
Highly Sensitive Measurement of Horseradish Peroxidase Using Surface-Enhanced Raman Scattering of 2,3-Diaminophenazine
 , , and
, , and
Abstract
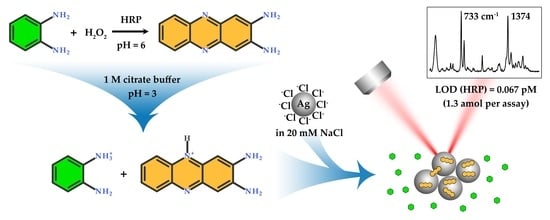
:
1. Introduction
2. Results
2.1. Synthesis, Characterization, and Standardization of Silver Nanoparticles
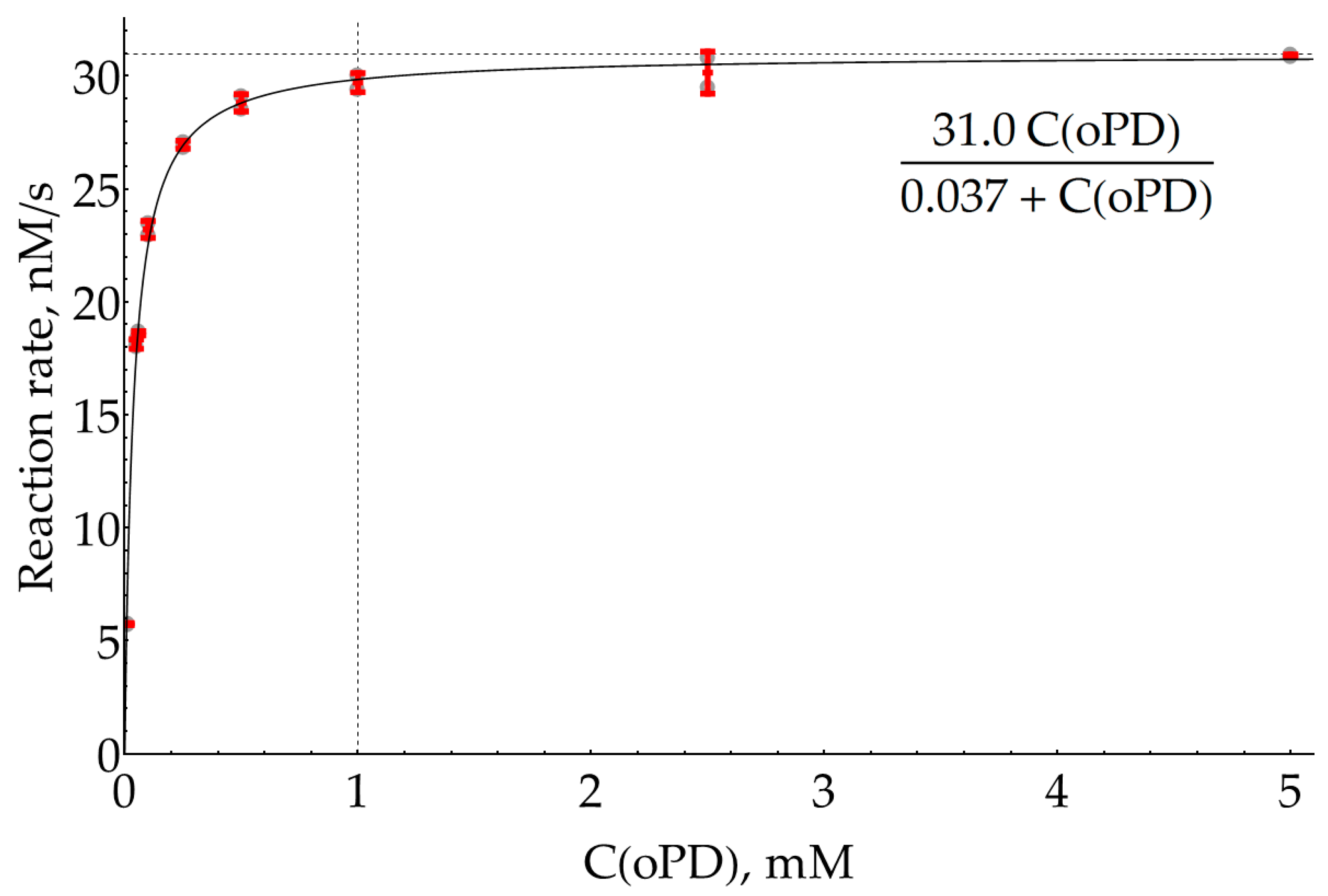
2.2. Estimation of HRP Effective KM for oPD
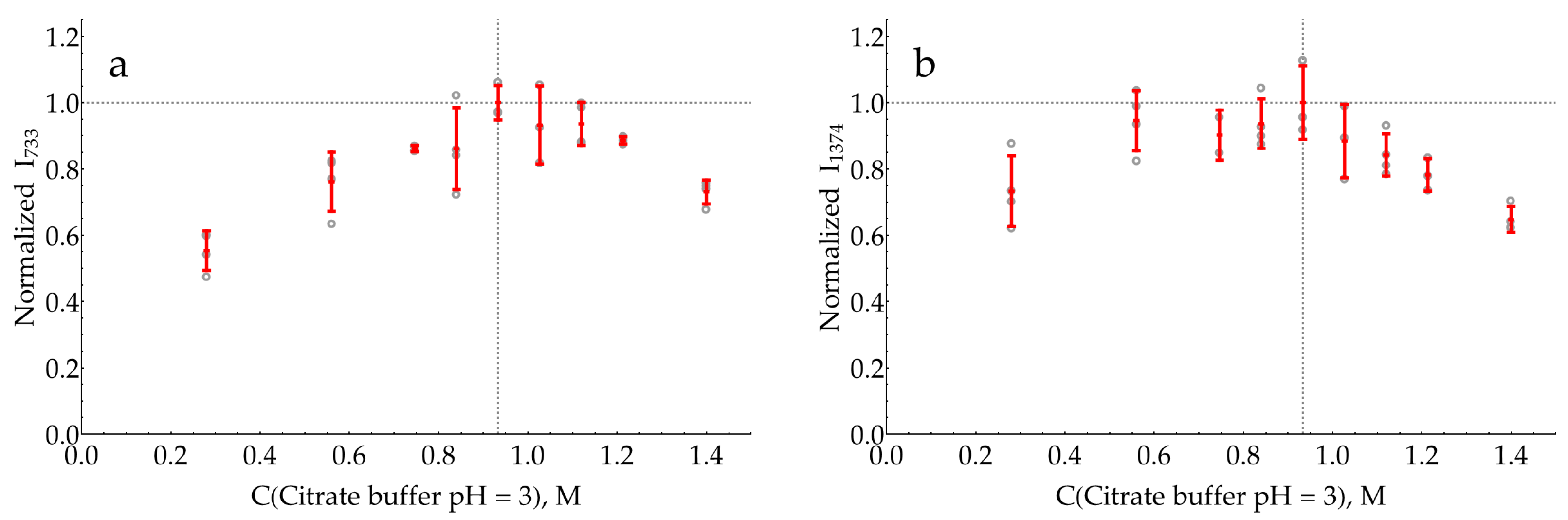
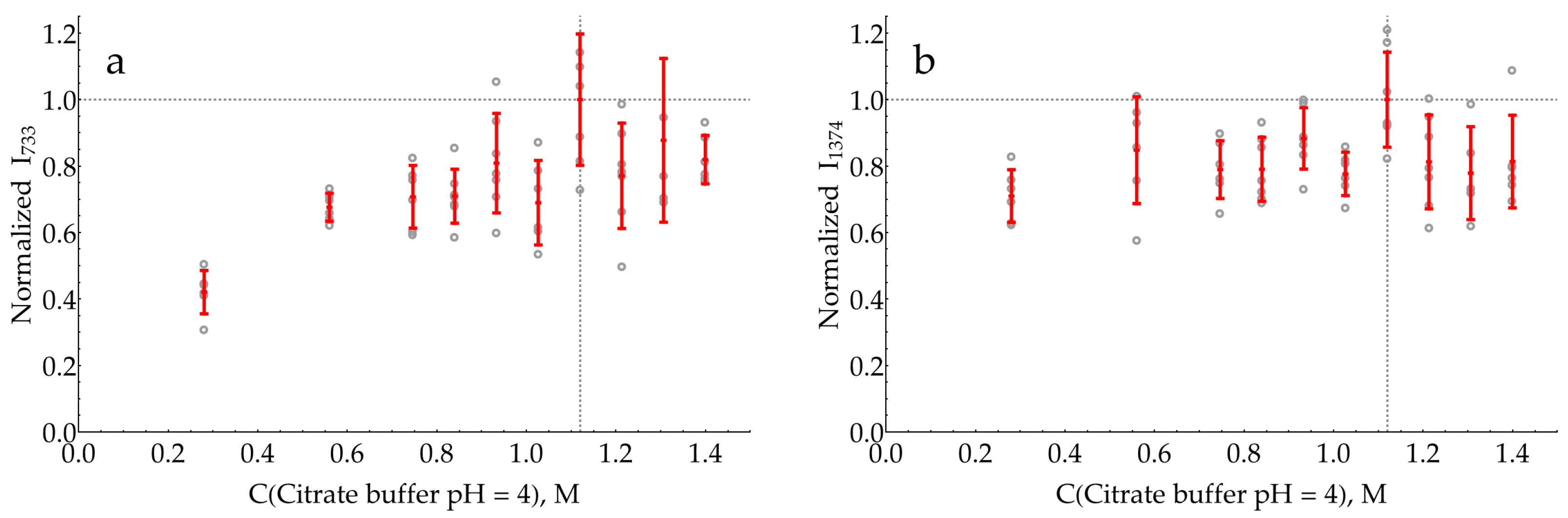
2.3. Selection of pH for SERS Detection of DAP
- The enzymatic reaction is stopped by an acid or base, and this mixture is further added to the AgNP colloid for SERS measurements. The stop reagent should create the optimal pH for DAP binding to the silver surface and the optimal ionic strength for the proper aggregation of AgNPs.
- If the sensitive detection of DAP in its mixtures with oPD is possible at the pH optimum of HRP, the enzymatic reaction mixture could be directly added to the AgNP colloid, followed by SERS detection. The advantage of this approach is the absence of dilution by the stop reagent.
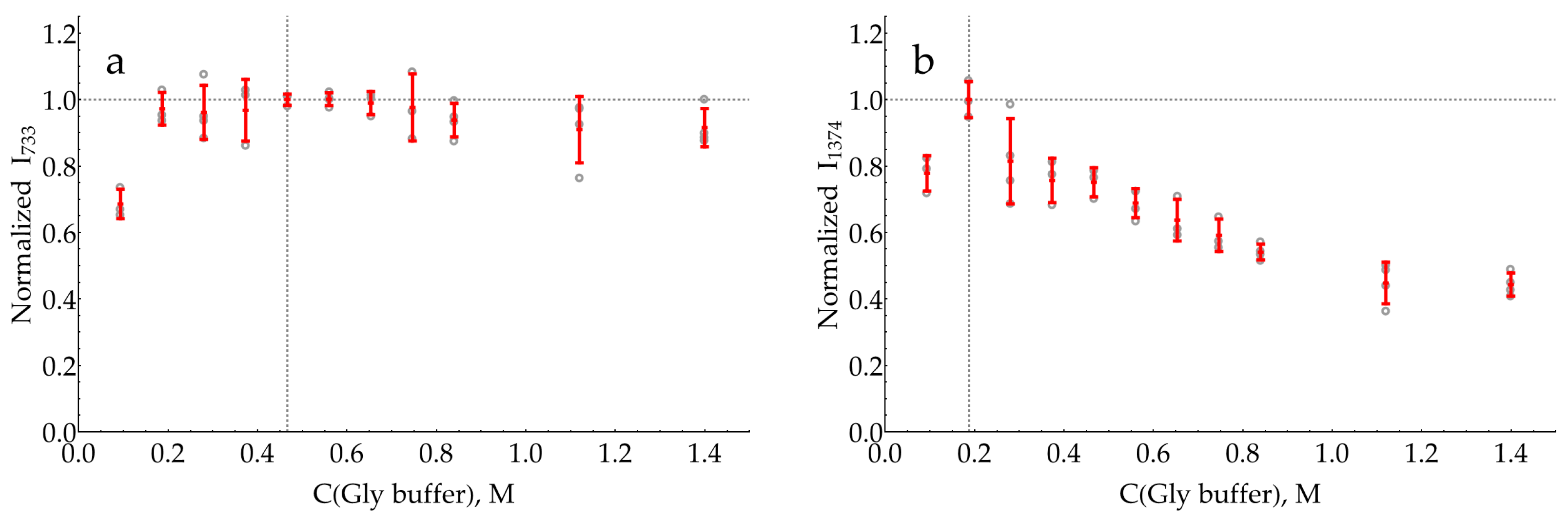
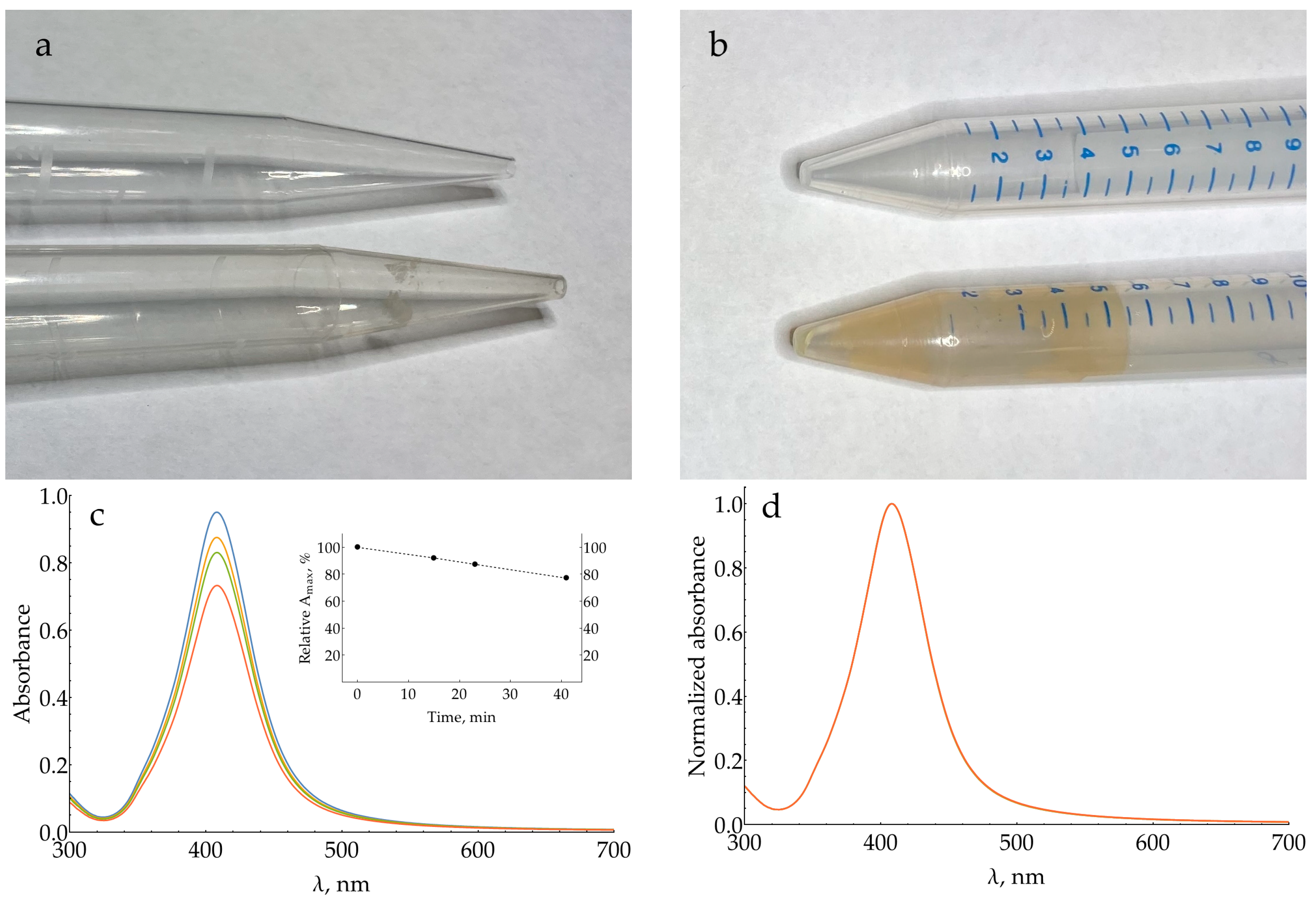
2.4. Influence of AgNP Chloride Stabilizer
2.5. Comparison of Colorimetric and SERS Detection Methods for DAP and HRP
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Synthesis and Standardization of AgNP Colloids
4.3. Determination of Mean Hydrodynamic Diameter and Total Particle Concentration of AgNPs
4.4. Transmission Electron Microscopy of AgNPs
4.5. Atomic Force Microscopy of AgNPs
4.6. Colorimetric Estimation of HRP Effective KM for oPD
4.7. Preparation of Standard DAP Solutions
4.8. Procedure for SERS Measurements
