
Natural Products–Pyrazine Hybrids: A Review of Developments in Medicinal Chemistry
by

Guo-Qing Chen
1,†, 
Hong-Yan Guo
1,†,
Zhe-Shan Quan
1,
Qing-Kun Shen
1,
Xiaoting Li
1,* and
Tian Luan
2,* 1
Key Laboratory of Natural Medicines of the Changbai Mountain, Ministry of Education, College of Pharmacy, Yanbian University, Yanji 133002, China
2
Department of Pharmacy, Shenyang Medical College, Shenyang 110034, China
*
Authors to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Molecules 2023, 28(21), 7440; https://doi.org/10.3390/molecules28217440
Submission received: 13 October 2023
/
Revised: 2 November 2023
/
Accepted: 3 November 2023
/
Published: 5 November 2023
(This article belongs to the Section Medicinal Chemistry)
Abstract
:Pyrazine is a six-membered heterocyclic ring containing nitrogen, and many of its derivatives are biologically active compounds. References have been downloaded through Web of Science, PubMed, Science Direct, and SciFinder Scholar. The structure, biological activity, and mechanism of natural product derivatives containing pyrazine fragments reported from 2000 to September 2023 were reviewed. Publications reporting only the chemistry of pyrazine derivatives are beyond the scope of this review and have not been included. The results of research work show that pyrazine-modified natural product derivatives have a wide range of biological activities, including anti-inflammatory, anticancer, antibacterial, antiparasitic, and antioxidant activities. Many of these derivatives exhibit stronger pharmacodynamic activity and less toxicity than their parent compounds. This review has a certain reference value for the development of heterocyclic compounds, especially pyrazine natural product derivatives.

1. Introduction
Heterocyclic structures are common in clinical drugs used to treat diseases. Such drugs typically contain nitrogen, oxygen, and sulfur, which can accept electrons and form hydrogen bonds. These properties enhance the target binding ability of the compound compared to that of hydrocarbons. Heterocyclic compounds are a common class of important compounds in medicinal chemistry and are often used in the synthesis of drugs and other active molecules [1,2,3,4]. Many natural products also contain different kinds of heterocyclic structures. The heterocyclic ring of pyrazine (Figure 1) consists of a six-membered aromatic structure containing two nitrogen atoms, arranged in a 1,4 orientation in the carbon skeleton.
The base of pyrazine (pKa 0.65) is weaker than both pyrazine (pKa 2.3) and pyrimidine (pKa 1.3). Pyrazine can be expressed as a resonance hybrid of some typical structures as shown in Figure 1, which has a resonance energy of 24.3 Kcal/mol and a dipole moment of zero due to the symmetry of the pyrazine molecule. The electron density data show that the electron density of nitrogen atoms increases while that of carbon atoms decreases [5]. Pyrazine is widely used in the synthesis of biologically active ingredients and catalysts, which makes pyrazine a hot topic in pharmaceutical chemistry research. In addition, pyrazine compounds include a variety of pharmacological effects, including antipyretic, anti-inflammatory, analgesic, anticancer, antibacterial, and antioxidant activities [6].
Pyrazine derivatives have been extensively studied as a disorder mediator, and Table 1 shows marketed drugs containing pyrazine structures that have been shown to have biological activity relevant to disease treatment. Many phenazine drugs and compounds containing fragments of pyrazine were also reported that have shown potential therapeutic value, including several that are clinically used to treat human diseases. These results suggest that pyrazine plays an important role in drug discovery [4,7].
Here, we review the pharmacological activities and mechanisms of action of natural products containing pyrazine structures. References are available at Web of Science, PubMed, Science Direct, and SciFinder Scholar. In this paper, the biological activities of natural product derivatives containing pyrazines were reviewed and their mechanism of action was also discussed.
2. Natural Product–Pyrazine Hybridization
2.1. Acrylic and Cinnamic Acid–Pyrazine Hybridization
Hepatitis C virus (HCV) is an RNA virus that is spread primarily through contaminated blood. Beaulieu et al. reported the discovery and optimization of specific allosteric inhibitors of NS5B RNA-dependent RNA polymerase (RdRp) encoded by the HCV virus. Derivative 1 (Figure 2) was obtained by introducing pyrazines into C-2 indole substituents. Compound 1 showed good inhibitory activity against RdRp (IC50 = 58 μM) and good permeability, solubility, and lipophilicity of caco-2 [69].
Rong et al. identified two cinnamate–pyrazine derivatives 2 and 3 with IC50 values of 0.69 and 1.2 μM by using HCV NS5B RdRp for compound library screening [70].
RhoA is a member of Rho GTPase, a subgroup of the Ras superfamily of small GTP-binding proteins. RhoA, as an important regulator of various cell signaling pathways, plays an important role in cytoskeletal organization, transcription, and cell cycle progression. RhoA may be a suitable therapeutic target for the treatment of cardiovascular disease. Ma et al. reported on RhoA inhibitors containing cinnamic acid. Compounds 4 and 5 showed high RhoA inhibitory activity with IC50 values of 1.51 and 1.81 μM [71].
Deng et al. reported the RhoA inhibitors of cinnamic acid. Compounds 6–9 showed high RhoA inhibitory activity with IC50 values of 1.51, 3.28, 2.58, and 2.62 μM. Pharmacological analysis showed that compound 6 had a significant vasodilation effect on PE-induced thoracic aortic ring constriction [72].
A series of Pim-2 kinase inhibitors were identified by Qian et al. through high-throughput screening. Compounds 10 and 11 showed stronger inhibition of Pim-2 kinase with IC50 values of 10 and 12 nM. Compound 11 had a stronger inhibitory effect on Pim-1 kinase with an IC50 value of 13 nM [73].
Zhang et al. synthesized cinnamic acid–pyrazine derivatives to enhance the bioactivity of cinnamic acid derivatives in neural function and neurovascular protection. The activity of the human microvascular endothelial cell line (HMEC-2) and the human neuroblastoma cell line (SH-SY5Y) against free radical damage increased under the action of compounds 12–15. Compound 15 showed the strongest activity in HBMEC-2 cells with EC50 values of 3.55 μM, respectively. Compounds 12–14 showed the strongest activity in SH-SY5Y cells, with EC50 values of 3.68, 3.74, and 3.62 μM, respectively [74].
Compounds 16 and 17 showed strong inhibitory activity against cholinesterase (ChE). Compound 16 showed the strongest inhibitory effect on BuChE with an IC50 of 2.3 nM. Compound 17 had the strongest inhibitory effect on AchE with an IC50 of 2.6 nM. Unfortunately, compound 17 had weak inhibition on the self-aggregation of Aβ42 [75].
Wang et al. synthesized a series of ligustrazine–cinnamic acid derivatives as potential neuroprotective agents. Among them, 18 and 19 showed good neuroprotective activity (EC50 = 5.44 and 3.68 μM). Compound 19 can inhibit the apoptosis of PC12 cells by blocking the mitochondrial apoptosis pathway by up-regulating the ratio of Bcl-2/Bax, down-regulating the expression of cytochrome-C (Cyt-c), and inhibiting the activities of caspase-9 and caspase-3 [76].
Chen et al. synthesized a series of novel ligustrazine acyloxy cinnamic acid derivatives and studied their in vitro inhibitory effect on adenosine diphosphate (ADP)-induced platelet aggregation and their protective effect on H2O2-induced oxidative damage of ECV-304 cells. Compounds 20 and 21 (Figure 3) had the highest protective effect on the proliferation of injured ECV-304 cells (EC50 = 0.046 and 0.020 μM), and compound 22 had the highest antiplatelet aggregant activity (EC50 = 0.054 μM) [77].
Chen et al. evaluated the inhibitory effect of compound 23–25 on ADP-induced platelet aggregation in vitro and investigated the protective effect of H2O2-induced oxidative damage in Ea.hy926 cells. Compounds 24 and 25 showed the highest protective effect on the proliferation of injured Ea.hy926 cells (EC50 = 2.2 and 1.7 μM). Compound 23 was the most active antiplatelet aggregator (IC50 = 9.6 μM) [78].
Chen et al. synthesized a series of ligustrazine–cinnamic acid derivatives based on the structural characteristics of platelet aggregation inhibitor ozagrel. In particular, compounds 26–28 (IC50 between 57–161 μM) have a higher platelet aggregation activity than ozagrel (IC50 = 360 μM) [77].
To further investigate the antiplatelet aggregation activity of trimethylpyrazine-2-carbonyloxy-cinnamic acids and esters. Chen et al. designed, synthesized, and evaluated a series of new compounds (24, 25, 29, and 30); 25 and 29 were the most effective platelet aggregation inhibitors with IC50 values of 9.6 and 24.4 μM, respectively, much higher than ozagrel (IC50 = 144.1 μM). Chen et al. then tested the protective effect of the compound against hydrogen peroxidation-damaged Ea.hy 926 cells. The corresponding ligustrazine–cinnamic acids/ethyl esters (17, 31–33) had higher activity (IC50 = 2.2, 8.8, 21.4, and 1.7 μM) than ligustrazine (IC50 = 83.4 μM) [78].
The cinnamic acid–ligustrazine derivative 34 (Figure 4) showed significant inhibitory effects on BEL-7402 and A549 cell lines with IC50 values of 9.400 and 7.833 μM [79].
Li et al. reported that ligustrazine–cinnamic acid derivatives showed protective effects against CoCl2-induced neurotoxicity in differentiated PC12 cells. The most active compound is 35 (EC50 = 25 μM), which exceeds the activity of ligustrazine (EC50 = 60 μM) [80].
Balasubramaniam et al. reported the design, synthesis, and evaluation of pyrimidine-based histone deacetylase inhibitors (HDACis). Compound 36 proved to be the most potent inhibitor, producing 100% inhibition at 100 µM, 90% inhibition at 10 µM, and 44% inhibition at 1 µM [81].
Paeonol has been shown to have anti-inflammatory activity, but its anti-inflammatory activity is poor, with only 14.74% inhibitory activity at 20 μM. Hu et al. designed and synthesized a series of paeonol derivatives and screened their anti-inflammatory activities. Compound 37 containing pyrazine structure showed 56.32% inhibitory activity against lipopolysaccharide (LPS)-induced nitric oxide (NO) overexpression in RAW264.7 macrophages at 20 μM [82].
Piperlongumine selectively targets a wide range of cancer cells and induces their death by triggering multiple pathways, including apoptosis, necrosis, and autophagy. Zuo et al. synthesized its analog 38–40 by substituting the pyrazine ring for trimethoxyphenyl. These compounds improved water solubility and showed potent anticancer activity against the HCT116 cell line with IC50 values of 3.19–8.90 μM [83].
Piperlongumine and ligustrazine have anti-proliferative effects on various types of cancer cells by up-regulating the levels of reactive oxygen species (ROS). Qian et al. designed and synthesized piperlongumine–ligustrazine derivatives and evaluated their bioactivities in vitro and in vivo. Derivative 41 had a more prominent inhibitory effect on the proliferation of drug-sensitive/drug-resistant cancer cells, and the IC50 value was lower than that of piperlongumine. The IC50 value of 41 pairs of resistant BEL-7402/5-FU cells was 0.9 μM, which was about 9 times higher than that of piperlongumine (IC50 = 8.4 μM). Mechanism studies have shown that derivative 41 regulates the DNA damage protein H2AX and autophagy-related proteins LC3, beclin-1, and p62 in drug-resistant BEL-7402/5-FU cells. TrxR activity was inhibited, ROS levels increased, mitochondrial transmembrane potential decreased, and DNA damage and autophagy were dose dependent. Finally, compound 41 showed strong in vivo antitumor activity at 5 mg/kg, with a tumor inhibition rate of 76% (w/w) [84].
Piperlongumine increases the levels of reactive ROS and induces apoptosis in cancer cells by triggering different pathways. However, the poor solubility of Piperlongumine has limited its further research and clinical application. Ligustrazine has a water-soluble pyrazine skeleton, which can inhibit the proliferation and metastasis of cancer cells. The solubility of compounds 42–45 in colorectal cancer HCT116 cells was 8.9–26.2 times higher than that of piperlongumine. Compounds 42–45 showed significant inhibitory effects on U87MG, HCT116, A549, and K562 cell lines with IC50 values ranging from 0.25 to 8.73 μM. Compound 43 also increased ROS levels. Additionally, compound 43 preferentially inhibited the proliferation, migration, invasion, and heteroadhesion of HCT116 cells. Compound 43 inhibits tumor growth and lung metastasis in vivo and prolongs the survival of tumor-bearing mice. Furthermore, compound 43 mitigated TGF-β1-induced epithelial-mesenchymal transformation and Wnt/βxcatenin activation by inhibiting Akt and GSK-3β phosphorylation in HCT116 cells. Compound 43 has significant anti-proliferation and anti-metastasis activities, which is superior to piperlongumine [85].
2.2. Chalcone–Pyrazine Hybridization
Compound 46 (Figure 5) showed good activity against BPH-1 and MCF-7, with IC50 values of 10.4 and 9.1 μM, respectively, comparable to adriamycin (IC50 values of 14.1 and 9.2 μM). Compound 47 showed the strongest activity against the PC12 cell line with an IC50 value of 16.4 μM [86].
Compound 48 showed the strongest inhibitory effect on the BEL-7402 cell line with an IC50 value of 10.74 μM and no toxicity to HUVEC-12 (IC50 > 40 μM). Fluorescence staining and flow cytometry analysis showed that compound 48 could induce apoptosis of BEL-7402 cells [87].
Srilaxmi et al. designed and synthesized a series of chalcone–pyazine derivatives and tested the anticarcinogenic activity of all derivatives against five human cancer cell lines (MCF-7, A549, Colo-205, A2780, and DU-145) using a MTT assay. Compound 49 showed significant inhibitory effects on A549 and Colo-205 cell lines with IC50 values of 0.13 and 0.19 μM. Compound 50 showed a significant inhibitory effect on the MCF-7 cell line with an IC50 value of 0.18 μM. Compound 51 showed significant inhibitory effects on MCF-7, A549, and DU-145 cell lines with IC50 values of 0.012, 0.045, and 0.33 μM [88].
The 50% effective concentration (EC50) values of compound 52 against Xanthomonas axonopodis pv.Citri (Xac), Xanthomonas oryzae pv.oryzae (Xoo), and Ralstonia solanacearum (Rs) were 6.72, 15.17, and 9.29 μg/cm3, respectively, which were better than those of Bismerthiazol (44.31, 42.46, and 62.36 μg/cm3, respectively) [89].
Compounds 53 and 54 showed good antibacterial activity against M. luteus, with a MIC value of 31.25 µg/mL, similar to that of tetracycline (MIC = 31.25 µg/mL) [90].
Luo et al. synthesized a series of ligustrazine–chalcone hybrids and evaluated their antitumor activity in vitro and in vivo. Compounds 55–60 showed significant cytotoxicity to MDA-MB-231, MCF-7, A549, and HepG-2 cell lines in vitro, with IC50 values ranging from 0.99 to 9.99 μM. Compounds 57 and 60 showed significant effects on the MDA-MB-231 cell line (IC50: 57, 1.60 μM; 60, 1.67 μM) and MCF-7 cell line (IC50: 57, 1.41 μM; 60, 1.54 μM) had a good anti-proliferation effect. Compounds 57 and 60 showed strong inhibition of colony formation in both MDA-MB-231 and MCF-7 cell lines, and also showed strong inhibition of migration of these two cell lines in wound healing tests. It should be noted that compound 57 can significantly induce apoptosis of MDA-MB-231 cells in a concentration-dependent manner, inhibit the transformation of the MDA-MB-231 cell growth cycle, and block the cell growth cycle in the G0/G1 phase. Furthermore, compound 57 showed significant antitumor growth efficacy in in vivo anti-proliferation experiments in the NBC model, with a wide safety window. Immunohistochemical analysis showed that compound 57 could significantly reduce the positive rate of Ki-67 in a dose-dependent manner [91].
Wang et al. designed and synthesized six series of ligustrazine–chalketone-modified platinum (IV) complexes and evaluated their anti-proliferation activities. Compounds 61–67 showed significant inhibitory effects on A549, PANC-1, MDA-MB-231, HCT116, and SGC-7901 cell lines, with an IC50 ranging from 0.93 to 7.29 μM. Among them, 66 showed higher cytotoxicity to cancer cell lines than the cisplatin (CDDP) or combination group, and lower cytotoxicity to normal human cells than the CDDP or combination group. Mechanism studies have shown that 66 effectively induces DNA damage and initiates mitochondrial-dependent apoptosis pathways. In addition, 66 regulates the expression level of nuclear factor erythroid 2-related factor 2, glutathione peroxidase 4, and solute carrier family 7 member 11 expression level, significantly induced iron sag. Furthermore, in pancreatic cancer anti-CDDP xenotransplantation models, 66 achieved better antitumor efficiency in vivo than CDDP, but without significant side effects [92].
2.3. Polyphenols–Pyrazine Hybridization
Du et al. designed and synthesized a series of heterocyclic analogs of resveratrol and evaluated their inhibitory effects on MCF-7 cells. Among them, compound 67 (Figure 6) linked to pyrazine showed a certain inhibitory effect on MCF-7 with an IC50 value of 70.9 μM. The activity exceeded that of resveratrol (IC50 = 80.0 μM) [93].
Resveratrol is widely used as a vasodilator, free radical scavenger, and antioxidant, as well as an anti-platelet aggregator and anti-atherosclerotic agent for the prevention and treatment of cardiovascular diseases and ischemia [94]. Deng et al. designed and synthesized a series of ligustrazine–stilbene hybrid derivatives. Derivatives 69–72 showed high protective effects on human umbilical cord vascular endothelial cells (HUVECs) damaged by hydrogen peroxide, with values of EC50 ranging from 0.0249 to 28.9 μM. Among them, the EC50 value of compound 71 is 0.0249 μM, which is 30,000 times higher than that of tetramethylpyrazine (EC50 = 788 μM) [95].
Chen et al. designed and synthesized a series of pyrazole–pyrimidine derivatives, and screened their anti-NO activity and toxicity to normal hepatocytes (L02). Compounds 73 and 74 have low toxicity (against L02: IC50 = 786.31 and >1000 μM, respectively) and strong anti-NO release effect (IR = 68.82%, 63.44%, at 10 μM, respectively) [96].
