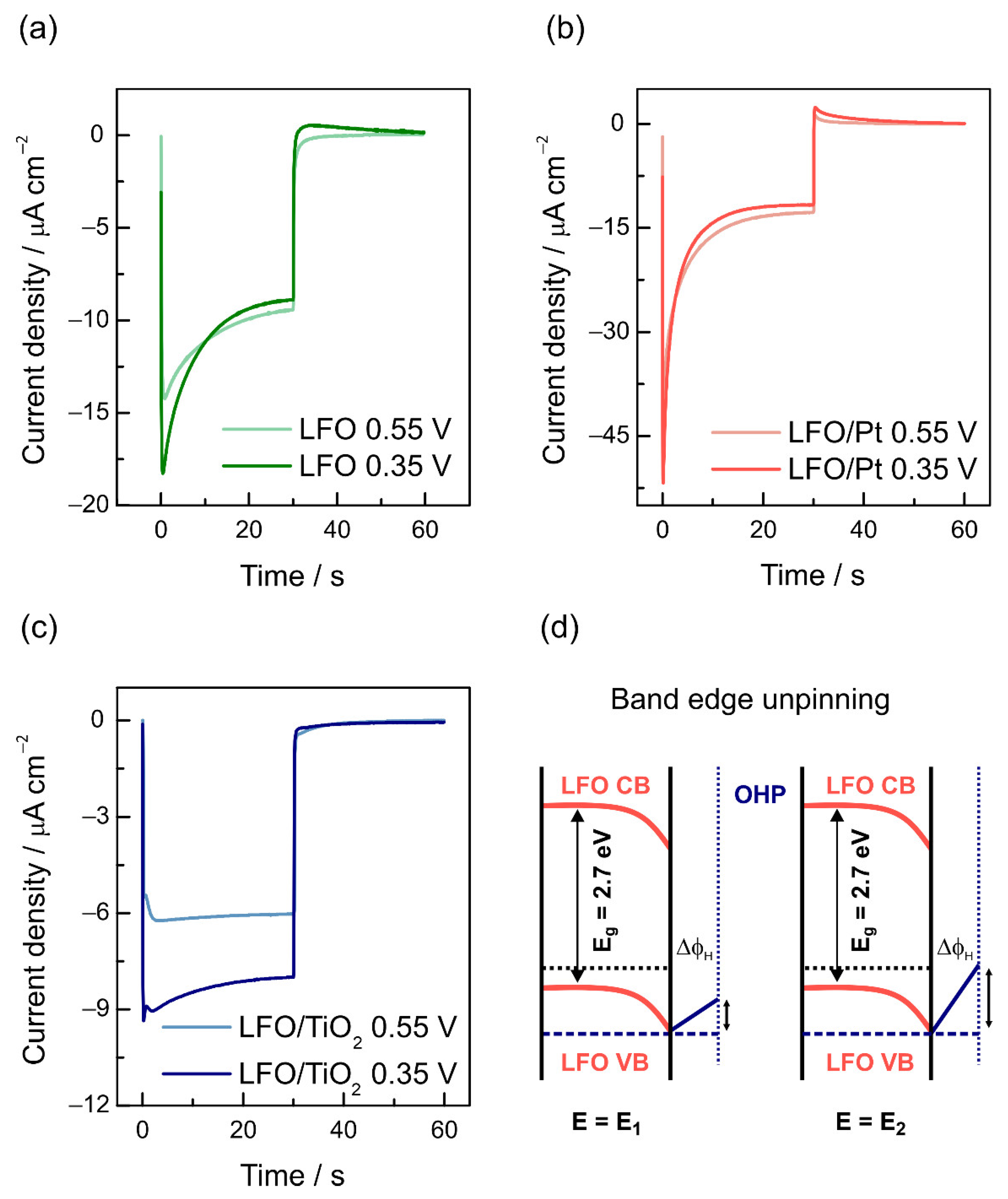
3.2. Photoelectrochemical Properties
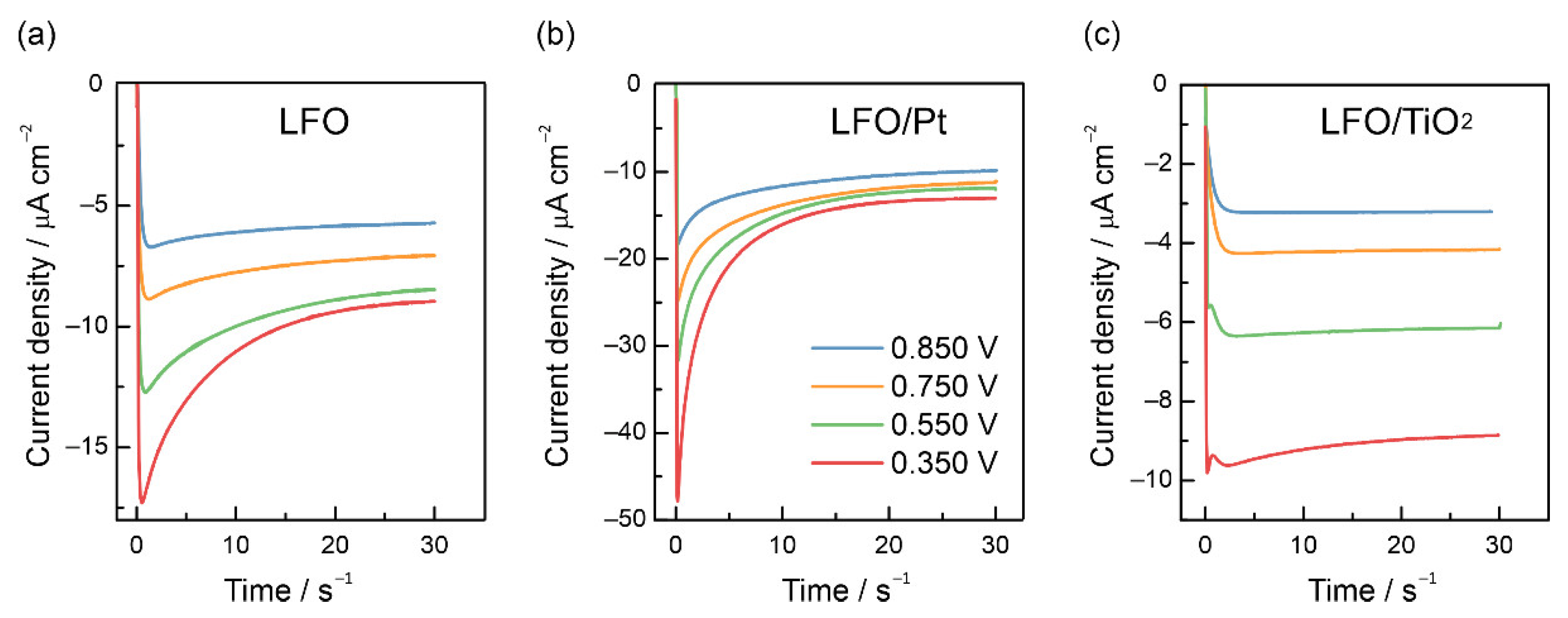
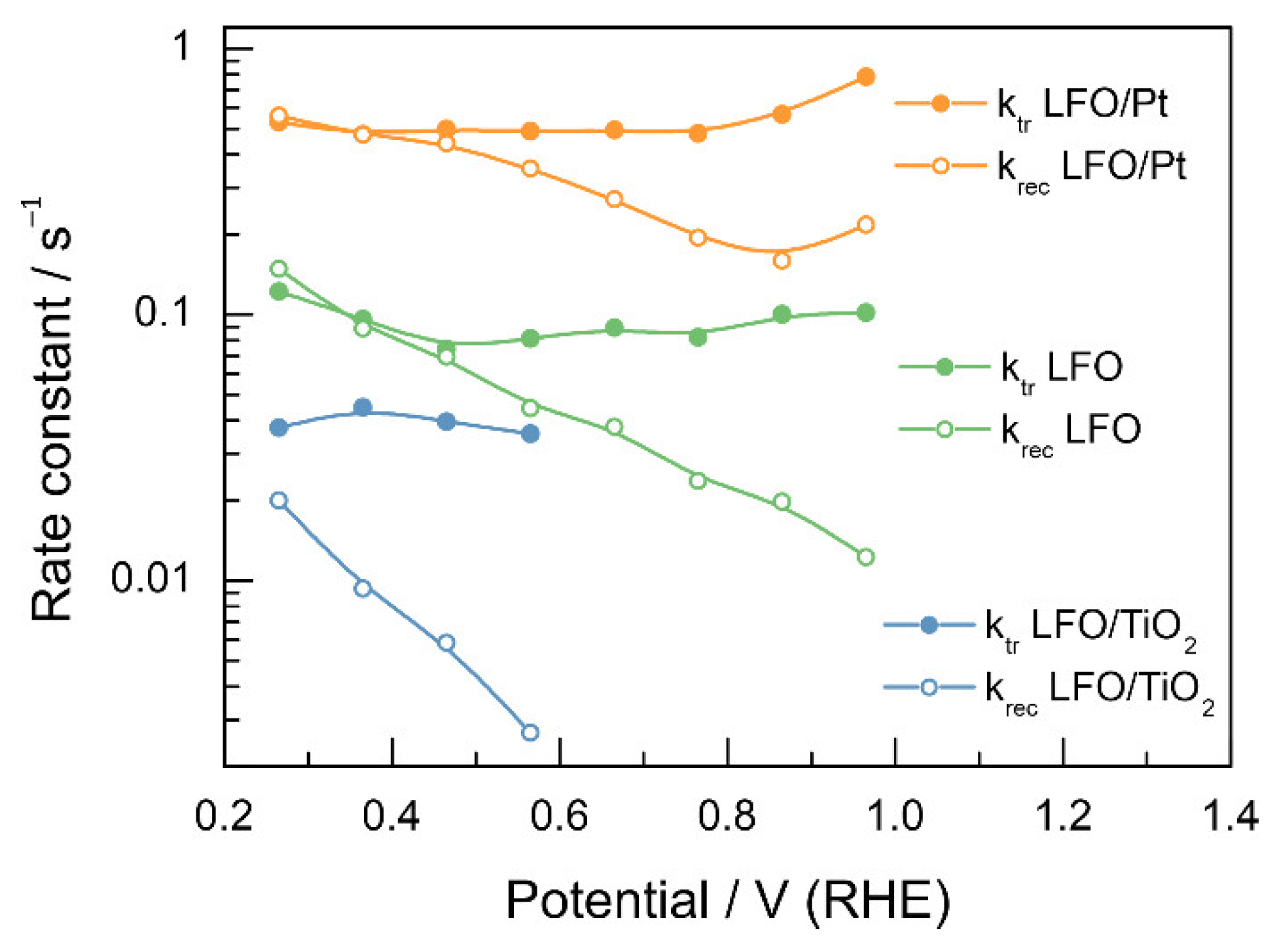
3.3. Recombination and Charge Transfer Kinetics
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Corby, S.; Rao, R.R.; Steier, L.; Durrant, J.R. The kinetics of metal oxide photoanodes from charge generation to catalysis. Nat. Rev. Mater. 2021, 6, 1136–1155. [Google Scholar] [CrossRef]
- Lee, D.K.; Lee, D.; Lumley, M.A.; Choi, K.S. Progress on ternary oxide-based photoanodes for use in photoelectrochemical cells for solar water splitting. Chem. Soc. Rev. 2019, 48, 2126–2157. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Prabhakar, R.R.; Tan, J.; Tilley, S.D.; Moon, J. Strategies for enhancing the photocurrent, photovoltage, and stability of photoelectrodes for photoelectrochemical water splitting. Chem. Soc. Rev. 2019, 48, 4979–5015. [Google Scholar] [CrossRef] [PubMed]
- Tang, P.; Arbiol, J. Engineering surface states of hematite based photoanodes for boosting photoelectrochemical water splitting. Nanoscale Horiz. 2019, 4, 1256–1276. [Google Scholar] [CrossRef]
- Roger, I.; Shipman, M.A.; Symes, M.D. Earth-abundant catalysts for electrochemical and photoelectrochemical water splitting. Nat. Rev. Chem. 2017, 1, 0003. [Google Scholar] [CrossRef]
- Ros, C.; Andreu, T.; Morante, J.R. Photoelectrochemical water splitting: A road from stable metal oxides to protected thin film solar cells. J. Mater. Chem. A 2020, 8, 10625–10669. [Google Scholar] [CrossRef]
- Hwang, J.; Rao, R.R.; Giordano, L.; Katayama, Y.; Yu, Y.; Shao-Horn, Y. Perovskites in catalysis and electrocatalysis. Science 2017, 358, 751–756. [Google Scholar] [CrossRef] [Green Version]
- Si, C.; Zhang, W.; Lu, Q.; Guo, E.; Yang, Z.; Chen, J.; He, X.; Luo, J. Recent Advances in Perovskite Catalysts for Efficient Overall Water Splitting. Catalysts 2022, 12, 601. [Google Scholar] [CrossRef]
- Xiao, M.; Luo, B.; Wang, Z.; Wang, S.; Wang, L. Recent Advances of Metal-Oxide Photoanodes: Engineering of Charge Separation and Transportation toward Efficient Solar Water Splitting. Solar RRL 2020, 4, 1900509. [Google Scholar] [CrossRef]