4.9. Colorimetric and SERS Measurements of DAP at pH 3
4.10. Colorimetric and SERS Measurements of HRP
4.11. Processing of Concentration Curves
4.12. Processing of Raman and SERS Spectra
4.13. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AgNPs | silver nanoparticles |
| CV | coefficient of variation |
| DAP | 2,3-diaminophenazine |
| DAPH+ | protonated 2,3-diaminophenazine |
| DAPH22+ | doubly protonated 2,3-diaminophenazine |
| ELISA | enzyme-linked immunosorbent assay |
| HRP | horseradish peroxidase |
| LOD | limit of detection |
| NTA | nanoparticle tracking analysis |
| oPD | o-phenylenediamine |
| oPDH+ | protonated o-phenylenediamine |
| oPDH22+ | doubly protonated o-phenylenediamine |
| SERS | surface-enhanced Raman scattering |
Appendix A




Appendix B



Appendix C
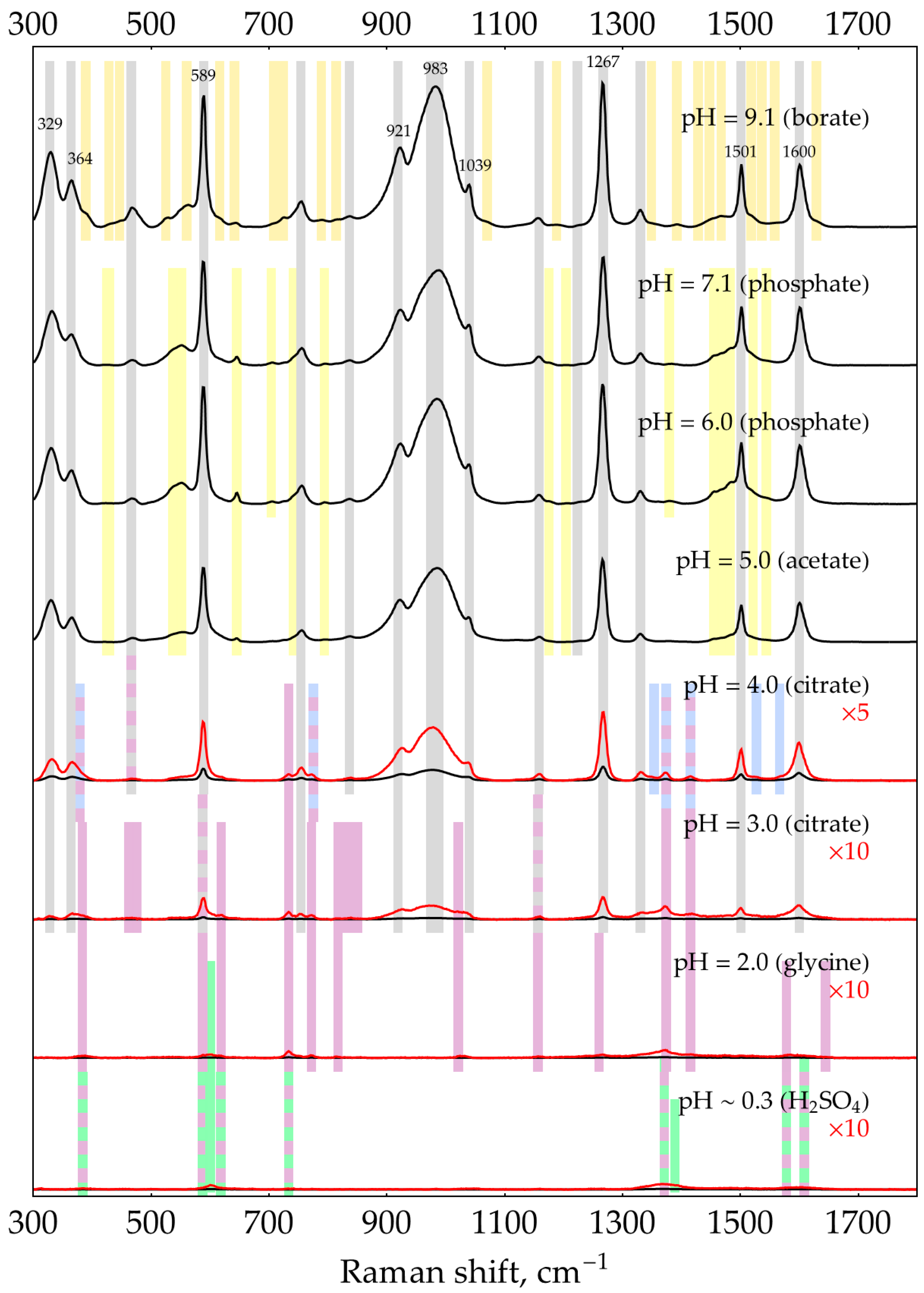
- At pHs from 2 to 3, the dominant DAP form is DAPH+, with ring nitrogen being protonated as evidenced by XRD [52,53] instead of the NH2 group as proposed by Brown and coauthors [41]. The SERS spectrum of DAPH+ corresponds well with the normal Raman spectrum of crystalline DAP×HCl [31]. Notably, the bands at 733 and 773 cm−1 are strongly enhanced in the SERS spectrum compared to a normal Raman spectrum. DAPH+ is shaded purple in Figure A8. The table of shifts is provided in [31].
- At pHs from 0.3 to 2, the second protonation occurs in agreement with DAPH22+ pKa of around 1 [41]. According to the law of acid–base equilibrium, a relatively pure (>99%) acidic form appears at pH = pKa–2 ≈ −1. Thus, the pure spectrum of DAPH22+ may be acquired in a concentrated acid only, which is corrosive to AgNPs. As a result, most