Liang et al. synthesized the folate receptor (FR)–targeted rhaponticin conjugate FRHA (75) using a hydrophilic peptide separator linked to folate and a disulfide linker. FRHA (75) maintains a high affinity for FR-positive cells and produces specific dose-responsive activity in vitro. Treatment of FRHA (75) with a reducing agent shows that the amino reactive derivatives of rhaponticin will be released spontaneously after the reduction of disulfide bonds in the nucleosome. In vivo, FRHA (75) has also been shown to have specific activity against FR-positive allograft and xenograft models, and possible therapeutic activity leads to mild to moderate toxicity [97].
Nkepang et al. designed and prepared A series of folate–combretastatin A-4 conjugated prodrugs. Prodrugs 76 and 77, with longer PEG intervals and greater hydrophilicity, enhance the uptake of colon 26 cells by FR-mediated mechanisms and specifically target SC colon 26 tumors in Balb/c mice [93].
Curcumin is a polyphenolic compound extracted from Curcuma longa, which has been extensively studied for its potential anticancer effects [98]. Wang et al. synthesized a series of ligustrazine–curcumin derivatives by coupling antitumor bioactive compounds with ether bonds. Among them, compound 78 (Figure 7) pairs BEL-7402, A549, HCT-8, BGC-823, and A2780 cells cell lines were 6.391, 5.890, 7.106, 5.472, and 5.540 μM [99].
Ai et al. designed and synthesized a series of ligustrazine–curcumin hybrids [71]. Compounds 79–81 showed significant inhibitory effects on A549 and A549/DDP cell lines with IC50 ranging from 0.60 to 2.85 μM. Pharmacological studies showed that compound 79 inhibited the expression of thioredoxin reductase (TrxR), promoted the accumulation of ROS in cells, and significantly inhibited the apoptosis of proliferation-sensitive (A549, SPCA-1, LTEP-G-2) and drug-resistant (A549/DDP) lung cancer cells. In addition, its antitumor activity was significantly weakened by active oxygen scavenger. In addition, 79 also inhibited NF-κB, AKT, and ERK signaling pathways, P-GP-mediated Rhodamine 123 efflux, P-gp ATPase activity, and P-gp expression in A549/DDP cells [100].
K. Singh et al. synthesized a series of curcumin bioconjugations and tested them for antibacterial and antiviral activity. The antibacterial activity of compounds 82 and 83 against Gram-positive (S. viridans) and Gram-negative (E. coli, K. pneumoniae, and P. miraleilis) ranged from 0.09 to 0.54 μM [101].
Curcumin, as a cell imaging and photodynamic therapy (PDT) agent, showed significant photocytotoxicity at visible wavelengths of 400–700 nm with IC50 = 8.2 μM. Its degradation is prevented by the formation of phototoxic dipyridophenazine (dppz) complex 84 (IC50 = 3.3 μM). However, both compounds are less toxic in the dark (IC50 > 50 μM) [102].
Banerjee et al. synthesized a ternary vanadium oxide complex of O-phenanthroline with curcumin or disaccharide curcumin, an anticancer compound based on curcumin. Complexes 84 and 85 showed significant phototoxicity at visible light (400–700 nm), with IC50 values < 5 μM in HeLa, HaCaT, and MCF-7 cells, and without significant dark toxicity. The DNA ladder, membrane VzFITC/PI, and DCFDA data showed that these complexes lead to apoptotic cell death by forming ROS under light exposure while remaining inert in the dark. Confocal microscopy showed that the complex was mainly located in the cytoplasm, and complex 84 had significant mitochondrial uptake [103].
Banaspati et al. prepared a series of curcumin–nickel (II) complexes and studied their photoinduced anticancer activity in vitro. Curcumin complexes 86 and 87 have REDOX activity in the nickel center, have considerable affinity for binding with calf thymus DNA (ct-DNA), and have moderate affinity for interacting with human serum albumin (HSA). Complexes 86 and 87 exhibit significant photoinduced in vitro cytotoxicity in HeLa and A549 involving reactive ROS with very low dark toxicity. Complexes 86 and 87 are much less toxic to immortalized normal lung epithelial cells (HPL1D). Confocal microscopy images of complexes 86 and 87 show that they are mainly localized in the cytoplasm of A549 cells. JC-1 experiment showed that under visible light irradiation, the sub-G1 cell cycle process of A549 cells was blocked, the mitochondrial membrane potential was significantly lost, and the main mechanism of cell death was apoptosis [104].
2.4. Flavono–Pyrazine Hybridization
Wang et al. designed and synthesized a series of derivatives using ligustrazine and flavonoids as raw materials and tested the antitumor activities of these derivatives. Compounds 88 and 90 (Figure 8) showed the strongest inhibitory effects on HT-29 cell lines, with IC50 values of 10.67 and 10.90 μM. Compound 89 showed the strongest inhibitory effect on the MCF-7 cell line with an IC50 value of 10.43 μM [87].
Vančo et al. prepared a series of heterologous fish meal containing copper complexes and evaluated their antitumor activity. Complex 91 has significant in vitro cytotoxicity against a variety of human cancer cells (MCF-7, HOS, A549, PC-3, A2780, A2780R, Caco-2, and THP-1) with IC50 values of 2.2–3.3 µM. Additionally, complex 91 was less toxic to healthy human hepatocytes, with IC50 > 100 µM. Complex 91 is capable of inducing the destruction of intracellular life molecules and subsequent cell death, primarily through the initiation or progression of oxidative stress. Complex 91, on the other hand, has shown the ability to inhibit inflammation-related signaling pathways (NF-κB/AP-1 activity, NF-κB translocation, and TNF-α secretion) [105].
Poly(ADP-ribose) polymerase (PARP) inhibitors are a class of anticancer drugs that block the catalytic activity of PARP protein. The nerone derivative 92 containing pyrazine showed a significant inhibitory effect on PARP, IC50 = 77 nM [106].
Compounds 93–95 showed stronger thrombin inhibitory activity than baicalin and TMP, all of which prolonged TT, APTT, and PT to varying degrees, and significantly reduced plasma FIB content at the same concentration. Compounds 93–95 showed enhanced neuroprotective and antithrombotic activity against H2O2-induced PC12 cell death. Compound 93 was used in cerebral ischemia–reperfusion experiments in the middle cerebral artery occlusion (MCAO) model. The results showed that compound 93 could significantly reduce the infarct size of CA1 pyramidal neurons and reduce the damage to neuron cells. Therefore, compound 93 has obvious antioxidant, anticoagulant, and protective effects on brain I/R injury [107].
2.5. Coumarin–Pyrazine Hybridization
The coumarin derivative 96 (Figure 9) containing sulfonamide showed moderate anticancer activity against the breast cancer cell line (T47D) with an IC50 of 86.9 μM [108].
Compound 97 showed significant inhibitory effects in HCT116, C-Raf, and MEK1 cell lines with IC50 values of 0.9, 0.056, and 0.65 μM. The effects of the interaction between the derivative 97 and its on-target and off-target proteins (Raf/MEK, CYPs, and hERG channels) were also detected, but the interaction was weaker [109].
The GI inhibition rate of the coumarin derivative 98 in the MALME-M cell line was 55.75% at a concentration of 10 μM [110].
Compounds 99 and 100 showed high visible-light trigger cytotoxicity against HeLa and MCF-7 cancer cells, producing significantly low micromolar IC50 values (IC50 = 1.1–10.0 μM) and much lower toxicity under dark conditions (IC50 > 50 μM). Confocal microscopy showed that compound 100 accumulated in HeLa cells’ mitochondria and induced apoptosis by ROS generation through type 1 photosynthesis [111].
Goel et al. found that arylated imidazo [1,2-α] pyrazine–coumarin hybrids 101 and 102 exhibited significant antitumor activity at a concentration of 10 μM [112].
H. Halawa et al. synthesized a series of new 4-arylamino-3-nitrocoumarin and evaluated the cytotoxic activity of the KB-3-1 cell line in vitro using the resazurin method. Among them, KB-3-1 cells containing the pyrazine derivative 103 showed moderate cytotoxicity with an IC50 value of 43 μM [113].
L. El-Ansary et al. prepared a new Schiff base SCH (104) using 8-acetyl-7-hydroxy-4-ethylcoumarin and sulfaclozine as raw materials. Its silver complex SCH-Ag (105) was also synthesized. The inhibitory effects of SCH (104) and SCH-AG (105) on a variety of bacteria and fungi and the antitumor activity against MCF-7 cell lines in vitro were evaluated. SCH (104) and SCH-AG (105) showed strong inhibitory activity against three species of bacteria (S. aureus, B. subtilis, and P. aeruginosa), but no activity against fungi (A. flavus and C. albicans). Furthermore, the antibacterial activity of SCH-Ag (105) was higher than that of SCH (104). The IC50 of SCH (104) for the MCF-7 cell line was 90.5 μg/mL, while that of SCH-Ag (105) was 9.3 μg/mL. SCH (104) and SCH-Ag (105) showed less antitumor activity than cisplatin (IC50 = 1.7 μg/mL) [114].
Compound 106 (Figure 10) has good antibacterial activity for strains of Salmonella typhi MTCC 537, Escherichia coli MTCC 64, and Candida albicans MTCC 3017, with a MIC value of 25 µg/mL. Compound 106 showed obvious inhibitory activity against chitinase with an IC50 value of 7.5 μM [115].
Chai et al. synthesized a series of 7-O-substituted pyridine-4-methyl coumarin derivatives and evaluated their antibacterial activity in vitro. Compound 107 showed significant inhibition against Candida tropicalis, Cryptococcus neoformans, and Trichophyton rubrum strains with MIC80 values of 1, 1, and 0.25 μg/mL. Compound 108 showed obvious inhibition against Candida tropicalis strain with MIC80 of 1 μg/mL [116].
Moosavi-Zare et al. synthesized a series of spiro–pyran derivatives and screened their antioxidant activities by a DPPH radical scavenging assay. Spiropyr derivative 109 showed good dose-dependent (0.2–1 mg/mL) free radical resistance (45.32–55.14%) [117].
Compound 110 showed stronger inhibitory activity against RANKL-induced osteoclast differentiation in RAW264.7 cells at 2 μM, with an inhibition rate of 60.6%. Compound 110 showed no cytotoxicity to the RAW264.7 cell line at a concentration of 10 μM [118].
Compound 111 showed significant protective activity against ECV-304 cells (EC50 = 0.14 μM), far superior to ligustrazine (EC50 = 0.60 μM) [119].
Priyanka et al. synthesized a series of 7-benzamidocoumarin derivatives and evaluated in vitro the antifilarial activity against the human lymphatic filarial parasite, Brugia malayi. There are also pyrazine compounds 112 and 113 with 95% and 70% inhibition of adult motility at 10 μM, which can permanently paralyze the nematode [120].
Ostrowska et al. designed a series of 6-acetyl-7-hydroxy-4-methyl coumarin derivatives containing piperazine groups. Pyrazine-containing derivative 114 showed weak activity against the 5-HT1A receptor with a Ki value of 25 (12.1–51.0) nM [121].
In order to search for potential drugs with good anti-aging effects, Tang et al. synthesized methylurolitin A and its amide derivatives. Caenorhabditis elegans (C. elegans) was used to evaluate its anti-aging effect and biosafety. Methylurolitin A has good biosecurity for the growth, reproduction, and activity of C. elegans. The derivative 115 has the best life-prolonging effect, the anti-aging effect is greater than methylurolitin A, and it has good biosafety [122].
Urolithin B is a natural metabolite that shows good activity in diseases such as obesity, diabetes, osteoporosis, cancer, learning, and memory disorders. Chen et al. designed and synthesized an amide derivative of urolithin B and verified its anti-aging and biosafety using C. elegans. The results showed that 116 had the best anti-aging activity among all derivatives, and the compound had good biosafety [123].
2.6. Anthraquinone- and 1,4-Naphtoquinone–Pyrazine Hybridization
The GI50 of compound 117 (Figure 11) in leukemia cell lines ranged from 0.07–3.65 μM. The GI50 of the breast cancer subgroups ranged from 0.72–19.1 μM. Compound 117 showed the strongest activity against K562 leukemia, with a GI50 of 0.07 μM, LC50, and TGI of >100 μM, respectively. HL-60 (TB) and MCF-7 cell lines followed, with GI50 values of 0.68 and 0.72 μM [124].
Compound 118 showed certain antiproliferative activity against MCF-7, HeLa, and A549 (IC50 = 53.5, 79.1, and 78.3 μM, respectively), and high cytotoxicity against L929 (IC50 = 49.6 μM) [125].
The derivative of tetramethylpyrazine–rhubaric acid 119 not only inhibited the proliferation of BEL-7402 cancer cells (IC50 = 26.4 μM), but also significantly inhibited the normal angiogenesis of the chicken chorionic allantoic bladder [126].
The IC50 of derivative 119 for CHMp (canine inflammatory mammary carcinoma cell line) and MDCK (Madin–Darby immortalized canine kidney cell line) is 42.59 μM and 79.37 μM, respectively. Derivative 119 mediates apoptosis through mitochondrial damage, and arrest of the S phase and G2/M phase by down-regulation of cyclin B1. In addition, derivative 119 reduces filamentous foot and inhibits cell migration by downregulating cadherin. In the CMIC lung metastasis model, derivative 119 can effectively inhibit lung tumor growth without obvious toxicity [127].
YM155 (120) is a potent broad-spectrum anticancer drug derived from phenotypic screening of inhibitors of survivin expression function. The anticancer drug YM155 (120) has been widely studied as a specific statin inhibitor. The IC50 value of YM155 (120) against the H1299 cell line was 0.0137 μM, and the SI value was 18. Furthermore, YM155 (120) has been found to induce DNA damage. Si-Han Sherman Ho et al. synthesized a series of YM155 (120)-linked pyrazine derivatives and tested their antitumor activity against H1299 cell lines. Compounds 37 and 38 showed little activity against H1299 cell lines, with IC50 values of 0.0377 and 0.0546 μM, but SI values of 38 and 30 were higher than YM155 (120) [128].
Iwai et al. found that YM155 (120) down-regulates survivin and exhibits strong antitumor activity. In the Caco-2 cell model, YM155 (120) was observed as a substrate for P-gp [99]. Premkumar et al. found that YM155 (120) at 25 nM down-regulates survivin in gliomas, down-regulates myeloid cell leukemia sequence 1 (Mcl-1), and up-regulates Noxa levels. These findings suggest that YM155 (120) negatively regulates Mcl-1 and survivin through endogenous and exogenous apoptotic pathways and amplifies mitochondrial signaling, thus inhibiting glioma cell resistance to TRAIL-induced apoptosis (TRAIL is a tumor necrosis factor-associated apoptosis-inducing ligand). YM155 (120) combined with TRAIL significantly increased antitumor activity and may have application value in the treatment of malignant glioma [129].
Ho et al. evaluated the DNA binding affinity of the test compound (120–125) by monitoring the displacement of thiazole oranges from herring sperm DNA [130,131]. The DC50 of YM155 (120) was 20.3 μM and that of adriamycin was 2.64 μM. Compounds 121, 124, and 125 showed higher activity than YM155 (120), with DC50 values of 18.4, 12.0, and 15.5 μM, respectively [128].
Glioblastoma (GBM) is the most common primary central nervous system (CNS) malignancy. Furthermore, YM155 (120) has clinical tolerance problems due to its lack of cell type selectivity. Thomas J. West et al. synthesized a prodrug of YM155 (120), named aYM155 (121). aYM155 (121) was used against GBM cancer stem-like cells (IC50 = 0.7–10 nM) from multiple patient sources. The EGFR variant III-expressing (EGFRvIII) cell line (IC50 = 3.8–36 nM) shows strong cell-killing activity and is activated in a cell type-dependent manner. The survivin inhibitory and apoptosis-inducing activity of YM155 (120) is related to its interaction with receptor-interacting protein kinase 2 (RIPK2). In an orthotopic intracranial GBM xenograft model, aYM155 (121) significantly inhibited brain tumor growth in vivo, which was related to the pharmacodynamics of selective survivin based on cytotypes [132].
Liu et al. designed and synthesized a series of 1-monosubstituted naphthoquinone imidazole derivatives and tested their antitumor activity in vitro. When the substituent was pyrazine, compound 127 (Figure 12) showed a weak inhibitory effect on MCF-7, HeLa, and A549 cell lines, with IC50 values ranging from 161–186 μM. However, compound 127 was more toxic to normal cell L929 with IC50 of 51 μM [133].
Bargiotti et al. synthesized a series of 3-arylnaphthalene [2,3] isoxazole-4,9-diones and tested the binding of these compounds to Hsp90 and their effects on Hsp90 client proteins expression in human tumor cell lines. The pyrazine-containing compounds 128 and 129 have a strong affinity for Hsp90 with IC50 values of 0.68 and 0.51 μM. Additionally, compounds 128 and 129 showed significant inhibitory effects on NCI-H460, A431, and STO cell lines, with IC50 values ranging from 0.017 to 0.22 μM [134].
Shanab et al. designed and synthesized a series of azanadione–pyrrolidinated derivatives and evaluated the anti-proliferative activity of all compounds in multiple cell lines. The pyrazine-containing compound 130 showed significant inhibitory activity against KB/HeLa, SF-268, NCI-H460, RKOp27, and RKOp27IND cell lines with IC50 values ranging from 1.121–2.973 μM [135].
Yu et al. designed and synthesized derivatives of indolizinoquinolinedione scaffold and tested the antitumor activity of these compounds. The MTT assay showed that compound 131 containing pyrazine showed significant inhibitory effects on HCT116, CCRF-CEM, A549, Huh7, and DU-145 cell lines, with IC50 values ranging from 1.61 to 13.15 μM [136].
Shen et al. designed and synthesized a series of new Indolizinoquinoxalin-5,12-dione derivatives. Compounds 132–134 showed significant inhibitory effects on the growth of four human tumor cell lines (GLC-82, NCI-H460, MCF-7, and MCF-ARD), with IC50 values ranging from 0.20 to 16.46 μM [137].
Devi et al. synthesized a series of new anthraquinone-based copper (II) complexes. Nuclear targeting complex 135 showed significant cytotoxicity to cancer cells in visible light (IC50 = 2.57–3.03 µM), but decreased dark toxicity (IC50 > 50 µM). Singlet oxygen produced by complex 135 photosensitization is a key cytotoxic substance that causes apoptosis damage in cancer cells. The S-coordination and anthraquinone moiety of complex 135 exhibit double photosensitivity, resulting in a significant PDT effect on cancer cells with minimal dark toxicity [138].