- Rodriguez-Gutierrez, I.; Bedin, K.C.; Mourino, B.; Souza Junior, J.B.; Souza, F.L. Advances in Engineered Metal Oxide Thin Films by Low-Cost, Solution-Based Techniques for Green Hydrogen Production. Nanomaterials 2022, 12, 1957. [Google Scholar] [CrossRef]
- Yao, Y.; Sang, D.; Zou, L.; Wang, Q.; Liu, C. A Review on the Properties and Applications of WO3 Nanostructure-Based Optical and Electronic Devices. Nanomaterials 2021, 11, 2136. [Google Scholar] [CrossRef] [PubMed]
- Nyarige, J.S.; Paradzah, A.T.; Kruger, T.P.J.; Diale, M. Mono-Doped and Co-Doped Nanostructured Hematite for Improved Photoelectrochemical Water Splitting. Nanomaterials 2022, 12, 366. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, B.; Wang, X.; Zhang, Y.; Huang, W. Nanoporous MoO3−x/BiVO4 photoanodes promoting charge separation for efficient photoelectrochemical water splitting. Nano Res. 2022, 15, 7026–7033. [Google Scholar] [CrossRef]
- Wang, S.; He, T.; Yun, J.H.; Hu, Y.; Xiao, M.; Du, A.; Wang, L. New Iron-Cobalt Oxide Catalysts Promoting BiVO4 Films for Photoelectrochemical Water Splitting. Adv. Funct. Mater. 2018, 28, 1802685. [Google Scholar] [CrossRef]
- Wang, S.; Wang, X.; Liu, B.; Guo, Z.; Ostrikov, K.K.; Wang, L.; Huang, W. Vacancy defect engineering of BiVO4 photoanodes for photoelectrochemical water splitting. Nanoscale 2021, 13, 17989–18009. [Google Scholar] [CrossRef]
- Wang, S.; Wang, L.; Huang, W. Bismuth-based photocatalysts for solar energy conversion. J. Mater. Chem. A 2020, 8, 24307–24352. [Google Scholar] [CrossRef]
- Díez-García, M.I.; Gómez, R. Progress in Ternary Metal Oxides as Photocathodes for Water Splitting Cells: Optimization Strategies. Solar RRL 2022, 6, 2100871. [Google Scholar] [CrossRef]
- Jang, Y.J.; Lee, J.S. Photoelectrochemical Water Splitting with p-Type Metal Oxide Semiconductor Photocathodes. ChemSusChem 2019, 12, 1835–1845. [Google Scholar] [CrossRef]