of the bands present in the spectrum at pH ~0.3 may correspond to either DAPH+ or DAPH22+. However, two unique bands at 603 and 1389 cm−1 as well as the shoulder at around 439 cm−1 could be univocally assigned to DAPH22+. They are shaded green in Figure A8.

Appendix D

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| pH | Buffer Type and Concentration |
|---|---|
| 3 | citrate 30 mM |
| 4 | acetate 30 mM |
| 5 | acetate 20 mM |
| 6 | MES 30 mM |
| 7 | HEPES 10 mM |
| 9 | borate 50 mM |


Appendix E
Appendix E.1. Normal Raman Spectra of oPD

- Raman spectra from solids may contain lattice modes;
- Some shifts in band positions and intensities may occur due to interactions between molecules in the crystal;
- For anisotropic crystals, slight shifts in the band positions and possibly large changes in the relative intensities may occur due to the preferred crystal orientation [58].

| System | pH | α (oPDH22+), % | α (oPDH+), % | α (oPD), % |
|---|---|---|---|---|
| oPD in 1 M HCl | 0 | 98 | 2 | 0 |
| oPD∙2HCl in water | 1.8 | 44 | 56 | 0 |
| oPD∙HCl in water | 3.3 | 2.5 | 93 | 4.5 |
| oPD in water | ≈6 | 0 | 3 | 97 |

- The Raman spectra of solid oPD (spectrum A in Figure A11, [37,55,56]) and the water solution of oPD (spectrum A in Figure A13) could be used as references for the neutral form of oPD.
- The Raman spectrum of the oPD∙HCl water solution (spectrum B in Figure A13) could be used as a reference for the oPDH+ form.
- The Raman spectra of solid oPD∙2HCl (spectra B and C in Figure A11) and the solution of oPD in 1 M HCl (spectrum D in Figure A13) could be used as references for the doubly protonated oPDH22+ form.