Kim et al. found that tetracyclic heteroquinone analogs containing pyrazine structures were highly cytotoxic to human tumor cell lines. Compound 136 (Figure 13) showed strong inhibitory effects on A549 and XF-498 cell lines with IC50 values of 1.64 and 2.26 μM. Compounds 137–142 showed significant inhibitory effects in A549, SK-OV-3, SK-MEL-2, XF-498, and HCT-15 cell lines, with IC50 values ranging from 0.06–1.01 μM. The IC50 value of compound 142 against the XF-498 cell line was 0.06 μM, 2.6 times that of doxorubicin (IC50 = 0.16 μM) [139].
Kim et al. designed and synthesized a series of pyrido [3,4-b] phenazinedione derivatives and evaluated their cytotoxic activity and topoisomerase II inhibitory activity. The derivative 143–147 showed significant inhibitory effects on human tumor cell lines (A549, SNU-638, Col2, HT1080, and HL-60), with IC50 values ranging from 0.12 to 1.26 μM. Compound 144 had the strongest effect on the SNU-638 cell line with an IC50 of 0.12 μM. It is 49.75 times that of ellipticine (IC50 = 5.97 μM). Compounds 143–147 showed inhibitory activity (39–100%) against topoisomerase II at 200 μM. The most active compound was 143, with an IC50 of 0.082 μM [140].
Lee et al. synthesized a series of benzo[g]quinoxalin-5,10-dione derivatives and evaluated in vitro cytotoxic activity against four human cancer cells (HCT-15, SK-OV-3, MD-MB-468, and T-47D). Compounds 148–155 (Figure 14) showed significant inhibitory activity against four cancer cells with IC50 values ranging from 0.005 to 10 μM. The cytotoxic activity of compound 153 against HCT-15 cells was similar to that of doxorubicin [141].
Kwak et al. synthesized a series of 2-alkyl-2, 3-dihydro-1h-2,6,9-triazacyclopenta[b]anthracene-5,10-diones. The cytotoxic activity of six human cancer cells (HCT-15, SK-OV-3, A549, SNB19, MCF-7, and MCF-7/ADR) was evaluated in vitro. Compounds 156–160 showed significant inhibitory effects on all human cancer cell lines, with IC50 values ranging from 0.035 to 0.381 μM [142].
Lee et al. synthesized pyridazo [2,3-b] phenazine-6,11-dione derivatives and evaluated their cytotoxic activity by the SRB (Sulforhodamine B) assay. Derivatives 161–173 (Figure 15) showed excellent cytotoxicity to human tumor cell lines (A549, SK-OV-3, SK-MEL-2, XF-498, and HCT-15) with IC50 values ranging from 0.004–0.361 μg/mL. The killing effect of 161 on HCT-15 (ED50 = 0.004 μg/mL) was 23 times that of adriamycin (ED50 = 0.093 μg/mL) [143].
Lee et al. designed and synthesized pyridazino [4,5-b]phenazine-5,12-diones. The cytotoxic activity of these compounds against human cancer cell lines was evaluated by a SRB (thiodan B) assay. The cytotoxicity of compound 7a–7j to cancer cells (A549, SK-OV-3, SK-MEL-2, XF498, and HCT-15) was higher (IC50 = 0.010–0.0.195 μM) than that of adriamycin (IC50 = 0.097–0.225 μM). The most active compounds 179 and 181 are about 10 times more cytotoxic than doxorubicin to all human cancer cell lines [144].
Tuyun et al. designed and synthesized a series of benzo[b]phenazine-6,11-dione derivatives and tested their antibacterial and antifungal activities in vitro. Among them, compound 184 (Figure 16) showed the strongest inhibition effect on S. epidermidis, and the MIC value was 156.2 μg/mL [145].
Kumar et al. designed and synthesized a series of benzoquinolin-5,10-dione compounds to test for in vitro antituberculosis activity against M. tuberculosis H37Rv. Compound 185 is the most active against M. tuberculosis, with a MIC of 12.5 μg/mL [146].
Kumar et al. designed and synthesized 2-amino-6-(5,10-dioxo-2,3-diphenyl-5,10-dihydrobenzo[g]quinoxalin-7-yl)-4-(substituted)phenylpyridine-3-carbonitrile. The antibacterial activity of newly synthesized compounds was screened by the L.J. Slope (conventional) method. Compound 186 has the strongest inhibitory effect against M. tuberculosis H37Rv with a MIC of 50 μg/mL [147].
S. Hammam and others designed and synthesized a series of diarylaminodiaminobenzoquinone, and studied the antifungal and antibacterial activities. Among them, compounds 187 and 188 had significant inhibitory effects on Fusarium solani, Fusarium oxysporum, and Aspergillus flavus, with MIC values of 20 μg/mL [148].
Morin et al. synthesized a series of various azotized analogs of 1, 4-naphthoquinone, The inhibitory activity of P. falciparum and human glutathione reductases and P. falciparum thioredoxin reductase was tested. Compounds 5,8-quinoxalinedione (189) and 190 (Figure 17) were the most specific TrxR inhibitors, with a stronger inhibitory effect than menadione [149].
The inhibitory activity of 5,8-quinolinedione (189) on the binding of BMAL1/CLOCK to Ebox DNA was concentration dependent, with an IC50 value of approximately 1 μM. 5,8-quinolinedione covalently reacts with protein(s) and may regulate dimer formation [150].
Keinan et al. designed and synthesized a series of Cdc25B quinone inhibitors. Among them, WDP1263 (191) containing pyrazine is the strongest Cdc25 inhibitor with an IC50 value of 0.5 μM, but in the presence of 0.8 mM DTT (EC50 = 1.4 μM). WDP1263 (191) showed inhibitory activity against the A549 cell line with an IC50 of 22.28 μM. WDP1263 (191) (E1/2 = 186 mV) prevents the redox cycle through its reducing state [151].
Besset et al. designed and synthesized a heteroquinone compound containing two methoxycarbonyl methyl sulfur groups in the benzoquinone ring and evaluated its Cdc25B phosphatase inhibitory activity. Compound 192 containing pyrazine showed a strong inhibitory effect on Cdc25B with an IC50 value of 5.40 μM. Furthermore, derivative 192 inhibited the pancreatic cell line (MiaPaCa-2) by 24% at 100 µM [152].
Yang et al. synthesized a series of furanoquinolinedione and isooxazolinequinolinedione derivatives and performed enzyme inhibition tests. Compounds 193 and 194 containing pyrazine have inhibitory activity of TDP2 with IC50 of 32 and 9.3 μM [153].
Ryu et al. reported that derivatives of 6-arylamino-quinoxalin-5,8-diones had inhibitory effects on the proliferation of rat aortic smooth muscle cells (RAoSMC). Compounds 195–208 significantly inhibited SMC proliferation, with IC50 values ranging from 1.0–5.5 μM. Compounds 196, 200, and 201 were the most active with IC50 values of 1.0 μM [154].
Chung et al. also reported that the 6-arylamino-quinoxalin-5,8-diones derivatives 195–202 and 209–210 had inhibitory effects on the proliferation of rat aortic smooth muscle cells (RAoSMC). The activity of compounds 195–202 was consistent with the literature. Additionally, compounds 209 and 210 significantly inhibited SMC proliferation with IC50 values of 1.1 and 1.2 μM. Furthermore, the inhibitory effect of compound 197 on SMC proliferation is mediated by the regulation of the kinase 1/2 signaling pathway regulated by extracellular signals [155].
Ye et al. synthesized folate–aminocaproate–doxorubicin (FA-AMA-DOX) and performed cytotoxicity and uptake tests on KB, HepG-2, and A549 cell lines. FA-AMA-DOX (211) (Figure 18) is more cytotoxic to KB and HepG-2 cells than DOX or AMA-DOX at the same concentration, and FA can reduce cytotoxicity in a dose-dependent manner. On the contrary, FA-AMA-DOX and AMA-DOX showed lower cytotoxicity to A549 cells than DOX at the same concentration, and FA could not reduce cytotoxicity. FA-AMA-DOX (211) increased DOX accumulation in KB cells compared to FA-AMA [156].
Huang et al. oxidized the phenol to O-naphthoquinone and tested its biological activity. Compound 4h effectively inhibited the proliferation of different AML (acute myelocytic leukemia) cell lines in vitro, with IC50 values ranging from 0.11 to 0.65 μM. In vivo antitumor studies have shown that compound 212 can cause tumor regression in MV4-11 xenograft tumor models at 40 mg/kg/d for 4 h, without obvious toxicity [157].
Sandilya et al. synthesized a series of xanthone derivatives containing 3,6-bis (3′-substituted propoxy) and 3,6-bis (5′-substituted pentyloxy). Anti-inflammation of Wistar albino rats was studied by carrageene-induced metatarsal edema in rats. Compounds 213 and 214 at 200 mg/kg body weight showed a slightly lower inhibitory effect than diclofenac sodium (10 mg/kg body weight dose, inhibition effect: 68.27%) in plantar edema after 6 h, with an inhibition effect of 63.32% and 62.75%, respectively [158].
2.7. Lignin–Pyrazine Hybridization
According to Zhao et al., 4′-demethylepipodophyllotoxin (DMEP) was prepared by a series of new types of podophyllum topoisomerase II (Topo II) inhibitors. The antitumor activity of compound 215 (Figure 19) against the tumor cell lines HeLa, A549, HepG-2, and BGC-823 was significantly improved with IC50 values of 0.88, 3.83, 1.21, and 4.15 μM, respectively. More than 4′-demethylepipodophyllotoxin antitumor activity (the IC50 values of HeLa: 15.96 μM; HepG-2: 18.74 μM; A549: 52.08 μM; and BGC-823: 21.26 μM). The antitumor activity of compound 216 against BGC-823 was significantly improved with an IC50 value of 1.50 μM. The amide derivatives 217 and 218 showed strong inhibitory effects in HepG-2, HeLa, A549, and BGC-823 cell lines, with IC50 values ranging from 3.49 to 18.71 μM. Compound 217 had the strongest killing ability against the BGC-823 cell line with an IC50 value of 3.49 μM. Compound 215 inhibited the G2/M cycle of HeLa cells and induced apoptosis by strongly attenuating Topo II DNA unshackling relaxations [159].
Wu et al. synthesized a series of podophyllotoxin (PPT) derivatives and evaluated the cytotoxicity of A549, MCF-7, HepG-2, and L02 cells. The IC50 values of compound 219 containing pyrazine for A549 and HepG-2 cell lines were 9.3 and 11.7 μM. The IC50 values of compound 220 against the A549 and MCF-7 cell lines were 8.1 and 11.3 μM [160].
Zhang et al. synthesized a series of poxylotoxin aromatic heterocyclic esters and evaluated the anticancer effects of two human chronic myeloid leukemia cell lines (K562 and K562/ADR). The IC50 values of compound 221 containing pyrazine for the K562 and K562/ADR cell lines were 0.034 and 0.022 μM [161].
Li et al. designed and synthesized podophyllotoxin derivatives and evaluated their anticancer activity in vitro against several human cancers. The pyrazinyl derivative 222 inhibited the HL60, SGC-7901, and A549 cell lines with IC50 values of 6.71, 12.72, and 11.15 μM [162].
Castro et al. designed and synthesized podophyllotoxin e-ring-modified derivatives and evaluated their cytotoxicity. The IC50 value of compound 223–226 containing pyrazine against the P-388, A549, HT-29, and MEL-28 cell lines was 0.56–2.8 μM, and the antitumor activity of compound 223–226 was lower than that of podophyllotoxin (IC50 = 0.012 μM) [163].
Compounds 227–230 (Figure 20) had a corrected mortality rate of 51.7%, 51.7%, 55.2%, and 55.2% in vivo against Mythimna separata (M. separata) at 1 mg/mL, higher than or equivalent to the activity of Toosendanin (51.7%) [164].
Zhi et al. conducted an in vivo insecticidal activity test on the pre-third-instar larva of M. separata (Walker) at 1 mg/mL. Compounds 231 and 232 exhibited the best potent insecticidal activity with the final mortality rate of 53.3% and 63.3%, the activity was higher than Toosendanin (46.7%) [165].
Zhi et al. synthesized C-ring, D-ring, and E-ring modified phenazines oxme derivatives of podophyllotoxin and performed 1 mg/mL in vivo insecticide on the pre-third-instar larva of the oriental armyworm M. separata (Walker). Compounds 233–235 exhibited the best potent insecticidal activity with a final mortality rate of 51.7%, 62.1%, and 58.6%, the activity was higher than Toosendanin (48.3%) [166].
In vivo insecticidal activity against the pre-third instar larva of M. separata (Walker) was measured at 1 mg/mL. Derivatives 236–238 exhibited the most promising insecticidal activity with the final mortality rate of 62.1, 62.1%, and 72.4%, The activity was higher than toosendanin (48.3%). Depending on the symptoms of M. separata tested, the derivative 238 May shows anti-melting hormone effects [167].
Hou et al. connected methotrexate (MTX) with the hydrophobic drug podophyllotoxin (PPT) via a disulfide bond to obtain the amphiphilic drug–drug coupling prodrug (MTX-SS-PPT). The first two parent molecules of the drug self-assemble into stable nanoaggregates (NAs) in an aqueous solution, which realizes the self-delivery of the drug. Additionally, the presence of disulfide bonds in MTX-SS-PPT (239) can be controlled by using high concentrations of dithiothreitol (DTT). Intracellular mercaptan breaks disulfide bonds in MTX-SS-PPT (239), releasing drugs and killing tumor cells. Methotrexate-covered NAs can also target folate receptor-positive KB cells. Animal experiments have shown that methotrexate-covered NAs prodrug has good blood compatibility, and MTX-SS-PPT (239) NAs can reduce the size of xenograft tumors with few side effects [168].
2.8. Steroidal–Pyrazine Hybridization
Amelie Talbot et al. designed and synthesized acetyne-based steroid derivatives 240 and 241 (Figure 21) and evaluated the antitumor activity of these two compounds. The inhibition rates of compounds 240 and 241 reached 98% and 97% at the concentration of 10 μM. The inhibition rates of Jurkat cells reached 93% and 91% at the same concentration [169].
Compounds 242 and 243 showed significant inhibitory effects on PC-3 cell lines, with IC50 values of 6.88 and 0.93 μM. THLE-2 cells of compound 243 showed low cytotoxicity (IC50 = 26.70 μM, SI = 28.71). Compound 243 induced apoptosis of PC-3 cells in a dose-dependent manner and led to cell cycle stagnation in the G2/M phase [170].
Tao et al. reported the synthesis and antitumor activity of DHEA derived from C-16 ropyrrolidine. Compounds 244 and 245 showed the best activity with LC50 values less than 6.19 and 9.92 µg/mL, exceeding dehydroepiandrosterone activity (LC50 > 200 µg/mL) by using the brine shrimp test [171].
The D-ring fused 1,2,3-thiadiazole dehydroepiandrosterone derivative 246 showed moderate inhibitory activity in T-47D cells with an IC50 value of 3.04 μM. Compound 246 was not as active as dehydroepiandrosterone (IC50 = 2.55 μM) [172].
Steroidal C-17 pyrazine (247) showed moderate inhibitory activity against PC3-AR cell lines with IC50 of 366 nM. Compound 247 is also a potent CYP17 inhibitor with an IC50 value of 3.81 μM for CYP17 [173].
The 16-position aryl or heteraryl side chain of estrone is a potent inhibitor of 17β-HSD1. Among them, compound 248 (Figure 22) containing the pyrazine group showed an inhibitory effect on 17β-HSD1 with an IC50 value of 3.62 μM [174].
Ivanov et al. prepared a series of estrone-derived quinolines. Acetylenated estrone and its derivatives have significant biological activity as alkaline phosphatase inhibitors. Compounds 249, 250, and 252 were more potent TNAP inhibitors with IC50 values of 0.52, 0.48, and 0.25 µM, exceeding the activity of Levamisole (IC50 = 19.21 µM). Compounds 249, 251, and 252 are potent IAP inhibitors with IC50 values of 0.32, 0.92, and 0.44 µM, which exceeds the activity of L-phenylalanine (IC50 = 80.21 µM) [175].
Benoît et al. synthesized bimetallic Au(III)/Au(I) complexes with 17α-ethylestradiol as the carrier. The toxicity of estradiol-conjugated AuC6Estra (253) to estrogen receptor-positive (ER+) cancer cells was greater than that of ER-cancer cells and non-cancer cells. AuC6Estra (253) tested MCF-7 (ER+), MDAMB-231 (ER−), and MRC-5 (healthy fibroblasts) cells. The anti-proliferation effect of AuC6Estra (253) on ER+ cells was slightly higher than that in ER- and non-cancer cells [176].
Ananthan et al. synthesized a series of estrogen-functionalized copper complexes and studied them as electrochemically active DNA binding and splitting agents. The cytotoxic activity of these compounds was evaluated against estrogen receptor-positive (ER+) and negative (ER−) human cancer cell lines, and compounds 254–256 showed inhibitory effects against A2780, 2008, A431, MCF-7, and HCT-15 cell lines with IC50 values ranging from 0.04–2.00 μM. Complex 256 has a high intercalation interaction with nuclear DNA in vitro and is a strong DNA-cutting agent. Finally, complex 256 is involved in cellular redox stress by stimulating ROS production [177].
The steroid–pyrazine derivative 257–260 (Figure 23) showed significant antibacterial activity against two Gram-positive bacteria and two Gram-negative bacteria. The MIC value of compound 257 against the E. coli strain was 0.39 μM. The MIC value of compound 258 against the S. typhimurium and E. coli strains was 0.39 μM. The MIC value of compound 259 against the S. pyogenes strain was 0.39 μM. The MIC value of compound 260 against the B. aureus, S. typhimurium, and E. coli strains was 0.39 μM. The antibacterial activity of compound 257–260 was higher than that of the standard drug amoxicillin (MIC = 3.12 μM) [178].
Salman Ahmad Khan et al. found that the MIC values of compounds 261 and 262 against E. coli strains were 64 and 32 mg/mL, higher than the activity of cholesterol (MIC = 512 mg/mL). It was comparable to positive control chloramphenicol (MIC = 32 mg/mL) [179].
Khan et al. found that compounds 263 and 264 had significant antibacterial activity against two Gram-positive and two Gram-negative bacteria. The MIC values of compound 263 for S. aureus, S. pyogenes, S. typhimurium, and E. coli strains were 0.78, 0.78, 0.78, and 0.39 mg/mL. The MIC values of compound 264 were 0.78, 0.78, 0.39, and 0.39 mg/mL [180].
Compound 265 (Figure 24) showed moderate inhibitory activity against H37RvMa in MB7H9/ADC medium with a MIC90 value of 17.49 μM and low toxicity to CHO cells (IC50 > 50 μM) [181].