- Kwon, J.; Cho, H.; Jung, J.; Lee, H.; Hong, S.; Yeo, J.; Han, S.; Ko, S.H. ZnO/CuO/M (M = Ag, Au) Hierarchical Nanostructure by Successive Photoreduction Process for Solar Hydrogen Generation. Nanomaterials 2018, 8, 323. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; He, J.; Xiao, Y.; Li, Y.; Delaunay, J.-J. Earth-abundant Cu-based metal oxide photocathodes for photoelectrochemical water splitting. Energy Environ. Sci. 2020, 13, 3269–3306. [Google Scholar] [CrossRef]
- Baran, T.; Visibile, A.; Busch, M.; He, X.; Wojtyla, S.; Rondinini, S.; Minguzzi, A.; Vertova, A. Copper Oxide-Based Photocatalysts and Photocathodes: Fundamentals and Recent Advances. Molecules 2021, 26, 7271. [Google Scholar] [CrossRef] [PubMed]
- Bae, D.; Seger, B.; Vesborg, P.C.; Hansen, O.; Chorkendorff, I. Strategies for stable water splitting via protected photoelectrodes. Chem. Soc. Rev. 2017, 46, 1933–1954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheeler, G.P.; Choi, K.-S. Photoelectrochemical Properties and Stability of Nanoporous p-Type LaFeO3 Photoelectrodes Prepared by Electrodeposition. ACS Energy Lett. 2017, 2, 2378–2382. [Google Scholar] [CrossRef]
- Celorrio, V.; Bradley, K.; Weber, O.J.; Hall, S.R.; Fermín, D.J. Photoelectrochemical Properties of LaFeO3 Nanoparticles. ChemElectroChem 2014, 1, 1667–1671. [Google Scholar] [CrossRef]
- Freeman, E.; Kumar, S.; Celorrio, V.; Park, M.S.; Kim, J.H.; Fermin, D.J.; Eslava, S. Strategies for the deposition of LaFeO3 photocathodes: Improving the photocurrent with a polymer template. Sustain. Energy Fuels 2020, 4, 884–894. [Google Scholar] [CrossRef] [Green Version]
- Pawar, G.S.; Tahir, A.A. Unbiased Spontaneous Solar Fuel Production using Stable LaFeO3 Photoelectrode. Sci. Rep. 2018, 8, 3501. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Tan, B.; Ma, W.; Liu, B.; Sun, M.; Cooper, J.K.; Han, W. BiFeO3 photocathodes for efficient H2O2 production via charge carrier dynamics engineering. Mater. Horiz. 2022, 9, 1999–2006. [Google Scholar] [CrossRef]
- Ida, S.; Yamada, K.; Matsunaga, T.; Hagiwara, H.; Matsumoto, Y.; Ishihara, T. Preparation of p-type CaFe2O4 photocathodes for producing hydrogen from water. J. Am. Chem. Soc. 2010, 132, 17343–17345. [Google Scholar] [CrossRef]