Appendix E.2. SERS Spectra of oPD with AgNPs




| Raman 785 nm, Solid oPD | Raman 785 nm, 27.7 mM oPD in Water | SERS 785 nm of oPD with AgNPs, Present Work | SERS 647 nm of oPD with a Gold Colloid at pH = 5 [37] | SERS 647 nm of oPD with a Silver Colloid at pH = 5 [37] * |
|---|---|---|---|---|
| 329 (13) | 331b (16) | 329 (61) | 350 | 330 |
| 343 (13) | ||||
| 369 (2) | Shoulder of 331 | 364 (34) | 382 | 362 |
| 398 (4) | ||||
| 453 (2) | 449 (2) | |||
| 472 (3) | 467 (4) | 480 | 476 | |
| 492 (1) | ||||
| 543 (16) | 526 (14) | Shoulder of 589 | ||
| 551 (31) | ||||
| 579 (21) | 586 (28) | 589 (90) | 586 | 588 |
| 718b (10) | ||||
| 761 (78) | 758 (100) | 753 (12) | 752 | 756 |
| 783b (33) | Shoulder of 758 | |||
| 835 (5) | 836 (5) | |||
| 861 (3) | ||||
| 928 (2) | 908b (2) | 921 (55) | 924 | 922 |
| 959 (1) | ||||
| 983b (98) | 1020b | 988b | ||
| 1034 (100) | 1039 (91) | 1039 (33) | 1040 | 1040 |
| 1115 (4) | ||||
| 1152 (17) | 1159 (20) | 1157 (4) | 1160 | 1156 |
| 1161 (24) | ||||
| 1246 (2) | 1236 (2) | 1225 (2) | ||
| 1276 (44) | 1279 (38) | 1267 (100) | 1262 | 1268 |
| 1324 (3) | 1332 (8) | 1330 (11) | 1324 | |
| 1337 (12) | 1340 | 1348 | ||
| 1460 (5) | 1461b (3) | |||
| 1504 (5) | 1506 (6) | 1501 (38) | 1500 | 1498 |
| 1594 (37) | 1600 (30) | 1600 (40) | 1596 | 1598 |
| 1618 (10) | Shoulder of 1600 | 1628 | ||
| 1652 (1) |


- “Pure” SERS spectra of oPD with AgNPs may be obtained only in reducing conditions if the oxidation of metal silver to silver (I) with atmospheric oxygen is suppressed. Under these conditions, the SERS spectrum of oPD corresponds well to a normal Raman spectrum of neutral oPD and the SERS spectrum of oPD with gold (Table A3).