Stephen Barrett et al. used planephroline-modified aromatic ligands and copper (II) complexes of steroids (ethinynoestradiol and ethyl ketone) and screened these compounds for antimicrobial resistance against Staphylococcus aureus and methicillin-resistant Staphylococcus aureus (MRSA). Testosterone derivative 266 showed the strongest inhibitory effect on S. aureus, with a MIC50 of 1.5 μM. Estradiol derivative 267 showed the strongest inhibitory effect on MRSA, with a MIC50 of 17.5 μM [182].
Wang et al. synthesized the gaudatin–pyrazine derivative 268 and evaluated its anti-hepatitis B virus (HBV) activity in HepG-2 cells. Compound 268 not only inhibited the secretion of HBsAg (IC50 = 95.52 μM) and HBeAg (IC50 < 50.28 μM), but also inhibited the replication of HBV DNA (IC50 = 47.92 μM). Compound 268 showed low toxicity to HepG-2 cells, with a value of CC50 of 61.34 μM [183].
Compound 269 showed a neuroprotective effect on H2O2-induced SH-SY5Y cells, with a cell protective activity of 22.3% at 10 µM, more than diosgenin activity (6.7%) [184].
2.9. Terpene–Pyrazine Hybridization
Betulinic acid-linked ligustrazine derivative 270 (Figure 25) showed good antitumor activity, with IC50 values of 4.19, 5.23, 4.48, 4.23, and 4.34 μM against BEL-7402, HT-29, HepG-2, MCF-7, and HeLa cells. The cytotoxicity selective assay showed that 270 had low cytotoxicity to MDCK cells (IC50 > 20 μM). Fluorescence staining and flow cytometry analysis showed that 270 could induce HepG-2 cell apoptosis. Further studies showed that 270-induced apoptosis was mediated by depolarizing mitochondrial membrane potential and increasing intracellular free Ca2+ concentration [185].
The IC50 values of compound 271 against the BEL-7402, HepG-2, and HeLa cell lines were 4.065, 8.475, and 4.419 μM [186].
Xu et al. designed and synthesized a series of betulinic acid-linked ligustrazine derivatives, and screened their selective cytotoxic activity against five cancer cell lines (HepG-2, HT-29, HeLa, BCG-823, and A549) and nonmalignant cell lines MDCK using a standard MTT assay. Compounds 272 and 273 showed the strongest inhibitory effect on the BGC-823 cell line, with IC50 values of 0.84 and 1.49 μM. Compound 274 showed the highest cytotoxic activity against tumor cell lines (mean IC50 = 2.31 μM), and the strongest cytotoxic activity against HT-29 and HeLa with IC50 values of 1.70 and 1.74 μM. Further mechanism studies showed that 274-induced apoptosis was associated with the loss of mitochondrial membrane potential and increased intracellular free Ca2+ concentration [187].
The betulinic acid derivative 275 (Figure 26) inhibited the osteoclast differentiation of RAW.264.7 cells induced by RANKL, and the inhibitory rate reached 100% at 5 μM. The activity of betulinic acid was higher than that of betulinic acid (5 μM: 0%). Compound 275 still had a certain inhibitory effect at a concentration of 1.0 μM, and the inhibitory rate was 14.5% [188].
Compound 276 showed an obvious inhibitory effect in the A549 cell line with an IC50 value of 0.25 μM. Compound 277 showed the strongest inhibitory effect on HT-29, K562, K562-TAX, and B2-4 cell lines with IC50 values of 2, 0.4, 4, and 0.3 μM [189].
Haavikko et al. determined the leishmania activity of compound 276 using the alamarBlue aseptic flagellate activity assay against leishmania donovani. Compound 276 showed an obvious inhibitory effect on leishmania donovani with an IC50 value of 13.2 μM [190].
The betulinic acid derivatives 276, 278, and 279 showed significant inhibitory effects on two cancer cells (CCRF-CEM and HCT116). The IC50 values of compound 276 against CCRF-CEM and HCT116 cell lines were 0.5 and 11.06 μM. The IC50 values of compound 278 against the CCRF-CEM and HCT116 cell lines were 12.25 and 20.5 μM. The IC50 values of compound 279 against CCRF-CEM and HCT116 cell lines were 5.87 and 18.01 μM [191].
Hodon et al. synthesized a series of betulinic acid–pyrazine compounds and tested the cytotoxicity of these compounds in multiple cancer cell lines. Compounds (276–277 and 280–284) were preferentially and highly cytotoxic to leukemia cell lines (CCRF-CEM, K562, CEM-DNR, and K562-TAX) (IC50 between 0.43 and 18 μM). Compound 283 showed a significant inhibitory effect in CIM-DNR cells with IC50 of 0.49 μM. The IC50 activity of compound 284 against K562 cells was 0.026 μM. In addition, compounds 276, 280, and 282 inhibited the growth and degradation of HCT116 and HeLa cells in sphere cultures [192].
Compounds 285 and 286 have an obvious inhibition effect on 4T1 and MIA-PaCa-2 cell lines. The IC50 value of compounds 285 and 286 against 4T1 cell lines was 2.88 μM, which exceeded the activity of betulinic acid (IC50 = 6.29 μM). The IC50 values of MIA-PaCa-2 cell lines were 3.87 and 4.36 μM, which exceeded the activity of betulinic acid (IC50 = 25.63 μM) [193].
Pyrazine-thickened 23-hydroxyl betulinic acid, further modified by replacing C-28 carboxyl with ester and amide bonds, increased its antitumor activity. Compound 109 (Figure 27) showed the strongest activity against the cell lines SF-763, B16, and HeLa, with IC50 values of 3.53, 4.42, and 5.13 µM, respectively. In a preliminary mechanism study, 109 induced G1 phase cell arrest and significantly induced apoptosis of B16 cells in a dose-dependent manner. Furthermore, the in vivo antitumor activity of 109 was demonstrated in mice with H22 liver cancer and B16 melanoma (tumor inhibition rates were 55.6% and 62.7%, respectively) [194].
Compound 288 (Figure 28) showed moderate antitumor activity, with GI50 values of 32.6 µg/mL for IMR 32 (neuroblastoma) cell lines [195].
Betulinic acid–pyrazine derivative 289 showed significant anti-proliferation activity against HeLa and HepG-2 cell lines with IC50 values of 42 and 19 μM [196].
Bhandari et al. synthesized the pyrazine derivative 289 from lupinol and evaluated its anti-inflammatory activity by inhibiting NO production in LPS-induced RAW264.7 and J774A.1 cells. The IC50 of compound 289 inhibited NO production in RAW264.7 and J774A.1 cells was 32.4 μM [197].
The cytotoxicity of compound 290 in cancer cells (mean IC50 = 4.86 μM) was three times higher than that of normal cells (mean IC50 = 16.11 μM). The IC50 values of compound 290 against the HeLa, HepG-2, BGC-823, and HT-29 cell lines were 4.91, 4.07, 4.24, and 6.21 μM. Additionally, 290 was more cytotoxic to tumor cells than the positive drug cisplatin. Furthermore, 290 was more cytotoxic to tumor cells than its lead compound TB (291) and positive control cisplatin. Subsequently, fluorescence staining, apoptosis detection, and cell cycle analysis showed that 290 induced early apoptosis of HepG-2 cells and blocked the G1 phase cell cycle [198].
The antitumor component ligustrazine was combined with oleanolic acid to form TOA (292) (Figure 29). TOA showed a good anticancer effect in vitro [79], with IC50 values of 21.45 and 8.683 μM for HepG-2 and HeLa cell lines [199]. The IC50 values of the BEL-7402, HepG-2, HT-29, and HeLa cell lines were 6.359, 23.75, 8.339, and 23.77 μM [186].
TOA (292) blocks the expression of the nuclear transcription factors NF-κB/p65 and COX-2 in S180 mice [200]. Furthermore, acute toxicity tests confirmed that the LD50 of TOA (292) exceeded 6.0 g/kg by intragastric administration in mice. However, the poor hydrophilicity of TOA (292) limits its oral bioavailability [200].
Compound 293 showed good antitumor activity, with an IC50 value of 4.273 μM against the BEL-7402 cell line [186].
Ursolic acid-linked ligustrazine derivative 294 showed good antitumor activity, with IC50 values of 9.28, 8.43, 7.94, 5.69, and 4.37 μM for BEL-7402, HT-29, HepG-2, MCF-7, and HeLa cells. The cytotoxicity selective assay showed that 294 had low cytotoxicity to MDCK cells (IC50 = 16.39 μM) [185].
Chu et al. linked amino acids to TOA (292) via ester bonds and evaluated their cytotoxicity in four cancer cell lines (HepG-2, HT-29, HeLa, and BGC-823) using a standard MTT assay. Compounds 295 and 296 not only showed good cytotoxicity (IC50 < 3.5 μM), but also showed better hydrophilicity than TOA (292). Compound 295 showed the strongest inhibitory effect on the HepG-2 cell line with an IC50 value of 1.999 μM. Compound 296 showed the strongest inhibitory effect on HT-29, HeLa, and BGC-823 cell lines with IC50 values of 2.347, 2.383, and 2.481 μM. Furthermore, the nephrotoxicity of 6a (IC50 = 4.884 μM) to MDCK cells was lower than that of 296 (IC50 = 2.310 μM) and cisplatin (IC50 = 3.691 μM). Combination 296 can induce HepG-2 apoptosis through nuclear division and has low nephrotoxicity [199].
A series of hawthorn acid derivatives were synthesized by introducing various thickened heterocycles at C-2 and C-3. Their inhibitory effects on PTP1B, TCPTP, and related PTPS were evaluated. Compounds 297–299 (Figure 30) showed significant increases in inhibitory power and selectivity, and the three most potent PTP1B inhibitors, 297 (IC50 = 1.43 μM), 298 (IC50 = 1.79 μM), and 299 (IC50 = 1.78 μM), were shown to be about two times stronger than hawkic acid. Furthermore, 297–299 are 4.1, 4.6, and 3.1 times more selective for PTP1B than for TCPTP [201].
Oleanolic acid has been found to have an anti-bone resorption effect. The oleanolic acid derivative 300 inhibited the formation of osteoclast-like multinucleated cells (OCL) and showed quite strong activity even at 200 nM. The formation of oleanolic acid was only 34.1% compared to the control group (100.0%) [202].
Compound 301 is a novel molecule with a strong anti-osteoporosis effect in vivo. To study the molecular mechanism of 301, a novel fluorescent-labeled chemical probe with biological activity was designed and synthesized. Compared to 302, fluorescence compounds 303 and 304 showed a stronger inhibitory activity against RANKL-induced osteoclast differentiation in RAW264.7 cells at 2 μM, with an inhibition rate of 95.0% and 87.8%. Compounds 303 and 304 did not show cytotoxicity for the RAW264.7 cell line at a concentration of 10 μM [118].
The inhibitory activity of pyrazine-fused oleanolic acid derivatives on osteoclast formation induced by the nuclear factor-κB ligand receptor activator (RANKL) was evaluated using a cell-based tartrate-resistant acid phosphatase (TRAP) assay. The most potent compound 305 had an IC50 of 62.4 nM, and cytotoxicity in marine-derived bone monocytes/macrophages (BMDMs) indicated that inhibition of 305 in osteoclast differentiation was not due to its cytotoxicity. More importantly, 305 mitigated bone loss in bilateral ovariectomy mice, and preliminary mechanism studies showed that 305 affected the early stages of osteoclast genesis [203]. Furthermore, compounds 305–308 showed considerable inhibitory activity, inhibiting osteoclast formation by more than 80% at lower concentrations (0.5 μM) compared to the control group.
The results of α-glucosidase inhibitory activity in vitro showed that compound 309 showed certain inhibitory activity with IC50 values of 3.14 μM, respectively. However, the activity was inferior to oleanolic acid (IC50 = 2.41 μM) [204].
Derivative 310 (Figure 31) is a hederagenin derivative that binds to paclitaxel at 10 µM with an IC50 value of 2.4 nM against drug-resistant KBV cells. Derivative 310 can activate P-gp ATPase, resulting in the inability of drug-resistant cells to remove the drug from the body. Therefore, the derivative 310 can enhance the antitumor activity of paclitaxel in KBV cells, and the reversal effect of the drug is stronger than that of verapamil (IC50 = 4.9 nM). Furthermore, in vivo experiments showed that under combined use of paclitaxel (30 mg/kg) and derivative 310 (10 mg/kg), the body weight of nude xenograft mice decreased slightly, and tumor weight decreased to 41.88%. The results showed that derivative 310 reversed multidrug resistance by stimulating the ATPase activity of P-gp and then competing with chemotherapeutic drugs for binding to P-gp, but was less soluble due to the benzyl group at C-28 [205].
The cell assay showed that derivative 311 had the strongest antitumor reversion activity. When the derivative 311 (10 μM) was combined with paclitaxel (100 nM), the survival rate of the KBV cells reached 18.60%, surpassing that of compound 310 (19.64%) and hederagenin (149.47%) [206].
Compound 312 at 5 µM significantly improved the cytotoxicity of paclitaxel in resistant KBV cells and sensitized cells to paclitaxel, thus preventing cells from entering the G2/M phase and inducing apoptosis. Compound 312 may block the efflux of P-gp drugs by stimulating the activity of P-gp ATPase. In vivo experiments demonstrated that compound 312 increased the efficacy of paclitaxel in KBV cancer cell-derived xenograft tumors [207].
In order to improve the water solubility and tumor multidrug resistance reversal activity of 309, Wang et al. designed and synthesized a new series of hederagenin derivatives. These derivatives significantly reversed the multidrug resistance phenotype of KBV cells to paclitaxel at a concentration of 10 μM. The water solubility of PEGylated derivatives 313 increased approximately 20 fold compared to 310, while maintaining tumor multidrug resistance reversal activity. Therefore, pegylation is an effective method to improve water solubility while maintaining tumor multidrug resistance reversal activity [208]. Compound 313, the most active compound in vitro, showed good chemical stability to esterases within 24 h and increased the sensitivity of KBV cells to paclitaxel and vincristine with IC50 values of 4.58 and 0.79 nM, respectively. Compound 313 also increased the sensitivity of MCF-7T cells to paclitaxel and vincristine with IC50 values of 0.89 and 0.04 nM, respectively. The combination of compound 313 and paclitaxel significantly increased the apoptosis rate of KBV cells. Compound 313 treatment increased the accumulation of rhodamine 123 and Flutax1 in KBV and MCF-7T cells at 5 and 10 μM concentrations, suggesting that compound 313 played a role in reversing tumor resistance by effectively inhibiting the efflux function of P-gp [208].
Fang et al. designed and synthesized a series of hederagenin–pyrazine derivatives and screened the in vitro cytotoxicity of five tumor cell lines. The antitumor activity of compound 314 against A549 (IC50 = 3.45 µM) was comparable to that of cisplatin (IC50 = 3.85 µM). Compound 314 induced early apoptosis of A549 cells in a concentration-dependent manner and induced cell arrest in the S phase [209].
The results of α-glucosidase inhibitory activity in vitro showed that compound 315 (Figure 32) showed certain inhibitory activity with IC50 values of 7.84 μM, respectively. However, the activity was less than 3-carbonyl ursolic acid (IC50 = 2.47 μM) [210].
Tryptophan hydroxylase 1 (Tph-1) is the main enzyme in the biosynthesis of peripheral blood albumin, providing a new target for the design of anabolic agents for osteoporosis. Fu et al. synthesized a series of ursolic acid derivatives and bioevaluated them using RBL2H3 cells and ovariectomized rats. Among these compounds, compound 316 showed effective inhibitory activity against serotonin biosynthesis. Further studies showed that 316, as an effective Tph-1 binder identified by SPR (estimated KD: 6.82 μM), inhibited the protein and mRNA expression of Tph-1 and reduced the serum and intestinal serotonin content, but had no effect on brain serotonin. In addition, in ovariectomized rats, oral administration of 316 increased serum levels of N-terminal propeptide (a marker of bone formation) of type 1 procollagen (P1NP) and improved bone microstructure without estrogenic side effects [211].
The pyrazine derivatives 317 and 318 of boswellic acid showed obvious antitumor effects. The IC50 values of compound 317 on A2780, HT-29, and A375 cells were 15.7, 22.7, and 12.8 μM. The IC50 values of compound 318 were 13.7, 12.2, and 2.1 μM [212].
Wu et al. synthesized 18β-glycyrtinic acid derivative 319 by introducing piperazine into C-2 after the hydroxyl group in C-3 was oxidized and evaluated its antibacterial activity. Compound 319 pairs of Staphylococcus aureus (ATCC 6538), Staphylococcus aureus (ATCC 29213), and the strain Staphylococcus epidermidis (ATCC 12228) showed obvious inhibition, and the MIC50 and MBC50 values were 6.25 and 12.5 μM, respectively [213].
Compound 320 showed a greater inhibitory effect on Gram-positive bacteria than glycyrrhetinic acid. Questions about Staphylococcus aureussubsp aureus (ATCC 29213), Staphylococcus epidermidis (ATCC 12228), and the MIC value of Staphylococcus aureus (ATCC 6538) was 2.72 μg/mL. The inhibitory activity was similar to that of ampicillin. In vivo, compound 320 was also shown to have anti-inflammatory effects, and 40.0 mg/mL gavage reduced approximately 59.69% of TPA-induced ear edema in mice. Immunohistochemical results showed that inhibition was related to inhibition of TPA-induced upregulation of the pro-inflammatory cytokines TNF-α and IL-1β. Furthermore, compound 320 significantly reduced the expression level of p65 in the NF-κB signaling pathway [214].
Xu et al. synthesized a series of C14 heterocyclic substituted epi-triptolide derivatives. Among them, the pyrazine derivatives 321 (Figure 33) showed certain inhibitory effects on SKOV-3 and PC-3 cell lines, with IC50 values of 368.7 nM and 157 nM. The activity was higher than epitriptolide (SKOV-3, IC50 = 790 nM; PC-3, IC50 = 1320 nM) [215].
Wei et al. obtained derivative 322 by esterification and etherification of 14-dehydroxy-11,12-didehydroandrographolide. Compound 322 showed obvious inhibitory effects on A549, DU145, KB, and KBVin cell lines with IC50 values of 4.87, 8.63, 8.24, and 9.19 μM. The activity of compound 322 exceeded that of andrographolide (A549, IC50 = 13.37 μM; DU145, IC50 = 15.99 μM; KB, IC50 = 13.18 μM; KBVin, IC50 = 13.82 μM) [216].