- Ismael, M.; Wark, M. Perovskite-type LaFeO3: Photoelectrochemical Properties and Photocatalytic Degradation of Organic Pollutants Under Visible Light Irradiation. Catalysts 2019, 9, 342. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wang, T.; Gao, B.; Fan, X.; Gong, H.; Xue, H.; Zhang, S.; Huang, X.; He, J. Efficient photocathode performance of lithium ion doped LaFeO3 nanorod arrays in hydrogen evolution. New J. Chem. 2021, 45, 3463–3468. [Google Scholar] [CrossRef]
- Sun, X.; Tiwari, D.; Fermin, D.J. Promoting Active Electronic States in LaFeO3 Thin-Films Photocathodes via Alkaline-Earth Metal Substitution. ACS Appl. Mater. Interfaces 2020, 12, 31486–31495. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; He, Y.; Mi, Y.; Zhu, J.; Zhang, F.; Liu, Y.; Yang, Y.; Chen, M.; Cao, D. Enhanced photoelectrochemical performance of LaFeO3 photocathode with Au buffer layer. RSC Adv. 2019, 9, 26780–26786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheeler, G.P.; Baltazar, V.U.; Smart, T.J.; Radmilovic, A.; Ping, Y.; Choi, K.-S. Combined Theoretical and Experimental Investigations of Atomic Doping To Enhance Photon Absorption and Carrier Transport of LaFeO3 Photocathodes. Chem. Mater. 2019, 31, 5890–5899. [Google Scholar] [CrossRef]
- Diez-Garcia, M.I.; Gomez, R. Metal Doping to Enhance the Photoelectrochemical Behavior of LaFeO3 Photocathodes. ChemSusChem 2017, 10, 2457–2463. [Google Scholar] [CrossRef] [Green Version]
- Bedin, K.C.; Muche, D.N.F.; Melo, M.A.; Freitas, A.L.M.; Gonçalves, R.V.; Souza, F.L. Role of Cocatalysts on Hematite Photoanodes in Photoelectrocatalytic Water Splitting: Challenges and Future Perspectives. ChemCatChem 2020, 12, 3156–3169. [Google Scholar] [CrossRef]
- Zhang, P.; Wang, T.; Gong, J. Current Mechanistic Understanding of Surface Reactions over Water-Splitting Photocatalysts. Chem 2018, 4, 223–245. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Cui, J.; Eslava, S. Oxygen Evolution Catalysts at Transition Metal Oxide Photoanodes: Their Differing Roles for Solar Water Splitting. Adv. Energy Mater. 2021, 11, 2003111. [Google Scholar] [CrossRef]