- If oxidized silver is allowed to form, multiple additional bands appear in the pH range of 5 to 9. They correspond to some oxidation product of oPD other than DAP. This is most likely some kind of intermediate(s) on a route from oPD to DAP.
- Only the neutral form of oPD has some affinity for silver. Binding occurs via at least one NH2 group (resulting in an intense broad band at around 983 cm−1). It is likely that both NH2 groups are involved in oPD binding to the silver surface.
- In acidic conditions and at low concentrations of oPD, its spectrum contains (or is even replaced with) the bands of DAP in neutral, DAPH+, or DAPH22+ forms, depending on the pH. This DAP is not a result of oPD oxidation by silver, but instead, is a minor impurity in the stock oPD and the product of spontaneous oPD oxidation upon storage in the solution.
Appendix F




Appendix G

Appendix H


| Band (Figure A26a,b) | Equation |
|---|---|
| I | |
| II | |
| III | |
| IV | |
| V | |
| VI | |
| VII |
References
- Mekler, V.M.; Bystryak, S.M. Application of O-Phenylenediamine as a Fluorogenic Substrate in Peroxidase-Mediated Enzyme-Linked Immunosorbent Assay. Anal. Chim. Acta 1992, 264, 359–363. [Google Scholar] [CrossRef]
- Acharya, A.P.; Nafisi, P.M.; Gardner, A.; MacKay, J.L.; Kundu, K.; Kumar, S.; Murthy, N. A Fluorescent Peroxidase Probe Increases the Sensitivity of Commercial ELISAs by Two Orders of Magnitude. Chem. Commun. 2013, 49, 10379–10381. [Google Scholar] [CrossRef]
- Zhang, Z.; Lai, J.; Wu, K.; Huang, X.; Guo, S.; Zhang, L.; Liu, J. Peroxidase-Catalyzed Chemiluminescence System and Its Application in Immunoassay. Talanta 2018, 180, 260–270. [Google Scholar] [CrossRef]
- Liang, J.; Liu, H.; Huang, C.; Yao, C.; Fu, Q.; Li, X.; Cao, D.; Luo, Z.; Tang, Y. Aggregated Silver Nanoparticles Based Surface-Enhanced Raman Scattering Enzyme-Linked Immunosorbent Assay for Ultrasensitive Detection of Protein Biomarkers and Small Molecules. Anal. Chem. 2015, 87, 5790–5796. [Google Scholar] [CrossRef]
- Pham, X.-H.; Hahm, E.; Kim, T.H.; Kim, H.-M.; Lee, S.H.; Lee, S.C.; Kang, H.; Lee, H.-Y.; Jeong, D.H.; Choi, H.S.; et al. Enzyme-Amplified SERS Immunoassay with Ag-Au Bimetallic SERS Hot Spots. Nano Res. 2020, 13, 3338–3346. [Google Scholar] [CrossRef]
- Lu, H.; Yang, Y.; Chen, R.; Huang, W.; Cheng, H.; Liu, X.; Kong, H.; Li, L.; Feng, J. Quantitative Evaluation of Human Carboxylesterase 1 by SERS-ELISA Using a Synergistic Enhancement Strategy Based on Gold Nanoparticles and Metal–Organic Framework. Microchem. J. 2022, 183, 108114. [Google Scholar] [CrossRef]
- Campbell, F.M.; Ingram, A.; Monaghan, P.; Cooper, J.; Sattar, N.; Eckersall, P.D.; Graham, D. SERRS Immunoassay for Quantitative Human CRP Analysis. Analyst 2008, 133, 1355–1357. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Cheng, H.; Tram, K.; Zhang, S.; Zhao, Y.; Han, L.; Chen, Z.; Huan, S. A Paper-Based Surface-Enhanced Resonance Raman Spectroscopic (SERRS) Immunoassay Using Magnetic Separation and Enzyme-Catalyzed Reaction. Analyst 2013, 138, 2624–2631. [Google Scholar] [CrossRef] [PubMed]
- Bozkurt, A.G.; Buyukgoz, G.G.; Soforoglu, M.; Tamer, U.; Suludere, Z.; Boyaci, I.H. Alkaline Phosphatase Labeled SERS Active Sandwich Immunoassay for Detection of Escherichia coli. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2018, 194, 8–13. [Google Scholar] [CrossRef]
- Li, N.; Chen, H.; Zhang, M.; Zha, Y.; Mu, Z.; Ma, Y.; Chen, P. A Universal Ultrasensitive Platform for Enzyme-Linked Immunoassay Based on Responsive Surface-Enhanced Raman Scattering. Sens. Actuators B Chem. 2020, 315, 128135. [Google Scholar] [CrossRef]
- Guo, W.; Hu, Y.; Wei, H. Enzymatically Activated Reduction-Caged SERS Reporters for Versatile Bioassays. Analyst 2017, 142, 2322–2326. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Liao, J.; Wang, D.; Li, G. Fabrication of Gold Nanoparticle-Embedded Metal–Organic Framework for Highly Sensitive Surface-Enhanced Raman Scattering Detection. Anal. Chem. 2014, 86, 3955–3963. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Jin, S.; Shi, W.; Oh, J.; Cao, H.; Jung, Y.M. Catalyzed Deposition of Signal Reporter for Highly Sensitive Surface-Enhanced Raman Spectroscopy Immunoassay Based on Tyramine Signal Amplification Strategy. Anal. Chem. 2018, 90, 13159–13162. [Google Scholar] [CrossRef] [PubMed]
- Dou, X.; Takama, T.; Yamaguchi, Y.; Yamamoto, H.; Ozaki, Y. Enzyme Immunoassay Utilizing Surface-Enhanced Raman Scattering of the Enzyme Reaction Product. Anal. Chem. 1997, 69, 1492–1495. [Google Scholar] [CrossRef]
- Fu, C.; Zhang, L.; Bao, M.; Zhang, Y.; Li, Y.; Wu, Y.; Jung, Y.M. Signal Amplification Surface-Enhanced Raman Scattering Immunosorbent Assay of Human Chorionic Gonadotrophin Based on Repeated Enzyme Biocatalytic Precipitation. Analyst 2022, 147, 5718–5724. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, R.; Ingram, A.; Leung, H.; McMillan, D.C.; Graham, D. Quantitative SERRS Immunoassay for the Detection of Human PSA. Analyst 2009, 134, 842–844. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Chen, L.; Wang, Y.; Wang, X.; Song, W.; Ruan, W.; Zhao, B.; Cong, Q. A SERS-Active Enzymatic Product Used for the Quantification of Disease-Related Molecules. J. Raman Spectrosc. 2014, 45, 75–81. [Google Scholar] [CrossRef]