Grigoropoulou et al. synthesized a series of dehydroabietic acid–chalketone heterozygotes. Compound 323 containing pyrazine showed certain inhibitory effects on MDA-MB-231 and Hs578T cell lines with IC50 values of 18.01 and 23.11 μM. However, compound 323 also showed some toxicity to fibroblasts with an IC50 value of 20.47 μM [217].
Zhao et al. synthesized a series of dehydroabietylamine derivatives containing pyrazine cycloheterocyclic rings. Anti-MCF-7 activity of compounds 324 and 325 increased (IC50: 8.81 and 6.66 μM) compared to dehydroabietylamine (IC50: 19.45 μM). However, the activity of 324 and 325 against other cells such as HeLa, HepG-2, A549, and HUVEC was lower than that of dehydroabietylamine [218].
Wang et al. designed and synthesized ligustrazine–deoxycholic acid/cholic acid derivatives as antitumor drugs. The IC50 values of compounds 326 and 327 against the BEL-7402, A549, HCT-8, BGC-823, and A2780 cell lines were 5.472–8.012 μg/mL [79].
Zhao et al. found that 2-Pyrazine-PPD (328) showed inhibitory activity in the gastric cancer BGC-823 cell line (IC50 = 11.52 μM). There is little toxicity to normal cells (human gastric epithelial cell line GES-1). Further studies showed that 2-Pyrazine-PPD (328) induced apoptosis of BGC-803 cancer cells through the mitochondrial pathway. Ros production was significantly increased in BGC-803 cancer cells treated with 2-Pyrazine-PPD (328). Therefore, 2-Pyrazine-PPD (328) exhibits anticancer activity through ROS-mediated apoptosis of gastric cancer cells and stress of the endoplasmic reticulum [219].
Xu et al. introduced piperazine in C-14 to synthesize Rabbesin derivatives for 329 (Figure 34) and evaluated their antibacterial activity. Compound 329 showed significant inhibitory effects on Mycobacterium phlei (ATCC 355), Mycobacterium smegmatis (ATCC 19420), and Mycobacterium marinum (ATCC 927). The MIC50 values are 4, 32, and 32 μM [220].
Xu et al. synthesized 330, an enmein derivative containing pyrazine, and studied its bacteriostatic effect. Compound 330 showed an obvious inhibitory effect on Mycobacterium phlei (ATCC 355) with a MIC50 of 2 μg/mL [221].
Chen et al. synthesized the C-16 carbonyl derivative 331 using isostevia as the raw material and evaluated its anticoagulant activity. In vitro activity of human FXa showed that 331 (Ki = 3.603 μM) showed relatively better inhibitory activity than isosteviol (Ki = 13.4 μM) [222].
Compound 332 showed obvious inhibitory activity against HIV replication in H9 lymphocytes with an IC50 value of 5.6 μg/mL more than linearol activity (IC50 = 56.5 μg/mL) [223].
Alla D. Zorina et al. tested the antiviral activity of pyrazine triterpenoids against the influenza virus A/Puerto Rico/8/34 (H1N1). The CC50 and IC50 values of compound 333 pairs of influenza virus A/Puerto Rico/8/34 (H1N1) were 21.2 and >11 μg mL−1. The CC50 and IC50 values of compound 334 were 34.2 and 8.2 μg mL−1. The IC50 values of compound 335 were 33 μg mL−1 [196].
The results of α-glucosidase inhibitory activity in vitro showed that compound 334 exhibited certain inhibitory activity, with IC50 values of 97.7 μM, respectively, which exceeded the activity of acarbose (IC50 = 397.6 μM) [224].
2.10. Alkaloid–Pyrazine Hybridization
Nishiyama et al. synthesized a 4-pyrazine substituted colchicine derivative 336 (Figure 35). Compound 336 showed moderate cytotoxicity against three human cancer cell lines (A549, HT-29, and HCT116), with IC50 values of 37.4, 19.4, and 33.0 μM [225].
In order to improve the antitumor activity of camptothecin, Li et al. designed and synthesized a series of 10 substituted camptothecin derivatives. Compound 337 showed the strongest inhibitory effect on the MCF-7 cell line with an IC50 value of 0.1 μM. Compound 338 showed the strongest inhibitory effect on HCT-8 and BEL-7402 cell lines with IC50 values of 0.08 and 0.26 μM [226].
Sinomenine derivatives showed stronger TNF-α inhibitory activity than sinomenine. Compounds 339–343 inhibited the production of TNF-α in mice peritoneal macrophages stimulated by LPS in vitro at a concentration of 10 μM, and the inhibitory rate was more than 95% [227].
Compound 344 inhibited TNF-α-induced NF-κB activation in a dose-dependent manner and showed a significant in vivo therapeutic effect on mice models of experimental autoimmune uveitis disease [228].
Watanabe et al. reported on a morphinan derivative of the oxazatricyclodecane skeleton and tested its opioid receptor agonist activity. Pyrazine-containing compound 345 (Figure 36) showed a high affinity for all types of receptors (DOR, MOR, and KOR) [229].
Ananthan et al. synthesized pyridomorphinans with aromatic or heterocyclic substitutions at the 5′ position of the morphinan pyridine ring and evaluated the binding and functional activity of opioid receptors δ, μ, and κ. Pyrazine-containing compounds 346 and 347 show significant affinity for these three receptors, with Ki values ranging from 2.3 to 16 nM.
The spinal selective mu-kappa agonist NNTA selectively activates the mu-kappa isomer in HEK-293 cells and produces an unusually potent antipain response after administration in the mouse intrasheath (i.t.) [230]. The quinoline analog 348 of NNTA is a potent agonist (ED50 values of 50.76 and 757.2 pmol, respectively) with a 15-fold increase in spinal cord efficacy in both in vitro and in vitro perfusion pathways. In particular, 348 showed no significant tolerance to either mode of administration [231].
3. Conclusions
Natural products derived from microorganisms, plants, and animals are a rich source of effective drugs with significant structural diversity and biological properties, which offer the possibility for researchers to develop new molecules to treat disease [232]. Over the past few decades, drugs that directly or indirectly replace natural product derivatives and analogs have played an important role in the fight against disease [233]. Natural products have a wide range of biological activities, but their activity is not strong and often has the shortcomings of low bioavailability and poor solubility [234]. Therefore, to improve their physical properties and ADME, it is important to make the necessary structural modifications to them. Through a literature review, it is found that pyrazine stents have considerable biological relevance in anti-osteoporosis, antiviral, anti-diabetic, anti-inflammatory, anti-thrombotic, anti-parasitic, anti-malaria, antibacterial, anticancer, and other studies, leading to the emergence of many strong bioactive pyrazine compounds.
Although much progress has been made in the pharmacochemistry of natural product–pyrazine complexes, the following research opportunities remain to be further investigated: Since many studies have not reported detailed studies of structural modifications of lead compounds, a complete SAR study of natural product–pyrazine complexes is needed. Detailed SAR studies may reveal more active compounds. Furthermore, because pyrazines have a variety of biological activities and multitarget properties, most natural product–pyrazine derivatives do not have specific drug adaptability, hindering the development of these derivatives from laboratory to clinical applications; therefore, the in vivo activity of the natural product–pyrazine hybrid needs to be evaluated. Additionally, most natural products–pyrazine derivatives have no clear targets. To solve this problem, using reasonable drug design strategies such as computer-aided drug design, the target identification of a powerful natural product–pyrazine hybrid was studied, and the compounds were optimized. Finally, pharmacophore combinations and structure-based drug design strategies should be widely used in the following studies to develop more novel, more active, and more specific natural products–pyrazine derivatives, and the mechanism of action of these compounds should be studied in detail.
In conclusion, this review provides a favorable reference for the study of compounds containing pyrazine fragments. Pyrazine derivatives of natural products still have a wide range of research prospects and are worth further research and development. With the rapid development of combinatorial chemistry, rational drug design, and chemical proteomics, we believe that researchers will find more novel derivatives of pyrazines with good biological activity and wide application.
Author Contributions
Conceptualization and review methodologies—G.-Q.C., H.-Y.G. and T.L.; Original draft writing—G.-Q.C. and T.L.; Editing—All authors; Figure creation—All authors; Revision—T.L. and X.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was financially supported by the Educational Department of Liaoning Province (No. LJKMZ20221801), the Doctoral Research Foundation of Shenyang Medical College (No. 20205041), the Higher Education Discipline Innovation Project (D18012), the National Natural Science Foundation of China (Nos. 81960626, 82060628, 82204310), the Key Projects of Jilin Province Science and Technology Development Plan (No. 20200404130YY), the Jilin Scientific and Technological Development Program (Nos. YDZJ202301ZYTS440, YDZJ202301ZYTS143), and the Education Department Project of Jilin (JJKH20220559KJ, JJKH20220563KJ).
Conflicts of Interest
The authors declare no conflict of interest.
List of Abbreviations
| HCV | Hepatitis C virus |
| RdRp | RNA-dependent RNA polymerase |
| SRB | Sulforhodamine B |
| HMEC-2 | Human microvascular endothelial cell line |
| SH-SY5Y | Human neuroblastoma cell line |
| ChEs | Cholinesterase |
| ROS | reactive oxygen species |
| Cyt-c | Cytochrome-C |
| ADP | Adenosine diphosphate |
| PARP | Poly(ADP-ribose) polymerase |
| MCAO | Middle cerebral artery occlusion |
| HBV | Hepatitis B virus |
| Tph-1 | Tryptophan hydroxylase 1 |
| ADME | Absorption, distribution, metabolism, excretion |
| LPS | Lipopolysaccharide |
| NO | Nitric oxide |
| NAs | Nanoaggregates |
| DTT | Dithiothreitol |
| PPT | Podophyllotoxin |
References
- Saleh, S.S.; AL-Salihi, S.S.; Mohammed, I.A. Biological activity Study for some heterocyclic compounds and their impact on the gram positive and negative bacteria. Energy Procedia 2019, 157, 296–306. [Google Scholar] [CrossRef]
- Barreca, M.; Spanò, V.; Rocca, R.; Bivacqua, R.; Gualtieri, G.; Raimondi, M.V.; Gaudio, E.; Bortolozzi, R.; Manfreda, L.; Bai, R.; et al. Identification of pyrrolo[3′,4’:3,4]cyclohepta[1,2-d][1,2]oxazoles as promising new candidates for the treatment of lymphomas. Eur. J. Med. Chem. 2023, 254, 115372. [Google Scholar] [CrossRef] [PubMed]
- Bivacqua, R.; Barreca, M.; Spanò, V.; Raimondi, M.V.; Romeo, I.; Alcaro, S.; Andrei, G.; Barraja, P.; Montalbano, A. Insight into non-nucleoside triazole-based systems as viral polymerases inhibitors. Eur. J. Med. Chem. 2023, 249, 115136. [Google Scholar] [CrossRef] [PubMed]
- Huigens, R.W., III; Brummel, B.R.; Tenneti, S.; Garrison, A.T.; Xiao, T. Pyrazine and Phenazine Heterocycles: Platforms for Total Synthesis and Drug Discovery. Molecules 2022, 27, 1112. [Google Scholar] [CrossRef]
- Miniyar, P.B.; Murumkar, P.R.; Patil, P.S.; Barmade, M.A.; Bothara, K.G. Unequivocal Role of Pyrazine Ring in Medicinally Important Compounds: A Review. Mini-Rev. Med. Chem. 2013, 13, 1607–1625. [Google Scholar] [CrossRef]
- Choudhary, D.; Garg, S.; Kaur, M.; Sohal, H.S.; Malhi, D.S.; Kaur, L.; Verma, M.; Sharma, A.; Mutreja, V. Advances in the Synthesis and Bio-Applications of Pyrazine Derivatives: A Review. Polycycl. Aromat. Compd. 2023, 43, 4512–4578. [Google Scholar] [CrossRef]
- Masaret, G.S.; Farghaly, T.A.; Al-Hussain, S.A.; Zaki, M.E.A.; Alsaedi, A.M.R.; Muhammad, Z.A. Site-Selectivity of the Reaction of 3-Amino-4-Cyano-5-Phenyl-1H-Pyrrole-2-Carboxylic Acid Amide with α-Halocarbonyl Compounds. Antimicrobial Activity and Docking Study for COVID-19 of the Products. Polycycl. Aromat. Compd. 2023, 43, 7110–7126. [Google Scholar] [CrossRef]
- Christie, A.W.; McCormick, D.K.T.; Emmison, N.; Kraemer, F.B.; Alberti, K.G.M.M.; Yeaman, S.J. Mechanism of anti-lipolytic action of acipimox in isolated rat adipocytes. Diabetologia 1996, 39, 45–53. [Google Scholar] [CrossRef]
- Loffing, J.; Kaissling, B. Sodium and calcium transport pathways along the mammalian distal nephron: From rabbit to human. Am. J. Physiol. 2003, 284, F628. [Google Scholar] [CrossRef]
- Chalfant, M.L.; Peterson-Yantorno, K.; O’Brien, T.G.; Civan, M.M. Regulation of epithelial Na+ channels from M-1 cortical collecting duct cells. Am. J. Physiol. 1996, 271, F861. [Google Scholar] [CrossRef]
- Adams, J.; Kauffman, M. Development of the Proteasome Inhibitor Velcade (Bortezomib). Cancer Investig. 2004, 22, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Curran, M.P.; McKeage, K. Bortezomib: A review of its use in patients with multiple myeloma. Drugs 2009, 69, 859–888. [Google Scholar] [CrossRef]
- Kouroukis, T.C.; Baldassarre, F.G.; Haynes, A.E.; Imrie, K.; Reece, D.E.; Cheung, M.C. Bortezomib in multiple myeloma: Systematic review and clinical considerations. Curr. Oncol. 2014, 21, e573–e603. [Google Scholar] [CrossRef]
- Prendergast, B.D. Glyburide and glipizide, second-generation oral sulfonylurea hypoglycemic agents. Clin. Pharm. 1984, 3, 473–485. [Google Scholar] [PubMed]
- Bonanni, G.; Ciccariello, M.; Mancini, P.; Pace, V.; Sagliaschi, G. Concomitant ceco-appendicular and urinary tuberculosis. Description of two rare cases: Physiopathological and diagnostic remarks. Riv. Eur. Sci. Med. Farmacol. 1993, 15, 171–174. [Google Scholar] [PubMed]
- Zimhony, O.; Cox, J.S.; Welch, J.T.; Vilcheze, C.; Jacobs, W.R., Jr. Pyrazinamide inhibits the eukaryotic-like fatty acid synthetase I (FASI) of Mycobacterium tuberculosis. Nat. Med. 2000, 6, 1043–1047. [Google Scholar] [CrossRef]
- Iida, K.; Itoh, K.; Kumagai, Y.; Oyasu, R.; Hattori, K.; Kawai, K.; Shimazui, T.; Akaza, H.; Yamamoto, M. Nrf2 Is Essential for the Chemopreventive Efficacy of Oltipraz against Urinary Bladder Carcinogenesis. Cancer Res. 2004, 64, 6424–6431. [Google Scholar] [CrossRef]
- Barth, F. CB1 cannabinoid receptor antagonist. Annu. Rep. Med. Chem. 2005, 40, 103–118. [Google Scholar]
- Foreman, M.M.; Hanania, T.; Stratton, S.C.; Wilcox, K.S.; White, H.S.; Stables, J.P.; Eller, M. In vivo pharmacological effects of JZP-4, a novel anticonvulsant, in models for anticonvulsant, antimania and antidepressant activity. Pharmacol. Biochem. Behav. 2008, 89, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.J.; Wu, P.; Spurden, D.; Ebbert, J.O.; Wilson, K. Efficacy of pharmacotherapies for short-term smoking abstinance: A systematic review and meta-analysis. Harm. Reduct. J. 2009, 6, 25. [Google Scholar] [CrossRef]
- James, N.D.; Growcott, J.W. ZD4054. Drugs Future 2009, 34, 624–633. [Google Scholar] [CrossRef]