- Ma, Y.; Le Formal, F.; Kafizas, A.; Pendlebury, S.R.; Durrant, J.R. Efficient suppression of back electron/hole recombination in cobalt phosphate surface-modified undoped bismuth vanadate photoanodes. J. Mater. Chem. A 2015, 3, 20649–20657. [Google Scholar] [CrossRef] [Green Version]
- Barroso, M.; Cowan, A.J.; Pendlebury, S.R.; Gratzel, M.; Klug, D.R.; Durrant, J.R. The role of cobalt phosphate in enhancing the photocatalytic activity of alpha-Fe2O3 toward water oxidation. J. Am. Chem. Soc. 2011, 133, 14868–14871. [Google Scholar] [CrossRef]
- Thorne, J.E.; Jang, J.W.; Liu, E.Y.; Wang, D. Understanding the origin of photoelectrode performance enhancement by probing surface kinetics. Chem. Sci. 2016, 7, 3347–3354. [Google Scholar] [CrossRef]
- Lee, J.; Seo, D.; Won, S.; Chung, T.D. Understanding the role of nickel–iron (oxy)hydroxide (NiFeOOH) electrocatalysts on hematite photoanodes. Sust. Energy Fuels 2021, 5, 501–508. [Google Scholar] [CrossRef]
- Lo Vecchio, C.; Trocino, S.; Giacoppo, G.; Barbera, O.; Baglio, V.; Díez-García, M.I.; Contreras, M.; Gómez, R.; Aricò, A.S. Water Splitting with Enhanced Efficiency Using a Nickel-Based Co-Catalyst at a Cupric Oxide Photocathode. Catalysts 2021, 11, 1363. [Google Scholar] [CrossRef]
- Morales-Guio, C.G.; Tilley, S.D.; Vrubel, H.; Gratzel, M.; Hu, X. Hydrogen evolution from a copper(I) oxide photocathode coated with an amorphous molybdenum sulphide catalyst. Nat. Commun. 2014, 5, 3059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, Y.J.; Park, Y.B.; Kim, H.E.; Choi, Y.H.; Choi, S.H.; Lee, J.S. Oxygen-Intercalated CuFeO2 Photocathode Fabricated by Hybrid Microwave Annealing for Efficient Solar Hydrogen Production. Chem. Mater. 2016, 28, 6054–6061. [Google Scholar] [CrossRef]
- Wu, G.W.; He, S.B.; Peng, H.P.; Deng, H.H.; Liu, A.L.; Lin, X.H.; Xia, X.H.; Chen, W. Citrate-capped platinum nanoparticle as a smart probe for ultrasensitive mercury sensing. Anal. Chem. 2014, 86, 10955–10960. [Google Scholar] [CrossRef]