- Zhan, L.; Zhen, S.J.; Wan, X.Y.; Gao, P.F.; Huang, C.Z. A Sensitive Surface-Enhanced Raman Scattering Enzyme-Catalyzed Immunoassay of Respiratory Syncytial Virus. Talanta 2016, 148, 308–312. [Google Scholar] [CrossRef]
- Kudryashova, A.M.; Galstian, A.G.; Faizuloev, E.B.; Olenin, A.Y.; Lisichkin, G.V.; Zverev, V.V.; Borisova, O.V. Detection of adenovirus antigen by a surface-enhanced Raman scattering enzyme-linked immunosorbent assay. J. Microbiol. Epidemiol. Immunobiol. 2018, 95, 25–31. [Google Scholar] [CrossRef]
- Feng, J.; Lu, H.; Yang, Y.; Huang, W.; Cheng, H.; Kong, H.; Li, L. SERS-ELISA Determination of Human Carboxylesterase 1 Using Metal-Organic Framework Doped with Gold Nanoparticles as SERS Substrate. Microchim. Acta 2021, 188, 280. [Google Scholar] [CrossRef]
- Larmour, I.A.; Faulds, K.; Graham, D. The Past, Present and Future of Enzyme Measurements Using Surface Enhanced Raman Spectroscopy. Chem. Sci. 2010, 1, 151–160. [Google Scholar] [CrossRef]
- Kurochkin, I.N.; Vasilyeva, A.D.; Evtushenko, E.G.; Eremenko, A.V.; Pergushov, D.V.; Sigolaeva, L.V. Enzymes in the Development of Physico-Chemical Methods for Biomedical Research. Moscow Univ. Chem. Bull. 2023, 78, 201–219. [Google Scholar] [CrossRef]
- Wu, Z.-S.; Zhou, G.-Z.; Jiang, J.-H.; Shen, G.-L.; Yu, R.-Q. Gold Colloid-Bienzyme Conjugates for Glucose Detection Utilizing Surface-Enhanced Raman Scattering. Talanta 2006, 70, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Hawi, S.R.; Rochanakij, S.; Adar, F.; Campbell, W.B.; Nithipatikom, K. Detection of Membrane-Bound Enzymes in Cells Using Immunoassay and Raman Microspectroscopy. Anal. Biochem. 1998, 259, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Nithipatikom, K.; McCoy, M.J.; Kajdacsy-Balla, A.; Kaul, S.; Lindholm, P.F.; Hawi, S.R.; Rochanakij, S.; Adar, F.; Campbell, W.B. Detection of Prostaglandin H Synthase (Cyclooxygenase) -1 and -2 Expression in Single Cancer Cells by Confocal Raman Microspectroscopy. In Biomedical Applications of Raman Spectroscopy; SPIE: Bellingham, WA, USA, 1999; Volume 3608, pp. 20–27. [Google Scholar] [CrossRef]
- Nithipatikom, K.; Borscheid, C.L.; Kajdacsy-Balla, A.; Kaul, S.; Lindholm, P.F.; Pytynia, K.B.; Campbell, W.B. Determination of Cyclooxygenase and Arachidonic Acid Metabolites in Invasive Human Prostate Cancer Cells. In Eicosanoids and Other Bioactive Lipids in Cancer, Inflammation, and Radiation Injury; Honn, K.V., Marnett, L.J., Nigam, S., Dennis, E., Serhan, C., Eds.; Springer New York: New York, NY, USA, 2002; pp. 269–274. ISBN 978-1-4615-0193-0. [Google Scholar] [CrossRef]
- Fu, C.; Wang, Y.; Tian, X.; Wu, Y.; Cao, H.; Li, Y.; Jung, Y.M. Horseradish Peroxidase-Repeat Assay Based on Tyramine Signal Amplification for Highly Sensitive H2O2 Detection by Surface-Enhanced Raman Scattering. Analyst 2021, 146, 7320–7326. [Google Scholar] [CrossRef]
- Song, Y.; Wang, D.; Li, Z.; Wang, L.; Fan, C.; He, X.; Xu, T.; Zhang, X. Jigsaw-like Mini-Pillar Platform for Multi-Mode Biosensing. Chin. Chem. Lett. 2022, 33, 3879–3882. [Google Scholar] [CrossRef]
- Tarcha, P.J.; Chu, V.P.; Whittern, D. 2,3-Diaminophenazine Is the Product from the Horseradish Peroxidase-Catalyzed Oxidation of o-Phenylenediamine. Anal. Biochem. 1987, 165, 230–233. [Google Scholar] [CrossRef]
- Hempen, C.; van Leeuwen, S.M.; Luftmann, H.; Karst, U. Liquid Chromatographic/Mass Spectrometric Investigation on the Reaction Products in the Peroxidase-Catalyzed Oxidation of o-Phenylenediamine by Hydrogen Peroxide. Anal. Bioanal. Chem. 2005, 382, 234–238. [Google Scholar] [CrossRef]
- Evtushenko, E.G.; Gavrilina, E.S.; Gusarova, D.Y.; Vasil’eva, A.D.; Yurina, L.V.; Kurochkin, I.N. Application of Hydroxylamine Sols of Silver Nanoparticles to Obtain Reference SERS Spectra. Bull. Lebedev Phys. Inst. 2023, 50, S547–S551. [Google Scholar] [CrossRef]
- Qi, G.; Fu, C.; Chen, G.; Xu, S.; Xu, W. Highly Sensitive SERS Sensor for Mercury Ions Based on the Catalytic Reaction of Mercury Ion Decorated Ag Nanoparticles. RSC Adv. 2015, 5, 49759–49764. [Google Scholar] [CrossRef]
- Bovaird, J.H.; Ngo, T.T.; Lenhoff, H.M. Optimizing the O-Phenylenediamine Assay for Horseradish Peroxidase: Effects of Phosphate and PH, Substrate and Enzyme Concentrations, and Stopping Reagents. Clin. Chem. 1982, 28, 2423–2426. [Google Scholar] [CrossRef]
- Gallati, H.; Brodbeck, H. Peroxidase Aus Meerrettich: Kinetische Studien Und Optimierung Der Aktivitätsbestimmung Mit Den Substraten H2O2 Und o-Phenylendiamin. J. Clin. Chem. Clin. Biochem. 1982, 20, 221–226. [Google Scholar] [CrossRef]
- Fornera, S.; Walde, P. Spectrophotometric Quantification of Horseradish Peroxidase with O-Phenylenediamine. Anal. Biochem. 2010, 407, 293–295. [Google Scholar] [CrossRef]