- De la Rosa, D.A.; Canessa, C.M.; Fyfe, G.K.; Zhang, P. Structure and regulation of amiloride-sensitive sodium channels. Annu. Rev. Physiol. 2000, 62, 573–594. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Serby, M.; Reed, A.; Lee, A.J.; Zhang, X.; Marsh, K.; Khatri, A.; Menon, R.; Kavetskaia, O.; Fischer, V. Metabolism and disposition of the hepatitis C protease inhibitor paritaprevir in humans. Drug Metab. Dispos. 2016, 44, 1164–1173. [Google Scholar] [CrossRef]
- Monti, J.M.; Pandi-Perumal, S.R. Eszopiclone: Its use in the treatment of insomnia. Neuropsychiatr. Dis. Treat. 2007, 3, 441–453. [Google Scholar] [PubMed]
- McCrae, C.S.; Ross, A.; Stripling, A.; Dautovich, N.D. Eszopiclone for late-life insomnia. Clin. Investig. Aging 2007, 2, 313–326. [Google Scholar]
- Balogh, A. Drug for the treatment of sleep disorders—Review. Z. Arztl. Fortbild. Qualitatssich. 2001, 95, 11–16. [Google Scholar]
- Lee, S.; LaCour, T.G.; Fuchs, P.L. Chemistry of trisdecacyclic pyrazine antineoplastics: The cephalostatins and ritterazines. Chem. Rev. 2009, 109, 2275–2314. [Google Scholar] [CrossRef] [PubMed]
- Fukuzawa, S.; Matsunaga, S.; Fusetani, N. Isolation and structure elucidation of ritterazines B and C, highly cytotoxic dimeric steroidal alkaloids, from the tunicate Ritterella tokioka. J. Org. Chem. 1995, 60, 608. [Google Scholar] [CrossRef]
- Guo, Q.; Xu, M.; Guo, S.; Zhu, F.; Xie, Y.; Shen, J. The complete synthesis of favipiravir from 2-aminopyrazine. Chem. Pap. 2019, 73, 1043–1051. [Google Scholar] [CrossRef]
- Agrawal, U.; Raju, R.; Udwadia, Z.F. Favipiravir: A new and emerging antiviral option in COVID-19. Med. J. Armed Forces India 2020, 76, 370–376. [Google Scholar] [CrossRef]
- Manabe, T.; Kambayashi, D.; Akatsu, H.; Kudo, K. Favipiravir for the treatment of patients with COVID-19: A systematic review and meta-analysis. BMC Infect. Dis. 2021, 21, 489. [Google Scholar] [CrossRef] [PubMed]
- Chill, L.; Aknin, M.; Kashman, Y. Barrenazine A and B; two new cytotoxic alkaloids from an unidentified tunicate. Org. Lett. 2003, 5, 2433–2435. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.H.; Chen, C.; Wang, H.; Ye, W.C.; Zhou, G.X. Indole alkaloids from Alocasia macrorrhiza. Chem. Pharm. Bull. 2012, 60, 670–673. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, A.; Hermanson, M.; Wickström, M.; Lindhagen, E.; Ekholm, C.; Jenmalm Jensen, A.; Löthgren, A.; Lehmann, F.; Larsson, R.; Parrow, V.; et al. The novel tyrosine kinase inhibitor AKN-028 has significant antileukemic activity in cell lines and primary cultures of acute myeloid leukemia. Blood Cancer J. 2012, 2, e81. [Google Scholar] [CrossRef]
- Bremberg, U.; Eriksson-Bajtner, J.; Lehmann, F.; Oltner, V.; Soelver, E.; Wennerberg, J. Development of a Synthesis of Kinase Inhibitor AKN028. Org. Process Res. Dev. 2018, 22, 1360–1364. [Google Scholar] [CrossRef]
- Duran, R.; Zubia, E.; Ortega, M.J.; Naranjo, S.; Salva, J. Novel alkaloids from the red ascidian Botryllus leachi. Tetrahedron 1999, 55, 13225–13232. [Google Scholar] [CrossRef]
- Saito, R.; Tokita, M.; Uda, K.; Ishikawa, C.; Satoh, M. Synthesis and in vitro evaluation of botryllazine B analogues as a new class of inhibitor against human aldose reductase. Tetrahedron 2009, 65, 3019–3026. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, Y.; Qiao, J.; Yu, W.; Pan, X.; Zhang, T.; Liu, Y.; Lu, S.E. Phenazine-1-carboxylic Acid Produced by Pseudomonas chlororaphis YL-1 Is Effective against Acidovorax citrulli. Microorganisms 2021, 9, 2012. [Google Scholar] [CrossRef]
- Conda-Sheridan, M.; Marler, L.; Park, E.J.; Kondratyuk, T.P.; Jermihov, K.; Mesecar, A.D.; Pezzuto, J.M.; Asolkar, R.N.; Fenical, W.; Cushman, M. Potential chemopreventive agents based on the structure of the lead compound 2-bromo-1-hydroxyphenazine, isolated from Streptomyces species, strain CNS284. J. Med. Chem. 2010, 53, 8688–8699. [Google Scholar] [CrossRef]
- Fotso, S.; Santosa, D.A.; Saraswati, R.; Yang, J.; Mahmud, T.; Zabriskie, T.M.; Proteau, P.J. Modified phenazines from an Indonesian Streptomyces sp. J. Nat. Prod. 2010, 73, 472–475. [Google Scholar] [CrossRef]
- Abouelhassan, Y.; Zhang, Y.; Jin, S.; Huigens, R.W., III. Transcript Profiling of MRSA Biofilms Treated with a Halogenated Phenazine Eradicating Agent: A Platform for Defining Cellular Targets and Pathways Critical to Biofilm Survival. Angew. Chem. Int. Ed. Engl. 2018, 57, 15523–15528. [Google Scholar] [CrossRef]
- Sletta, H.; Degnes, K.F.; Herfindal, L.; Klinkenberg, G.; Fjaervik, E.; Zahlsen, K.; Brunsvik, A.; Nygaard, G.; Aachmann, F.L.; Ellingsen, T.E.; et al. Anti-microbial and cytotoxic 1,6-dihydroxyphenazine-5,10-dioxide (iodinin) produced by Streptosporangium sp. DSM 45942 isolated from the fjord sediment. Appl. Microbiol. Biotechnol. 2014, 98, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Guiza Beltran, D.; Schacht, A.; Wright, S.; Zhang, L.; Du, L. Functional and Structural Analysis of Phenazine O-Methyltransferase LaPhzM from Lysobacter antibioticus OH13 and One-Pot Enzymatic Synthesis of the Antibiotic Myxin. ACS Chem. Biol. 2018, 13, 1003–1012. [Google Scholar] [CrossRef] [PubMed]
- Avtsyn, A.P.; Pershin, G.N.; Trager, R.S.; Shakhlamov, V.A.; Padeĭskaia, E.N. Activity of di-N-hydroxyquinoxaline and depot-sulfanilamide derivatives in experimental NAG infection. Biull. Eksp. Biol. Med. 1982, 93, 76–78. [Google Scholar] [CrossRef]
- Samata, K.; Yamagishi, T.; Ichihara, T.; Nanaumi, K.; Ikeda, T.; Ikeya, H.; Kuraishi, A.; Nakaike, S.; Kashiwagi, K.; Igarashi, K. Establishment and characterization of a mouse FM3A cell mutant resistant to topoisomerase II-inhibitor NC-190. Cancer Chemother. Pharmacol. 2002, 50, 367–372. [Google Scholar] [CrossRef]
- Tarui, M.; Doi, M.; Ishida, T.; Inoue, M.; Nakaike, S.; Kitamura, K. DNA-binding characterization of a novel antitumor benzo[a]phenazine derivative NC-182: Spectroscopic and viscometric studies. Biochem. J. 1994, 304, 271. [Google Scholar] [CrossRef]
- Montazeri, K.; Bellmunt, J. Erdafitinib for the treatment of metastatic bladder cancer. Expert Rev. Clin. Pharmacol. 2020, 13, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Weng, W.; Hong, J.; Owusu-Ansah, K.G.; Chen, B.; Zheng, S.; Jiang, D. Pralatrexate mediates effective killing of gemcitabine-resistant pancreatic cancer: Role of mTOR/4E-BP1 signal pathway. Heliyon 2022, 8, e12064. [Google Scholar] [CrossRef]
- Widemann, B.C.; Adamson, P.C. Understanding and managing methotrexate nephrotoxicity. Oncologist 2006, 11, 694–703. [Google Scholar] [CrossRef]
- Bogani, G.; Monk, B.J.; Coleman, R.L.; Vergote, I.; Oakin, A.; Ray-Coquard, I.; Mariani, A.; Scambia, G.; Raspagliesi, F.; Bolognese, B. Selinexor in patients with advanced and recurrent endometrial cancer. Curr. Probl. Cancer 2023, 100963. [Google Scholar] [CrossRef]
- Mizuta, H.; Okada, K.; Araki, M.; Adachi, J.; Takemoto, A.; Kutkowska, J.; Maruyama, K.; Yanagitani, N.; Oh-Hara, T.; Watanabe, K.; et al. Gilteritinib overcomes lorlatinib resistance in ALK-rearranged cancer. Nat. Commun. 2021, 12, 1261. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.J.; Huang, C.F.; Yu, M.L. Elbasvir and grazoprevir for the treatment of hepatitis C. Expert Rev. Anti-Infect. Ther. 2021, 19, 1071–1081. [Google Scholar] [CrossRef]
- Muir, A.J. Telaprevir for the treatment of chronic hepatitis C infection. Expert Rev. Anti-Infect. Ther. 2011, 9, 1105–1114. [Google Scholar] [CrossRef] [PubMed]
- Horisberger, J.D.; Giebisch, G. Potassium-sparing diuretics. Kidney Blood Press. Res. 1987, 10, 198–220. [Google Scholar] [CrossRef] [PubMed]
- van Gool, J.D.; Hirche, H.; Lax, H.; De Schaepdrijver, L. Folic acid and primary prevention of neural tube defects: A review. Reprod. Toxicol. 2018, 80, 73–84. [Google Scholar] [CrossRef]
- Sitbon, O.; Channick, R.; Chin, K.M.; Frey, A.; Gaine, S.; Galiè, N.; Ghofrani, H.A.; Hoeper, M.M.; Lang, I.M.; Preiss, R.; et al. Selexipag for the Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2015, 373, 2522–2533. [Google Scholar] [CrossRef]
- Hussain, Z.; Zhu, J.; Ma, X. Metabolism and Hepatotoxicity of Pyrazinamide, an Antituberculosis Drug. Drug Metab. Dispos. 2021, 49, 679–682. [Google Scholar] [CrossRef]
- Fiori, G.; Saglini, V.; Bertini, F.; Domenighetti, G.; Mombelli, G. Severe poisoning with the organophosphorus insecticide thionazine--2 cases with the development of adult respiratory distress syndrome (ARDS). Schweiz. Med. Wochenschr. 1987, 117, 399–401. [Google Scholar]
- Chen, W.; Chen, W.; Zhu, J.; Chen, N.; Lu, Y. Potent Anti-Inflammatory Activity of Tetramethylpyrazine Is Mediated through Suppression of NF-k. Iran. J. Pharm. Res. IJPR 2016, 15, 197–204. [Google Scholar]
- Lidin-Janson, G. Sulphonamides in the treatment of acute Escherichia coli infection of the urinary tract in women. Clinical and ecological effects of sulphasomidine and sulphalene. Scand. J. Infect. Dis. 1977, 9, 211–217. [Google Scholar] [CrossRef]
- Oh, D.J.; Chen, J.L.; Vajaranant, T.S.; Dikopf, M.S. Brimonidine tartrate for the treatment of glaucoma. Expert Opin. Pharmacother. 2019, 20, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Foster, B.J.; Clagett-Carr, K.; Shoemaker, D.D.; Suffness, M.; Plowman, J.; Trissel, L.A.; Grieshaber, C.K.; Leyland-Jones, B. Echinomycin: The first bifunctional intercalating agent in clinical trials. Investig. New Drugs 1985, 3, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Fisherman, J.S.; Osborn, B.L.; Chun, H.G.; Plowman, J.; Smith, A.C.; Christian, M.C.; Zaharko, D.S.; Shoemaker, R.H. Chloroquinoxaline sulfonamide: A sulfanilamide antitumor agent entering clinical trials. Investig. New Drugs 1993, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Clanner-Engelshofen, B.M.; Bernhard, D.; Dargatz, S.; Flaig, M.J.; Gieler, U.; Kinberger, M.; Klövekorn, W.; Kuna, A.C.; Läuchli, S.; Lehmann, P.; et al. S2k guideline: Rosacea. J. Dtsch. Dermatol. Ges. 2022, 20, 1147–1165. [Google Scholar] [CrossRef]
- Moore, D.J.; Brodfuehrer, J.I.; Wilke, T.J.; Powis, G. Disposition and metabolism of the antitumor agent pyrazine-2-diazohydroxide in mouse and beagle dog. Cancer Chemother. Pharmacol. 1988, 21, 269. [Google Scholar] [CrossRef]
- Mori, M.; Supuran, C.T. Acipimox inhibits human carbonic anhydrases. J. Enzym. Inhib. Med. Chem. 2022, 37, 672–679. [Google Scholar] [CrossRef]
- Williams, R.L.; Trenholme, G.M.; Carson, P.E.; Frischer, H.; Rieckmann, K.H. The influence of acetylator phenotype on the response to sulfalene in individuals with chloroquine-resistant falciparum malaria. Am. J. Trop. Med. Hyg. 1978, 27, 226–231. [Google Scholar] [CrossRef] [PubMed]
- France, C.P.; Winger, G.; Medzihradsky, F.; Seggel, M.R.; Rice, K.C.; Woods, J.H. Mirfentanil: Pharmacological profile of a novel fentanyl derivative with opioid and nonopioid effects. J. Pharmacol. Exp. Ther. 1991, 258, 502–510. [Google Scholar]
- Beaulieu, P.L.; Gillard, J.; Jolicoeur, E.; Duan, J.; Garneau, M.; Kukolj, G.; Poupart, M.A. From benzimidazole to indole-5-carboxamide Thumb Pocket I inhibitors of HCV NS5B polymerase. Part 1: Indole C-2 SAR and discovery of diamide derivatives with nanomolar potency in cell-based subgenomic replicons. Bioorg. Med. Chem. Lett. 2011, 21, 3658–3663. [Google Scholar] [CrossRef] [PubMed]
- Rong, F.; Chow, S.; Yan, S.; Larson, G.; Hong, Z.; Wu, J. Structure-activity relationship (SAR) studies of quinoxalines as novel HCV NS5B RNA-dependent RNA polymerase inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 1663–1666. [Google Scholar] [CrossRef]
- Ma, S.; Deng, J.; Li, B.; Li, X.; Yan, Z.; Zhu, J.; Chen, G.; Wang, Z.; Jiang, H.; Miao, L.; et al. Development of Second-Generation Small-Molecule RhoA Inhibitors with Enhanced Water Solubility, Tissue Potency, and Significant in vivo Efficacy. ChemMedChem 2015, 10, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Feng, E.; Ma, S.; Zhang, Y.; Liu, X.; Li, H.; Huang, H.; Zhu, J.; Zhu, W.; Shen, X.; et al. Design and synthesis of small molecule RhoA inhibitors: A new promising therapy for cardiovascular diseases? J. Med. Chem. 2011, 54, 4508–4522. [Google Scholar] [CrossRef] [PubMed]
- Qian, K.; Wang, L.; Cywin, C.L.; Farmer, B.T.; Hickey, E.; Homon, C.; Jakes, S.; Kashem, M.A.; Lee, G.; Leonard, S.; et al. Hit to Lead Account of the Discovery of a New Class of Inhibitors of Pim Kinases and Crystallographic Studies Revealing an Unusual Kinase Binding Mode. J. Med. Chem. 2009, 52, 1814–1827. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.X.; Wang, H.; Cui, H.R.; Guo, W.B.; Zhou, F.; Cai, D.S.; Xu, B.; Jia, X.H.; Huang, X.M.; Yang, Y.Q.; et al. Design, synthesis and biological evaluation of cinnamic acid derivatives with synergetic neuroprotection and angiogenesis effect. Eur. J. Med. Chem. 2019, 183, 111695. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Hong, G.; Li, X.; Zhang, Y.; Xu, Z.; Mao, L.; Feng, X.; Liu, T. Synthesis and activity towards Alzheimer’s disease in vitro: Tacrine, phenolic acid and ligustrazine hybrids. Eur. J. Med. Chem. 2018, 148, 238–254. [Google Scholar] [CrossRef]
- Wang, P.; Zhao, R.; Yan, W.; Zhang, X.; Zhang, H.; Xu, B.; Chu, F.; Han, Y.; Li, G.; Liu, W.; et al. Neuroprotection by new ligustrazine-cinnamon acid derivatives on CoCl2-induced apoptosis in differentiated PC12 cells. Bioorg. Chem. 2018, 77, 360–369. [Google Scholar] [CrossRef]
- Chen, H.; Li, G.; Zhan, P.; Liu, X. Ligustrazine derivatives. Part 5: Design, synthesis and biological evaluation of novel ligustrazinyloxy-cinnamic acid derivatives as potent cardiovascular agents. Eur. J. Med. Chem. 2011, 46, 5609–5615. [Google Scholar] [CrossRef]
- Chen, H.; Li, G.; Zhan, P.; Li, H.; Wang, S.; Liu, X. Design, synthesis and biological evaluation of novel trimethylpyrazine-2-carbonyloxy-cinnamic acids as potent cardiovascular agents. MedChemComm 2014, 5, 711–718. [Google Scholar] [CrossRef]
- Wang, P.; She, G.; Yang, Y.; Li, Q.; Zhang, H.; Liu, J.; Cao, Y.; Xu, X.; Lei, H. Synthesis and biological evaluation of new ligustrazine derivatives as anti-tumor agents. Molecules 2012, 17, 4972–4985. [Google Scholar] [CrossRef]
- Li, G.; Xu, X.; Xu, K.; Chu, F.; Song, J.; Zhou, S.; Xu, B.; Gong, Y.; Zhang, H.; Zhang, Y.; et al. Ligustrazinyl amides: A novel class of ligustrazine-phenolic acid derivatives with neuroprotective effects. Chem. Cent. J. 2015, 9, 9. [Google Scholar] [CrossRef]
- Balasubramaniam, S.; Vijayan, S.; Goldman, L.V.; May, X.A.; Dodson, K.; Adhikari, S.; Rivas, F.; Watkins, D.L.; Stoddard, S.V. Design and synthesis of diazine-based panobinostat analogues for HDAC8 inhibition. Beilstein J. Org. Chem. 2020, 16, 628–637. [Google Scholar] [CrossRef]
- Hu, Y.S.; Han, X.; Yu, P.J.; Jiao, M.M.; Liu, X.H.; Shi, J.B. Novel paeonol derivatives: Design, synthesis and anti-inflammatory activity in vitro and in vivo. Bioorg. Chem. 2020, 98, 103735. [Google Scholar] [CrossRef]