- McMurdie, H.F.; Morris, M.C.; Evans, E.H.; Paretzkin, B.; Wong-Ng, W.; Hubbard, C.R. Standard X-Ray Diffraction Powder Patterns from The JCPDS Research Associateship. Powder Diffr. 2013, 1, 265–275. [Google Scholar] [CrossRef]
- Zhu, J.; Li, H.; Zhong, L.; Xiao, P.; Xu, X.; Yang, X.; Zhao, Z.; Li, J. Perovskite Oxides: Preparation, Characterizations, and Applications in Heterogeneous Catalysis. ACS Catal. 2014, 4, 2917–2940. [Google Scholar] [CrossRef]
- Joy, J.; Mathew, J.; George, S.C. Nanomaterials for photoelectrochemical water splitting—Review. Int. J. Hydrogen Energy 2018, 43, 4804–4817. [Google Scholar] [CrossRef]
- Gunasekaran, N.; Rajadurai, S.; Carberry, J.J.; Bakshi, N.; Alcock, C.B. Surface characterization and catalytic properties of La1−xAxMO3 perovskite type oxides. Part I. Studies on La0.95Ba0.05MO3 (M = Mn, Fe or Co) oxides. Solid State Ion. 1994, 73, 289–295. [Google Scholar] [CrossRef]
- Yamazoe, N.; Teraoka, Y.; Seiyama, T. Tpd and xps study on thermal behavior of absorbed oxygen in La1−xSrxCoO3. Chem. Lett. 1981, 10, 1767–1770. [Google Scholar] [CrossRef]
- Wu, Y.; Cordier, C.; Berrier, E.; Nuns, N.; Dujardin, C.; Granger, P. Surface reconstructions of LaCo1−xFexO3 at high temperature during N2O decomposition in realistic exhaust gas composition: Impact on the catalytic properties. Appl. Catal. B 2013, 140–141, 151–163. [Google Scholar] [CrossRef]
- Korjenic, A.; Raja, K.S. Electrochemical Stability of Fluorine Doped Tin Oxide (FTO) Coating at Different pH Conditions. J. Electrochem. Soc. 2019, 166, C169–C184. [Google Scholar] [CrossRef]
- Ryabova, A.S.; Napolskiy, F.S.; Poux, T.; Istomin, S.Y.; Bonnefont, A.; Antipin, D.M.; Baranchikov, A.Y.; Levin, E.E.; Abakumov, A.M.; Kéranguéven, G.; et al. Rationalizing the Influence of the Mn(IV)/Mn(III) Red-Ox Transition on the Electrocatalytic Activity of Manganese Oxides in the Oxygen Reduction Reaction. Electrochim. Acta 2016, 187, 161–172. [Google Scholar] [CrossRef]
- Peter, L.M. Kinetics and Mechanisms of Light-Driven Reactions at Semiconductor Electrodes: Principles and Techniques. In Photoelectrochemical Water Splitting: Materials, Processes and Architectures; Lewerenz, H.-J., Peter, L.M., Eds.; The Royal Society of Chemistry: Cambridge, UK, 2013. [Google Scholar]