- Leopold, N.; Lendl, B. A New Method for Fast Preparation of Highly Surface-Enhanced Raman Scattering (SERS) Active Silver Colloids at Room Temperature by Reduction of Silver Nitrate with Hydroxylamine Hydrochloride. J. Phys. Chem. B 2003, 107, 5723–5727. [Google Scholar] [CrossRef]
- Koh, T.Y.; Greaves, S.J.; Griffith, W.P. Vibrational Spectra of 1,2-Diaminobenzene, 4,5-Dimethyl-1,2-Diaminobenzene and Catechol and Their SER Spectra. Spectrochim. Acta Part A Mol. Spectrosc. 1994, 50, 857–873. [Google Scholar] [CrossRef]
- Ouyang, L.; Li, D.; Zhu, L.; Yang, W.; Tang, H. A New Plasmonic Pickering Emulsion Based SERS Sensor for in Situ Reaction Monitoring and Kinetic Study. J. Mater. Chem. C 2016, 4, 736–744. [Google Scholar] [CrossRef]
- He, D.; Jones, A.M.; Garg, S.; Pham, A.N.; Waite, T.D. Silver Nanoparticle–Reactive Oxygen Species Interactions: Application of a Charging–Discharging Model. J. Phys. Chem. C 2011, 115, 5461–5468. [Google Scholar] [CrossRef]
- Sigg, L.; Lindauer, U. Silver Nanoparticle Dissolution in the Presence of Ligands and of Hydrogen Peroxide. Environ. Pollut. 2015, 206, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.C.; Corbett, J.F.; Loveless, N.P. Spectrophotometric Studies on the Protonation of Hydroxy and Aminophenazines in Aqueous Solution. Spectrochim. Acta Part A Mol. Spectrosc. 1979, 35, 421–423. [Google Scholar] [CrossRef]
- He, X.; Zhang, L.; Chua, R.; Wong, P.K.J.; Arramel, A.; Feng, Y.P.; Wang, S.J.; Chi, D.; Yang, M.; Huang, Y.L.; et al. Selective Self-Assembly of 2,3-Diaminophenazine Molecules on MoSe2 Mirror Twin Boundaries. Nat. Commun. 2019, 10, 2847. [Google Scholar] [CrossRef]
- Iseminger, P.W.; Gregory, M.; Weakley, T.J.R.; Caple, G.; Sykes, A.G. Characterization of 3-Aminophenazin-2-Ol Isolated from the Chemical Oxidation of o-Phenylenediamine. J. Org. Chem. 1997, 62, 2643–2645. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-E.; Chen, Y.-T. Migration Behavior and Separation of Benzenediamines, Aminophenols and Benzenediols by Capillary Zone Electrophoresis. J. Chromatogr. A 2000, 871, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, H.; Nakabayashi, S.; Ohno, K.; Maeda, M. New Fluorimetric Assay of Horseradish Peroxidase Using Sesamol as Substrate and Its Application to EIA. J. Pharm. Anal. 2012, 2, 156–159. [Google Scholar] [CrossRef] [PubMed]
- Marzocchi, E.; Grilli, S.; Della Ciana, L.; Prodi, L.; Mirasoli, M.; Roda, A. Chemiluminescent Detection Systems of Horseradish Peroxidase Employing Nucleophilic Acylation Catalysts. Anal. Biochem. 2008, 377, 189–194. [Google Scholar] [CrossRef]
- Iwata, R.; Ito, H.; Hayashi, T.; Sekine, Y.; Koyama, N.; Yamaki, M. Stable and General-Purpose Chemiluminescent Detection System for Horseradish Peroxidase Employing a Thiazole Compound Enhancer and Some Additives. Anal. Biochem. 1995, 231, 170–174. [Google Scholar] [CrossRef]
- Sakharov, I.Y.; Demiyanova, A.S.; Gribas, A.V.; Uskova, N.A.; Efremov, E.E.; Vdovenko, M.M. 3-(10′-Phenothiazinyl)Propionic Acid Is a Potent Primary Enhancer of Peroxidase-Induced Chemiluminescence and Its Application in Sensitive ELISA of Methylglyoxal-Modified Low Density Lipoprotein. Talanta 2013, 115, 414–417. [Google Scholar] [CrossRef]
- Orendorff, C.J.; Gearheart, L.; Jana, N.R.; Murphy, C.J. Aspect Ratio Dependence on Surface Enhanced Raman Scattering Using Silver and Gold Nanorod Substrates. Phys. Chem. Chem. Phys. 2006, 8, 165–170. [Google Scholar] [CrossRef]
- Zannotti, M.; Rossi, A.; Giovannetti, R. SERS Activity of Silver Nanosphere, Triangular Nanoplates, Hexagonal Nanoplates and Quasi-Spherical Nanoparticles: Effect of Shape and Morphology. Coatings 2020, 10, 288. [Google Scholar] [CrossRef]
- Doyle, R.P.; Kruger, P.E.; Mackie, P.R.; Nieuwenhuyzen, M. Phenazine-2,3-Diamine. Acta Crystallogr. Sect. C 2001, C57, 104–105. [Google Scholar] [CrossRef]
- Mei, L.; Tai, L.S.; Tao, F.H.; Jie, S.; Rong, L.Q. A Novel Synthesis of 2,3-Diaminophenazine. Res. Chem. Intermed. 2012, 38, 499–505. [Google Scholar] [CrossRef]
- Mahato, R.K.; Mahanty, A.K.; Kotakonda, M.; Prasad, S.; Bhattacharyya, S.; Biswas, B. A Hydrated 2,3-Diaminophenazinium Chloride as a Promising Building Block against SARS-CoV-2. Sci. Rep. 2021, 11, 23122. [Google Scholar] [CrossRef]
- Li, D.-J.; Li, X.-W.; Xie, Y.-X.; Cai, X.-Q.; Zou, G.-L. Identification of Intermediate and Product from Methemoglobin-Catalyzed Oxidation of o-Phenylenediamine in Two-Phase Aqueous—Organic System. Biochem. 2005, 70, 92–99. [Google Scholar] [CrossRef]
- Badawi, H.M.; Förner, W.; Ali, S.A. A Comparative Study of the Infrared and Raman Spectra of Aniline and O-, m-, p-Phenylenediamine Isomers. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2013, 112, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Kaya Kınaytürk, N.; Kalaycı, T.; Tunalı, B. Experimental and Computational Investigations on the Molecular Structure, Vibrational Spectra, Electronic Properties, and Molecular Electrostatic Potential Analysis of Phenylenediamine Isomers. Spectrosc. Lett. 2021, 54, 693–706. [Google Scholar] [CrossRef]