- Zou, Y.; Yan, C.; Zhang, H.; Xu, J.; Zhang, D.; Huang, Z.; Zhang, Y. Synthesis and evaluation of N-heteroaromatic ring-based analogs of piperlongumine as potent anticancer agents. Eur. J. Med. Chem. 2017, 138, 313–319. [Google Scholar] [CrossRef]
- Qian, J.; Xu, Z.; Zhu, P.; Meng, C.; Liu, Y.; Shan, W.; He, A.; Gu, Y.; Ran, F.; Zhang, Y.; et al. A Derivative of Piperlongumine and Ligustrazine as a Potential Thioredoxin Reductase Inhibitor in Drug-Resistant Hepatocellular Carcinoma. J. Nat. Prod. 2021, 84, 3161–3168. [Google Scholar] [CrossRef]
- Zou, Y.; Zhao, D.; Yan, C.; Ji, Y.; Liu, J.; Xu, J.; Lai, Y.; Tian, J.; Zhang, Y.; Huang, Z. Novel Ligustrazine-Based Analogs of Piperlongumine Potently Suppress Proliferation and Metastasis of Colorectal Cancer Cells In Vitro and In Vivo. J. Med. Chem. 2018, 61, 1821–1832. [Google Scholar] [CrossRef]
- Ma, X.Y.; Wang, D.P.; Wei, G.; Zhou, Q.D.; Gan, X.H. Synthesis and anticancer activity of chalcone-quinoxalin conjugates. Synth. Commun. 2021, 51, 1363–1372. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, W.; Cheng, Y.; Zhang, X.; Xue, N.; Wu, G.; Chen, M.; Fang, K.; Guo, W.; Zhou, F.; et al. Design, Synthesis and Biological Evaluation of Ligustrazine-Flavonoid Derivatives as Potential Anti-Tumor Agents. Molecules 2018, 23, 2187. [Google Scholar] [CrossRef] [PubMed]
- Srilaxmi, D.; Sreenivasulu, R.; Mak, K.K.; Pichika, M.R.; Jadav, S.S.; Ahsan, M.J.; Rao, M.V.B. Design, synthesis, anticancer evaluation and molecular docking studies of chalcone linked pyrido[4,3-b]pyrazin-5(6H)-one derivatives. J. Mol. Struct. 2021, 1229, 129851. [Google Scholar] [CrossRef]
- Xia, R.J.; Guo, T.; He, J.; Chen, M.; Su, S.J.; Jiang, S.C.; Tang, X.; Chen, Y.; Xue, W. Antimicrobial evaluation and action mechanism of chalcone derivatives containing quinoxaline moiety. Monatsh. Chem. 2019, 150, 1325–1334. [Google Scholar] [CrossRef]
- Besharati-Seidani, T.; Keivanloo, A.; Kaboudin, B.; Yoshida, A.; Yokomatsu, T. Regioselective synthesis of 2,3-disubstituted 1-alkyl pyrrolo[2,3-b] quinoxalines through palladium-catalyzed Heck reaction of chalcones and evaluation of their anti-bacterial activities. Tetrahedron 2018, 74, 2350–2358. [Google Scholar] [CrossRef]
- Luo, Y.; Wu, W.; Zha, D.; Zhou, W.; Wang, C.; Huang, J.; Chen, S.; Yu, L.; Li, Y.; Huang, Q.; et al. Synthesis and biological evaluation of novel ligustrazine-chalcone derivatives as potential anti-triple negative breast cancer agents. Bioorg. Med. Chem. Lett. 2021, 47, 128230. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Cao, G.; Zhou, J.; Cai, J.; Ma, X.; Liu, Z.; Huang, X.; Wang, H. Ligustrazine-Derived Chalcones-Modified Platinum(IV) Complexes Intervene in Cisplatin Resistance in Pancreatic Cancer through Ferroptosis and Apoptosis. J. Med. Chem. 2023, 66, 13587–13606. [Google Scholar] [CrossRef]
- Nkepang, G.; Bio, M.; Rajaputra, P.; Awuah, S.G.; You, Y. Folate Receptor-Mediated Enhanced and Specific Delivery of Far-Red Light-Activatable Prodrugs of Combretastatin A-4 to FR-Positive Tumor. Bioconjug. Chem. 2014, 25, 2175–2188. [Google Scholar] [CrossRef]
- Böhm, M.; Rosenkranz, S.; Laufs, U. Alcohol and red wine: Impact on cardiovascular risk. Nephrol. Dial. Transplant. 2004, 19, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Guo, X.; Zhai, L.; Song, Y.; Chen, H.; Zhan, P.; Wu, J.; Liu, X. Ligustrazine derivatives. Part 4: Design, synthesis, and biological evaluation of novel ligustrazine-based stilbene derivatives as potential cardiovascular agents. Chem. Biol. Drug Des. 2012, 79, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.Z.; Shu, H.Y.; Wu, J.; Yu, Y.L.; Ma, D.; Huang, X.; Liu, M.M.; Liu, X.H.; Shi, J.B. Discovery and development of novel pyrimidine and pyrazolo/thieno-fused pyrimidine derivatives as potent and orally active inducible nitric oxide synthase dimerization inhibitor with efficacy for arthritis. Eur. J. Med. Chem. 2021, 213, 113174. [Google Scholar] [CrossRef]
- Liang, X.; Sun, Y.; Zeng, W.; Liu, L.; Ma, X.; Zhao, Y.; Fan, J. Synthesis and biological evaluation of a folate-targeted rhaponticin conjugate. Bioorg. Med. Chem. 2013, 21, 178–185. [Google Scholar] [CrossRef]
- Shanmugam, M.K.; Rane, G.; Kanchi, M.M.; Arfuso, F.; Chinnathambi, A.; Zayed, M.E.; Alharbi, S.A.; Tan, B.K.; Kumar, A.P.; Sethi, G. The multifaceted role of curcumin in cancer prevention and treatment. Molecules 2015, 20, 2728–2769. [Google Scholar] [CrossRef]
- Iwai, M.; Minematsu, T.; Li, Q.; Iwatsubo, T.; Usui, T. Utility of P-Glycoprotein and Organic Cation Transporter 1 Double-Transfected LLC-PK1 Cells for Studying the Interaction of YM155 Monobromide, Novel Small-Molecule Survivin Suppressant, with P-Glycoprotein. Drug Metab. Dispos. 2011, 39, 2314–2320. [Google Scholar] [CrossRef]
- Ai, Y.; Zhu, B.; Ren, C.; Kang, F.; Li, J.; Huang, Z.; Lai, Y.; Peng, S.; Ding, K.; Tian, J.; et al. Discovery of New Monocarbonyl Ligustrazine-Curcumin Hybrids for Intervention of Drug-Sensitive and Drug-Resistant Lung Cancer. J. Med. Chem. 2016, 59, 1747–1760. [Google Scholar] [CrossRef]
- Singh, R.K.; Rai, D.; Yadav, D.; Bhargava, A.; Balzarini, J.; De Clercq, E. Synthesis, antibacterial and antiviral properties of curcumin bioconjugates bearing dipeptide, fatty acids and folic acid. Eur. J. Med. Chem. 2010, 45, 1078–1086. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Prasad, P.; Hussain, A.; Khan, I.; Kondaiah, P.; Chakravarty, A.R. Remarkable photocytotoxicity of curcumin in HeLa cells in visible light and arresting its degradation on oxovanadium(iv) complex formation. Chem. Commun. 2012, 48, 7702–7704. [Google Scholar] [CrossRef]
- Banerjee, S.; Pant, I.; Khan, I.; Prasad, P.; Hussain, A.; Kondaiah, P.; Chakravarty, A.R. Remarkable enhancement in photocytotoxicity and hydrolytic stability of curcumin on binding to an oxovanadium(IV) moiety. Dalton Trans. 2015, 44, 4108–4122. [Google Scholar] [CrossRef] [PubMed]
- Banaspati, A.; Raza, K.M.; Goswami, T.K. Ni(II) curcumin complexes for cellular imaging and photo-triggered in vitro anticancer activity. Eur. J. Med. Chem. 2020, 204, 112632. [Google Scholar] [CrossRef]
- Vanco, J.; Travnicek, Z.; Hosek, J.; Malina, T.; Dvorak, Z. Copper(II) Complexes Containing Natural Flavonoid Pomiferin Show Considerable In Vitro Cytotoxicity and Anti-inflammatory Effects. Int. J. Mol. Sci. 2021, 22, 7626. [Google Scholar] [CrossRef] [PubMed]
- Velagapudi, U.K.; Langelier, M.F.; Delgado-Martin, C.; Diolaiti, M.E.; Bakker, S.; Ashworth, A.; Patel, B.A.; Shao, X.; Pascal, J.M.; Talele, T.T. Design and Synthesis of Poly(ADP-ribose) Polymerase Inhibitors: Impact of Adenosine Pocket-Binding Motif Appendage to the 3-Oxo-2,3-dihydrobenzofuran-7-carboxamide on Potency and Selectivity. J. Med. Chem. 2019, 62, 5330–5357. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Zhang, X.; Liu, M.; Yang, Y.; Guo, T.; Mao, Y.; Zhang, J.; Fu, X.; Zhao, Y.; Chen, J.; et al. Hybrid molecules of scutellarein and tertramethylpyrazine’s active metabolites for ischemic stroke. Bioorg. Med. Chem. Lett. 2019, 29, 126608. [Google Scholar] [CrossRef] [PubMed]
- Ghorab, M.M.; Alsaid, M.S.; Al-Ansary, G.H.; Abdel-Latif, G.A.; Abou El Ella, D.A. Analogue based drug design, synthesis, molecular docking and anticancer evaluation of novel chromene sulfonamide hybrids as aromatase inhibitors and apoptosis enhancers. Eur. J. Med. Chem. 2016, 124, 946–958. [Google Scholar] [CrossRef]
- Aoki, T.; Hyohdoh, I.; Furuichi, N.; Ozawa, S.; Watanabe, F.; Matsushita, M.; Sakaitani, M.; Morikami, K.; Takanashi, K.; Harada, N.; et al. Optimizing the Physicochemical Properties of Raf/MEK Inhibitors by Nitrogen Scanning. ACS Med. Chem. Lett. 2014, 5, 309–314. [Google Scholar] [CrossRef]
- Kamble, A.A.; Kamble, R.R.; Kumbar, M.N.; Tegginamath, G. Pyridine-catalyzed synthesis of quinoxalines as anticancer and anti-tubercular agents. Med. Chem. Res. 2016, 25, 1163–1174. [Google Scholar] [CrossRef]
- Sarkar, T.; Kumar, A.; Sahoo, S.; Hussain, A. Mixed-Ligand Cobalt(III) Complexes of a Naturally Occurring Coumarin and Phenanthroline Bases as Mitochondria-Targeted Dual-Purpose Photochemotherapeutics. Inorg. Chem. 2021, 60, 6649–6662. [Google Scholar] [CrossRef]
- Goel, R.; Luxami, V.; Paul, K. Synthesis, in vitro anticancer activity and SAR studies of arylated imidazo[1,2-a]pyrazine-coumarin hybrids. RSC Adv. 2015, 5, 37887–37895. [Google Scholar] [CrossRef]
- Halawa, A.H.; Eliwa, E.M.; Hassan, A.A.; Nassar, H.S.; El-Eisawy, R.A.; Ismail, M.; Frese, M.; Shaaban, M.; El-Agrody, A.M.; Bedair, A.H.; et al. Synthesis, in vitro cytotoxicity activity against the human cervix carcinoma cell line and in silico computational predictions of new 4-arylamino-3-nitrocoumarin analogues. J. Mol. Struct. 2020, 1200, 127047. [Google Scholar] [CrossRef]
- El-Ansary, A.L.; Moustafa, H.; Abdel-Kader, N.S.; Farghaly, A.M. Effect of the formation of silver nano-binuclear complex on the refinement of cytotoxic and antibacterial potency of coumarin Schiff base: Spectroscopy, thermal, X-ray diffraction analyses and density functional theory calculations. Appl. Organomet. Chem. 2019, 33, e4968. [Google Scholar] [CrossRef]
- Sharma, R.K.; Singh, V.; Tiwari, N.; Butcher, R.J.; Katiyar, D. Synthesis, antimicrobial and chitinase inhibitory activities of 3-amidocoumarins. Bioorg. Chem. 2020, 98, 103700. [Google Scholar] [CrossRef] [PubMed]
- Chai, X.; Yu, S.; Wang, X.; Wang, N.; Zhu, Z.; Zhang, D.; Wu, Q.; Cao, Y.; Sun, Q. Synthesis and antifungal activity of novel 7-O-substituted pyridyl-4-methylcoumarin derivatives. Med. Chem. Res. 2013, 22, 4654–4662. [Google Scholar] [CrossRef]
- Moosavi-Zare, A.R.; Zolfigol, M.A.; Noroozizadeh, E.; Zarei, M.; Karamian, R.; Asadbegy, M. Synthesis and characterization of acetic acid functionalized poly (4-vinylpyridinium) salt as new catalyst for the synthesis of spiropyran derivatives and their biological activity. J. Mol. Catal. A Chem. 2016, 425, 217–228. [Google Scholar] [CrossRef]
- Wu, J.; Shen, Q.; Wang, Y.; Zhao, D.; Peng, C.; Li, J.X. Fluorescent Probes for Subcellular Localization during Osteclast Formation. ACS Med. Chem. Lett. 2014, 5, 911–914. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Li, G.; Zhan, P.; Guo, X.; Ding, Q.; Wang, S.; Liu, X. Design, synthesis and biological evaluation of novel ligustrazinylated derivatives as potent cardiovascular agents. MedChemComm 2013, 4, 827–832. [Google Scholar] [CrossRef]
- Priyanka; Neelabh; Tiwari, N.; Sharma, R.K.; Gupta, P.; Misra, S.; Misra-Bhattacharya, S.; Butcher, R.J.; Singh, K.; Katiyar, D. Synthesis, Structure Elucidation, Homology Modeling and Antifilarial Activity of 7-Benzamidocoumarin Derivatives. ChemistrySelect 2019, 4, 3300–3307. [Google Scholar] [CrossRef]
- Ostrowska, K.; Lesniak, A.; Gryczka, W.; Dobrzycki, L.; Bujalska-Zadrozny, M.; Trzaskowski, B. New Piperazine Derivatives of 6-Acetyl-7-hydroxy-4-methylcoumarin as 5-HT1A Receptor Agents. Int. J. Mol. Sci. 2023, 24, 2779. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Chen, X.; Kong, X.M.; Liu, T.W.; Feng, X.Q.; Chen, F.E.; Zhuang, Z.H. Anti-aging effect of methylurolithin A and its amide derivatives on nematode Caenorhabditis elegans. Tetrahedron Lett. 2023, 118, 154389. [Google Scholar] [CrossRef]
- Chen, X.; Feng, X.; Kong, X.; Wang, H.; Liu, T.; Tang, L.; Zhuang, Z. Design, synthesis, and anti-aging effect evaluation of urolithin B and its amide derivatives on nematode Caenorhabditis elegans. Med. Chem. Res. 2023, 32, 1087–1097. [Google Scholar] [CrossRef]
- Huang, H.S.; Chen, T.C.; Chen, R.H.; Huang, K.F.; Huang, F.C.; Jhan, J.R.; Chen, C.L.; Lee, C.C.; Lo, Y.; Lin, J.J. Synthesis, cytotoxicity and human telomerase inhibition activities of a series of 1,2-heteroannelated anthraquinones and anthra[1,2-d]imidazole-6,11-dione homologues. Bioorg. Med. Chem. 2009, 17, 7418–7428. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, Z.; Zhang, W.; Yan, D. 2-Substituted-1-(2-morpholinoethyl)-1H-naphtho[2,3-d]imidazole-4,9-diones: Design, synthesis and antiproliferative activity. Bioorg. Med. Chem. Lett. 2018, 28, 2454–2458. [Google Scholar] [CrossRef]
- Wang, P.L.; Cheng, Y.T.; Xu, K.; An, Y.W.; Wang, W.; Li, Q.S.; Han, Q.J.; Li, Q.; Zhang, H.G.; Lei, H.M. Synthesis and antitumor evaluation of one novel tetramethylpyrazine-rhein derivative. Asian J. Chem. 2013, 25, 4885–4888. [Google Scholar] [CrossRef]
- Li, X.; Lin, Z.; Wang, P.; Zhou, C.; Xu, J.; Lin, J.; Lin, D.; Zhang, D. Tetramethylpyrazine-Rhein Derivative inhibits the migration of canine inflammatory mammary carcinoma cells by mitochondrial damage-mediated apoptosis and cadherins downregulation. Biomed. Pharmacother. 2023, 162, 114731. [Google Scholar] [CrossRef]
- Ho, S.H.S.; Sim, M.Y.; Yee, W.L.S.; Yang, T.; Yuen, S.P.J.; Go, M.L. Antiproliferative, DNA intercalation and redox cycling activities of dioxonaphtho[2,3-d]imidazolium analogs of YM155: A structure-activity relationship study. Eur. J. Med. Chem. 2015, 104, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Premkumar, D.R.; Jane, E.P.; Foster, K.A.; Pollack, I.F. Survivin inhibitor YM-155 sensitizes tumor necrosis factor-related apoptosis-inducing ligand-resistant glioma cells to apoptosis through Mcl-1 downregulation and by engaging the mitochondrial death pathway. J. Pharmacol. Exp. Ther. 2013, 346, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Boger, D.L.; Tse, W.C. Thiazole orange as the fluorescent intercalator in a high resolution fid assay for determining DNA binding affinity and sequence selectivity of small molecules. Bioorg. Med. Chem. 2001, 9, 2511–2518. [Google Scholar] [CrossRef]
- Tse, W.C.; Boger, D.L. A fluorescent intercalator displacement assay for establishing DNA binding selectivity and affinity. Acc. Chem. Res. 2004, 37, 61–69. [Google Scholar] [CrossRef]
- West, T.J.; Bi, J.; Martinez-Pena, F.; Curtis, E.J.; Gazaniga, N.R.; Mischel, P.S.; Lairson, L.L. A Cell Type Selective YM155 Prodrug Targets Receptor-Interacting Protein Kinase 2 to Induce Brain Cancer Cell Death. J. Am. Chem. Soc. 2023, 145, 8355–8363. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Yuan, J.; Zhang, Z.; Yan, D.; Zhang, W. Design, synthesis and antitumor activity of 1-monosubstituted 1H-naphth[2,3-d]imidazole-4,9-diones and 1H-anthra[2,3-d]imidazole-4,11-diones. Chin. J. Org. Chem. 2018, 38, 3302–3317. [Google Scholar] [CrossRef]
- Bargiotti, A.; Musso, L.; Dallavalle, S.; Merlini, L.; Gallo, G.; Ciacci, A.; Giannini, G.; Cabri, W.; Penco, S.; Vesci, L.; et al. Isoxazolo(aza)naphthoquinones: A new class of cytotoxic Hsp90 inhibitors. Eur. J. Med. Chem. 2012, 53, 64–75. [Google Scholar] [CrossRef]
- Shanab, K.; Schirmer, E.; Knafl, H.; Wulz, E.; Holzer, W.; Spreitzer, H.; Schmidt, P.; Aicher, B.; Mueller, G.; Guenther, E. Synthesis and biological evaluation of new cytotoxic azanaphthoquinone pyrrolo-annelated derivatives. Bioorg. Med. Chem. Lett. 2010, 20, 3950–3952. [Google Scholar] [CrossRef]