- Sanjinés, R.; Tang, H.; Berger, H.; Gozzo, F.; Margaritondo, G.; Lévy, F. Electronic structure of anatase TiO2 oxide. J. Appl. Phys. 1994, 75, 2945–2951. [Google Scholar] [CrossRef]
- Ong, J.L.; Lucas, L.C.; Raikar, G.N.; Gregory, J.C. Electrochemical corrosion analyses and characterization of surface-modified titanium. Appl. Surf. Sci. 1993, 72, 7–13. [Google Scholar] [CrossRef]
- Salvador, P. Kinetic approach to the photocurrent transients in water photoelectrolysis at n-titanium dioxide electrodes. 1. Analysis of the ratio of the instantaneous to steady-state photocurrent. J. Phys. Chem. 1985, 89, 3863–3869. [Google Scholar] [CrossRef]
- Eisenberg, D.; Ahn, H.S.; Bard, A.J. Enhanced photoelectrochemical water oxidation on bismuth vanadate by electrodeposition of amorphous titanium dioxide. J. Am. Chem. Soc. 2014, 136, 14011–14014. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.D.; Sundaram, V.S.; Kleiman, G.G.; Castro, S.G.C.; Douglas, R.A.; Peterlevitz, A.C. High resolution study of the M45N67N67 and M45N45N67 Auger transitions in the 5d series. J. Phys. F Met. Phys. 1982, 12, 2097–2102. [Google Scholar] [CrossRef]
- Hammond, J.S.; Winograd, N. XPS spectroscopic study of potentiostatic and galvanostatic oxidation of Pt electrodes in H2SO4 and HClO4. J. Electroanal. Chem. Interfacial Electrochem. 1977, 78, 55–69. [Google Scholar] [CrossRef]
- Isaifan, R.J.; Ntais, S.; Baranova, E.A. Particle size effect on catalytic activity of carbon-supported Pt nanoparticles for complete ethylene oxidation. Appl. Catal. A 2013, 464–465, 87–94. [Google Scholar] [CrossRef]
- Drawdy, J.E.; Hoflund, G.B.; Gardner, S.D.; Yngvadottir, E.; Schryer, D.R. Effect of pretreatment on a platinized tin oxide catalyst used for low-temperature Co oxidation. Surf. Interface Anal. 1990, 16, 369–374. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.S.; Winograd, N.; Davis, R.E. Electron spectroscopy of platinum-oxygen surfaces and application to electrochemical studies. J. Am. Chem. Soc. 2002, 93, 6296–6297. [Google Scholar] [CrossRef]