- Martin, E.L. O-Phenylenediamine. Org. Synth. 1939, 19, 70. [Google Scholar] [CrossRef]
- Zhong, X.; Loges, A.; Roddatis, V.; John, T. Measurement of Crystallographic Orientation of Quartz Crystal Using Raman Spectroscopy: Application to Entrapped Inclusions. Contrib. Mineral. Petrol. 2021, 176, 89. [Google Scholar] [CrossRef]
- Wu, D.; Fang, Y. Study of Adsorptive Behavior of a Series of N-Aminobenzoic Acids on Silver Nanoparticles by SERS. Sci. Access 2004, 2, 286–287. [Google Scholar] [CrossRef]
- Yan, B.; Fang, Y.; Zhao, X.; Liang, L. A Comparative Study on the Adsorption Behaviors of PABA in the Silver Nano-Particles. J. Mol. Struct. 2014, 1074, 660–665. [Google Scholar] [CrossRef]
- Akbali, B.; Yagmurcukardes, M.; Peeters, F.M.; Lin, H.-Y.; Lin, T.-Y.; Chen, W.-H.; Maher, S.; Chen, T.-Y.; Huang, C.-H. Determining the Molecular Orientation on the Metal Nanoparticle Surface through Surface-Enhanced Raman Spectroscopy and Density Functional Theory Simulations. J. Phys. Chem. C 2021, 125, 16289–16295. [Google Scholar] [CrossRef]
- Chong, N.S.; Donthula, K.; Davies, R.A.; Ilsley, W.H.; Ooi, B.G. Significance of Chemical Enhancement Effects in Surface-Enhanced Raman Scattering (SERS) Signals of Aniline and Aminobiphenyl Isomers. Vib. Spectrosc. 2015, 81, 22–31. [Google Scholar] [CrossRef]
- Zhao, L.-B.; Huang, R.; Bai, M.-X.; Wu, D.-Y.; Tian, Z.-Q. Effect of Aromatic Amine–Metal Interaction on Surface Vibrational Raman Spectroscopy of Adsorbed Molecules Investigated by Density Functional Theory. J. Phys. Chem. C 2011, 115, 4174–4183. [Google Scholar] [CrossRef]
- Tao, S.; Yu, L.-J.; Pang, R.; Huang, Y.-F.; Wu, D.-Y.; Tian, Z.-Q. Binding Interaction and Raman Spectra of P–π Conjugated Molecules Containing CH2/NH2 Groups Adsorbed on Silver Surfaces: A DFT Study of Wagging Modes. J. Phys. Chem. C 2013, 117, 18891–18903. [Google Scholar] [CrossRef]
- Noto, R.; Leone, M.; La Manna, G.; Brugè, F.; Fornili, S.L. Ab Initio Calculations and Vibrational Spectroscopy on the Phenylenediamine Isomers. J. Mol. Struct. Theochem. 1998, 422, 35–48. [Google Scholar] [CrossRef]
- Li, F.; Liu, J.; Hu, Y.; Deng, N.; He, J. An Ultrasensitive Label-Free Colorimetric Assay for Glutathione Based on Ag+ Regulated Autocatalytic Oxidation of o-Phenylenediamine. Talanta 2018, 186, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Lian, F.; Yao, S.; Ge, L.; Wang, Y.; Zhao, Y.; Zhao, J.; Song, X.; Zhao, C.; Xu, K. Detection of Formaldehyde (HCHO) in Solution Based on the Autocatalytic Oxidation Reaction of o-Phenylenediamine (OPD) Induced by Silver Ions (Ag+). J. Iran. Chem. Soc. 2021, 18, 3387–3397. [Google Scholar] [CrossRef]
- Al-Onazi, W.A.; Abdel-Lateef, M.A. Catalytic Oxidation of O-Phenylenediamine by Silver Nanoparticles for Resonance Rayleigh Scattering Detection of Mercury (II) in Water Samples. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2022, 264, 120258. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Lateef, M.A. Utilization of the Peroxidase-like Activity of Silver Nanoparticles Nanozyme on O-Phenylenediamine/H2O2 System for Fluorescence Detection of Mercury (II) Ions. Sci. Rep. 2022, 12, 6953. [Google Scholar] [CrossRef] [PubMed]
- Kleinman, S.L.; Frontiera, R.R.; Henry, A.-I.; Dieringer, J.A.; Van Duyne, R.P. Creating, Characterizing, and Controlling Chemistry with SERS Hot Spots. Phys. Chem. Chem. Phys. 2013, 15, 21–36. [Google Scholar] [CrossRef]
- Pazos-Perez, N.; Wagner, C.S.; Romo-Herrera, J.M.; Liz-Marzán, L.M.; García de Abajo, F.J.; Wittemann, A.; Fery, A.; Alvarez-Puebla, R.A. Organized Plasmonic Clusters with High Coordination Number and Extraordinary Enhancement in Surface-Enhanced Raman Scattering (SERS). Angew. Chem. Int. Ed. 2012, 51, 12688–12693. [Google Scholar] [CrossRef]
- Edel, J.B.; Kornyshev, A.A.; Urbakh, M. Self-Assembly of Nanoparticle Arrays for Use as Mirrors, Sensors, and Antennas. ACS Nano 2013, 7, 9526–9532. [Google Scholar] [CrossRef]
- Velleman, L.; Sikdar, D.; Turek, V.A.; Kucernak, A.R.; Roser, S.J.; Kornyshev, A.A.; Edel, J.B. Tuneable 2D Self-Assembly of Plasmonic Nanoparticles at Liquid|liquid Interfaces. Nanoscale 2016, 8, 19229–19241. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Evtushenko, E.G.; Gavrilina, E.S.; Vasilyeva, A.D.; Yurina, L.V.; Kurochkin, I.N. Highly Sensitive Measurement of Horseradish Peroxidase Using Surface-Enhanced Raman Scattering of 2,3-Diaminophenazine. Molecules 2024, 29, 793. https://doi.org/10.3390/molecules29040793
Evtushenko EG, Gavrilina ES, Vasilyeva AD, Yurina LV, Kurochkin IN. Highly Sensitive Measurement of Horseradish Peroxidase Using Surface-Enhanced Raman Scattering of 2,3-Diaminophenazine. Molecules. 2024; 29(4):793. https://doi.org/10.3390/molecules29040793
Chicago/Turabian StyleEvtushenko, Evgeniy G., Elizaveta S. Gavrilina, Alexandra D. Vasilyeva, Lyubov V. Yurina, and Ilya N. Kurochkin. 2024. "Highly Sensitive Measurement of Horseradish Peroxidase Using Surface-Enhanced Raman Scattering of 2,3-Diaminophenazine" Molecules 29, no. 4: 793. https://doi.org/10.3390/molecules29040793