- Tijare, S.N.; Joshi, M.V.; Padole, P.S.; Mangrulkar, P.A.; Rayalu, S.S.; Labhsetwar, N.K. Photocatalytic hydrogen generation through water splitting on nano-crystalline LaFeO3 perovskite. Int. J. Hydrogen Energy 2012, 37, 10451–10456. [Google Scholar] [CrossRef]
- Bockris, J.O.M.; Reddy, A.K.N.; Gamboa-Aldeco, M. Modern Electrochemistry 2A: Fundamentals of Electrodics, 2nd ed.; Springer-Verlag: Berlin/Heidelberg, Germany, 2000. [Google Scholar]
- Peter, L.M.; Walker, A.B.; Bein, T.; Hufnagel, A.G.; Kondofersky, I. Interpretation of photocurrent transients at semiconductor electrodes: Effects of band-edge unpinning. J. Electroanal. Chem. 2020, 872, 114234. [Google Scholar] [CrossRef]
- Sun, X.; Tiwari, D.; Fermin, D.J. Nanostructured LaFeO3 Photocathodes with Onset Potentials for the Hydrogen Evolution Reaction Over 1.4 V vs. RHE. J. Electrochem. Soc. 2019, 166, H764–H768. [Google Scholar]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chertkova, V.P.; Iskortseva, A.N.; Pazhetnov, E.M.; Arkharova, N.A.; Ryazantsev, S.V.; Levin, E.E.; Nikitina, V.A. Evaluation of the Efficiency of Photoelectrochemical Activity Enhancement for the Nanostructured LaFeO3 Photocathode by Surface Passivation and Co-Catalyst Deposition. Nanomaterials 2022, 12, 4327. https://doi.org/10.3390/nano12234327
Chertkova VP, Iskortseva AN, Pazhetnov EM, Arkharova NA, Ryazantsev SV, Levin EE, Nikitina VA. Evaluation of the Efficiency of Photoelectrochemical Activity Enhancement for the Nanostructured LaFeO3 Photocathode by Surface Passivation and Co-Catalyst Deposition. Nanomaterials. 2022; 12(23):4327. https://doi.org/10.3390/nano12234327
Chicago/Turabian StyleChertkova, Victoria P., Aleksandra N. Iskortseva, Egor M. Pazhetnov, Natalia A. Arkharova, Sergey V. Ryazantsev, Eduard E. Levin, and Victoria A. Nikitina. 2022. "Evaluation of the Efficiency of Photoelectrochemical Activity Enhancement for the Nanostructured LaFeO3 Photocathode by Surface Passivation and Co-Catalyst Deposition" Nanomaterials 12, no. 23: 4327. https://doi.org/10.3390/nano12234327