- Yu, Q.; Yang, H.; Zhu, T.W.; Yu, L.M.; Chen, J.W.; Gu, L.Q.; Huang, Z.S.; An, L.K. Synthesis, cytotoxicity and structure-activity relationship of indolizinoquinolinedione derivatives as DNA topoisomerase IB catalytic inhibitors. Eur. J. Med. Chem. 2018, 152, 195–207. [Google Scholar] [CrossRef]
- Shen, D.Q.; Wu, N.; Li, Y.P.; Wu, Z.P.; Zhang, H.B.; Huang, Z.S.; Gu, L.Q.; An, L.K. Design, Synthesis, and Cytotoxicity of Indolizinoquinoxaline-5,12-dione Derivatives, Novel DNA Topoisomerase IB Inhibitors. Aust. J. Chem. 2010, 63, 1116–1121. [Google Scholar] [CrossRef]
- Devi, L.R.; Raza, K.M.; Musib, D.; Ramu, V.; Devi, J.; Roy, M. Nucleus targeting anthraquinone-based copper (II) complexes as the potent PDT agents: Synthesis, photo-physical and theoretical evaluation. Inorg. Chim. Acta 2020, 500, 119208. [Google Scholar] [CrossRef]
- Kim, Y.S.; Park, S.Y.; Lee, H.J.; Suh, M.E.; Schollmeyer, D.; Lee, C.O. Synthesis and cytotoxicity of 6,11-Dihydro-pyrido- and 6,11-Dihydro-benzo[2,3-b]phenazine-6,11-dione derivatives. Bioorg. Med. Chem. 2003, 11, 1709–1714. [Google Scholar] [CrossRef]
- Kim, J.S.; Rhee, H.K.; Park, H.J.; Lee, I.K.; Lee, S.K.; Suh, M.E.; Lee, H.J.; Ryu, C.K.; Choo, H.Y.P. Synthesis of 6-chloroisoquinoline-5,8-diones and pyrido[3,4-b]phenazine-5,12-diones and evaluation of their cytotoxicity and DNA topoisomerase II inhibitory activity. Bioorg. Med. Chem. 2007, 15, 451–457. [Google Scholar] [CrossRef]
- Lee, H.; Cho, S.; Namgoong, K.; Jung, J.K.; Cho, J.; Yang, S.I. Synthesis and in vitro evaluation of 7-dialkylaminomethylbenzo[g]quinoxaline-5,10-diones. Bioorg. Med. Chem. Lett. 2004, 14, 1235–1237. [Google Scholar] [CrossRef] [PubMed]
- Kwak, J.H.; Namgoong, K.; Jung, J.K.; Cho, J.; Kim, H.M.; Park, S.G.; Yoo, Y.A.; Kwon, J.H.; Lee, H. Synthesis and cytotoxic activities of 2-alkyl-2,3-dihydro-1H-2,6,9-triazacyclopenta[b]anthracene-5,10-diones. Arch. Pharmacal Res. 2008, 31, 995–998. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Kim, J.S.; Park, S.Y.; Suh, M.E.; Kim, H.J.; Seo, E.K.; Lee, C.O. Synthesis and cytotoxicity evaluation of 6,11-dihydro-pyridazo- and 6,11-dihydro-pyrido[2,3-b]phenazine-6,11-diones. Bioorg. Med. Chem. 2004, 12, 1623–1628. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-J.; Kim, J.S.; Suh, M.-E.; Park, H.J.; Lee, S.K.; Rhee, H.-K.; Kim, H.J.; Seo, E.-K.; Kim, C.; Lee, C.-O.; et al. Synthesis and cytotoxicity evaluation of substituted pyridazino[4,5-b]phenazine-5,12-diones and tri/tetra-azabenzofluorene-5,6-diones. Eur. J. Med. Chem. 2007, 42, 168–174. [Google Scholar] [CrossRef]
- Tuyun, A.F.; Bayrak, N.; Yildirim, H.; Onul, N.; Kara, E.M.; Celik, B.O. Synthesis and in vitro biological evaluation of aminonaphthoquinones and Benzo[b]phenazine-6,11-dione derivatives as potential antibacterial and antifungal compounds. J. Chem. 2015, 2015, 645902. [Google Scholar] [CrossRef]
- Kumar, S.; Kumar, N.; Drabu, S. Synthesis of benzo[g]quinoxaline-5,10-dione based pyrazoline derivatives and their antimycobacterial activity. Int. J. Pharm. Sci. Res. 2018, 9, 498–508. [Google Scholar]
- Kumar, S.; Kumar, N.; Drabu, S. Synthesis of benzo[g]quinoxaline-5,10-dione based pyridine derivatives and their antimycobacterial activity. Orient. J. Chem. 2017, 33, 821–828. [Google Scholar] [CrossRef]
- Hammam, A.S.; Youssef, M.S.K.; Atta, F.M.; Mohamed, T.A. Synthesis of new quinoxalinophenazinediones and tetrahydrobenzodipyrrolotetrones of biological interest. Chem. Pap. 2008, 62, 194–206. [Google Scholar] [CrossRef]
- Morin, C.; Besset, T.; Moutet, J.C.; Fayolle, M.; Brueckner, M.; Limosin, D.; Becker, K.; Davioud-Charvet, E. The aza-analogues of 1,4-naphthoquinones are potent substrates and inhibitors of plasmodial thioredoxin and glutathione reductases and of human erythrocyte glutathione reductase. Org. Biomol. Chem. 2008, 6, 2731–2742. [Google Scholar] [CrossRef]
- Hosoya, Y.; Nojo, W.; Kii, I.; Suzuki, T.; Imanishi, M.; Ohkanda, J. Identification of synthetic inhibitors for the DNA binding of intrinsically disordered circadian clock transcription factors. Chem. Commun. 2020, 56, 11203–11206. [Google Scholar] [CrossRef]
- Keinan, S.; Paquette, W.D.; Skoko, J.J.; Beratan, D.N.; Yang, W.; Shinde, S.; Johnston, P.A.; Lazo, J.S.; Wipf, P. Computational design, synthesis and biological evaluation of para-quinone-based inhibitors for redox regulation of the dual-specificity phosphatase Cdc25B. Org. Biomol. Chem. 2008, 6, 3256–3263. [Google Scholar] [CrossRef] [PubMed]
- Besset, T.; Braud, E.; Jarray, R.; Garbay, C.; Kolb, S.; Leo, P.M.; Morin, C. Preparation and evaluation of a set of bis(methoxycarbonylmethio) heteroquinones as CDC25B phosphatase inhibitors. Eur. J. Chem. 2011, 2, 433–440. [Google Scholar] [CrossRef]
- Yang, H.; Zhu, X.Q.; Wang, W.J.; Chen, Y.; Hu, Z.; Zhang, Y.; Hu, D.X.; Yu, L.M.; Agama, K.; Pommier, Y.; et al. The synthesis of furoquinolinedione and isoxazoloquinolinedione derivatives as selective Tyrosyl-DNA phosphodiesterase 2 (TDP2) inhibitors. Bioorg. Chem. 2021, 111, 104881. [Google Scholar] [CrossRef]
- Ryu, C.K.; Lee, Y.; Park, S.G.; You, H.J.; Lee, R.Y.; Lee, S.Y.; Choi, S. 3D-QSAR studies of heterocyclic quinones with inhibitory activity on vascular smooth muscle cell proliferation using pharmacophore-based alignment. Bioorg. Med. Chem. 2008, 16, 9772–9779. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.J.; Jung, O.J.; Chae, M.J.; Hong, S.Y.; Chung, K.H.; Lee, S.K.; Ryu, C.K. Synthesis and biological evaluation of quinoxaline-5,8-diones that inhibit vascular smooth muscle cell proliferation. Bioorg. Med. Chem. Lett. 2005, 15, 3380–3384. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.L.; Teng, Z.H.; Liu, D.Z.; Cui, H.; Liu, M.; Cheng, Y.; Yang, T.H.; Mei, Q.B.; Zhou, S.Y. Synthesis of a new pH-sensitive folate-doxorubicin conjugate and its antitumor activity in vitro. J. Pharm. Sci. 2013, 102, 530–540. [Google Scholar] [CrossRef]
- Huang, H.; Yan, M.; Chen, J.; Yuan, B.; Chen, G.; Cheng, S.; Huang, D.; Gao, Z.; Cao, C. Identification of ortho-naphthoquinones as anti-AML agents by highly efficient oxidation of phenols. Bioorg. Chem. 2019, 86, 97–102. [Google Scholar] [CrossRef]
- Sandilya, S.; Das, A.; Deka, S.; Kalita, R. Synthesis, in-vivo anti-inflammatory evaluation and molecular docking study of a series of substituted xanthone derivatives as novel COX-2 inhibitors. Int. J. Pharm. Sci. Res. 2021, 12, 4491–4503. [Google Scholar]
- Zhao, W.; Chen, L.; Li, H.M.; Wang, D.J.; Li, D.S.; Chen, T.; Yuan, Z.P.; Tang, Y.J. A rational design strategy of the novel topoisomerase II inhibitors for the synthesis of the 4-O-(2-pyrazinecarboxylic)-4’-demethylepipodophyllotoxin with antitumor activity by diminishing the relaxation reaction of topoisomerase II-DNA decatenation. Bioorg. Med. Chem. 2014, 22, 2998–3007. [Google Scholar] [CrossRef]
- Wu, G.R.; Xu, B.; Yang, Y.Q.; Zhang, X.Y.; Fang, K.; Ma, T.; Wang, H.; Xue, N.N.; Chen, M.; Guo, W.B.; et al. Synthesis and biological evaluation of podophyllotoxin derivatives as selective antitumor agents. Eur. J. Med. Chem. 2018, 155, 183–196. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Z.; Chen, F.; Chen, Y.; Lin, Y.; Wang, J. Aromatic heterocyclic esters of podophyllotoxin exert anti-MDR activity in human leukemia K562/ADR cells via ROS/MAPK signaling pathways. Eur. J. Med. Chem. 2016, 123, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Su, H.; Yu, W.; Li, X.; Cheng, H.; Liu, M.; Pang, X.; Zou, X. Design, synthesis and anticancer activities of novel otobain derivatives. Org. Biomol. Chem. 2016, 14, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.A.; Miguel del Corral, J.M.; Gordaliza, M.; Gomez-Zurita, M.A.; de la Puente, M.L.; Betancur-Galvis, L.A.; Sierra, J.; San Feliciano, A. Synthesis, cytotoxicity and antiviral activity of podophyllotoxin analogues modified in the E-ring. Eur. J. Med. Chem. 2003, 38, 899–911. [Google Scholar] [CrossRef] [PubMed]
- Zhi, X.Y.; Yang, C.; Zhang, R.; Hu, Y.; Ke, Y.Z.; Xu, H. Natural products-based insecticidal agents 13. Semisynthesis and insecticidal activity of novel phenazine derivatives of 4β-acyloxypodophyllotoxin modified in the E-ring against Mythimna separata Walker in vivo. Ind. Crops Prod. 2013, 42, 520–526. [Google Scholar] [CrossRef]
- Zhi, X.; Yu, X.; Yang, C.; Ding, G.; Chen, H.; Xu, H. Synthesis of 4β-acyloxypodophyllotoxin analogs modified in the C and E rings as insecticidal agents against Mythimna separata Walker. Bioorg. Med. Chem. Lett. 2014, 24, 765–772. [Google Scholar] [CrossRef]
- Zhi, X.; Yang, C.; Yu, X.; Xu, H. Synthesis and insecticidal activity of new oxime derivatives of podophyllotoxin-based phenazines against Mythimna separata Walker. Bioorg. Med. Chem. Lett. 2014, 24, 5679–5682. [Google Scholar] [CrossRef]
- Wang, J.; Zhi, X.; Yu, X.; Xu, H. Synthesis and insecticidal activity of new deoxypodophyllotoxin-based phenazine analogues against Mythimna separata Walker. J. Agric. Food Chem. 2013, 61, 6336–6343. [Google Scholar] [CrossRef]
- Hou, M.; Li, S.; Xu, Z.; Li, B. A Reduction-responsive Amphiphilic Methotrexate-Podophyllotoxin Conjugate for Targeted Chemotherapy. Chem. Asian J. 2019, 14, 3840–3844. [Google Scholar] [CrossRef]
- Talbot, A.; Maltais, R.; Kenmogne, L.C.; Roy, J.; Poirier, D. Solid-phase synthesis of libraries of ethynylated aminosteroid derivatives as potential antileukemic agents. Steroids 2016, 107, 55–64. [Google Scholar] [CrossRef]
- Wang, S.; Yuan, X.; Qian, H.; Li, N.; Wang, J. Design, synthesis, and biological evaluation of two series of novel A-ring fused steroidal pyrazines as potential anticancer agents. Int. J. Mol. Sci. 2020, 21, 1665. [Google Scholar] [CrossRef]
- Tao, H.W.; Peng, W.Y.; Yuan, J.C.; Li, Q.; Zeng, L.Y.; Yu, X.Y.; Yi, P.G. Facile preparation and preliminary cytotoxicity evaluation of dehydroepiandrosterone C-16 spiro-pyrrolidine derivatives. Chem. Pap. 2021, 75, 823–829. [Google Scholar] [CrossRef]
- Cui, H.W.; Peng, S.; Gu, X.Z.; Chen, H.; He, Y.; Gao, W.; Lv, F.; Wang, J.H.; Wang, Y.; Xie, J.; et al. Synthesis and biological evaluation of D-ring fused 1,2,3-thiadiazole dehydroepiandrosterone derivatives as antitumor agents. Eur. J. Med. Chem. 2016, 111, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Handratta, V.D.; Vasaitis, T.S.; Njar, V.C.O.; Gediya, L.K.; Kataria, R.; Chopra, P.; Newman, D., Jr.; Farquhar, R.; Guo, Z.; Qiu, Y.; et al. Novel C-17-Heteroaryl Steroidal CYP17 Inhibitors/Antiandrogens: Synthesis, In Vitro Biological Activity, Pharmacokinetics, and Antitumor Activity in the LAPC4 Human Prostate Cancer Xenograft Model. J. Med. Chem. 2005, 48, 2972–2984. [Google Scholar] [CrossRef] [PubMed]
- Vicker, N.; Lawrence, H.R.; Allan, G.M.; Bubert, C.; Smith, A.; Tutill, H.J.; Purohit, A.; Day, J.M.; Mahon, M.F.; Reed, M.J.; et al. Focused libraries of 16-substituted estrone derivatives and modified E-ring steroids: Inhibitors of 17β-hydroxysteroid dehydrogenase type 1. ChemMedChem 2006, 1, 464–481. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.; Boldt, S.; un Nisa, Z.; Ali Shah, S.J.; Ehlers, P.; Villinger, A.; Schneider, G.; Woelfling, J.; Rahman, Q.; Iqbal, J.; et al. Synthesis and phosphatase inhibitory activity of 3-alkynylestrones and their derivatives. RSC Adv. 2016, 6, 11118–11127. [Google Scholar] [CrossRef]
- Bertrand, B.; O’Connell, M.A.; Waller, Z.A.E.; Bochmann, M. A Gold(III) Pincer Ligand Scaffold for the Synthesis of Binuclear and Bioconjugated Complexes: Synthesis and Anticancer Potential. Chem. Eur. J. 2018, 24, 3613–3622. [Google Scholar] [CrossRef]
- Barrett, S.; De Franco, M.; Kellett, A.; Dempsey, E.; Marzano, C.; Erxleben, A.; Gandin, V.; Montagner, D. Anticancer activity, DNA binding and cell mechanistic studies of estrogen-functionalised Cu(II) complexes. JBIC J. Biol. Inorg. Chem. 2020, 25, 49–60. [Google Scholar] [CrossRef]
- Khan, S.A.; Asiri, A.M. Synthesis of novel steroidal oxazolo quinoxaline as antibacterial agents. Arab. J. Chem. 2010, 4, 349–354. [Google Scholar] [CrossRef]
- Khan, S.A.; Saleem, K.; Khan, Z. Synthesis, structure elucidation and antibacterial evaluation of new steroidal -5-en-7-thiazoloquinoxaline derivatives. Eur. J. Med. Chem. 2008, 43, 2257–2261. [Google Scholar] [CrossRef]
- Khan, S.A. Synthesis, characterization and in vitro antibacterial activity of new steroidal 5-en-3-oxazolo and thiazoloquinoxaline. Eur. J. Med. Chem. 2008, 43, 2040–2044. [Google Scholar] [CrossRef]
- Dziwornu, G.A.; Kamunya, S.; Ntsabo, T.; Chibale, K. Novel antimycobacterial C-21 amide derivatives of the antibiotic fusidic acid: Synthesis, pharmacological evaluation and rationalization of media-dependent activity using molecular docking studies in the binding site of human serum albumin. MedChemComm 2019, 10, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Barrett, S.; Delaney, S.; Kavanagh, K.; Montagner, D. Evaluation of in vitro and in vivo antibacterial activity of novel Cu(II)-steroid complexes. Inorg. Chim. Acta 2018, 479, 261–265. [Google Scholar] [CrossRef]
- Wang, L.J.; Geng, C.A.; Ma, Y.B.; Huang, X.Y.; Luo, J.; Chen, H.; Guo, R.H.; Zhang, X.M.; Chen, J.J. Synthesis, structure-activity relationships and biological evaluation of caudatin derivatives as novel anti-hepatitis B virus agents. Bioorg. Med. Chem. 2012, 20, 2877–2888. [Google Scholar] [CrossRef]
- Yang, G.X.; Huang, Y.; Zheng, L.L.; Zhang, L.; Su, L.; Wu, Y.H.; Li, J.; Zhou, L.C.; Huang, J.; Tang, Y.; et al. Design, synthesis and evaluation of diosgenin carbamate derivatives as multitarget anti-Alzheimer’s disease agents. Eur. J. Med. Chem. 2020, 187, 111913. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Chu, F.; Zhang, Y.; Wang, X.; Li, Q.; Liu, W.; Xu, X.; Xing, Y.; Chen, J.; Wang, P.; et al. A Series of New Ligustrazine-Triterpenes Derivatives as Anti-Tumor Agents: Design, Synthesis, and Biological Evaluation. Int. J. Mol. Sci. 2015, 16, 21035–21055. [Google Scholar] [CrossRef]
- Xu, K.; Xu, X.; Chu, F.H.; Wang, M.N.; Wang, P.L.; Li, G.L.; Song, J.X.; Zhang, Y.Z.; Lei, H.M. Synthesis and biological evaluation of T-OA analogues as the cytotoxic agents. Res. Chem. Intermed. 2015, 41, 6257–6269. [Google Scholar] [CrossRef]
- Xu, B.; Yan, W.Q.; Xu, X.; Wu, G.R.; Zhang, C.Z.; Han, Y.T.; Chu, F.H.; Zhao, R.; Wang, P.L.; Lei, H.M. Combination of amino acid/dipeptide with ligustrazine-betulinic acid as antitumor agents. Eur. J. Med. Chem. 2017, 130, 26–38. [Google Scholar] [CrossRef]
- Xu, J.; Li, Z.; Luo, J.; Yang, F.; Liu, T.; Liu, M.; Qiu, W.W.; Tang, J. Synthesis and biological evaluation of heterocyclic ring-fused betulinic acid derivatives as novel inhibitors of osteoclast differentiation and bone resorption. J. Med. Chem. 2012, 55, 3122–3134. [Google Scholar] [CrossRef]
- Urban, M.; Sarek, J.; Kvasnica, M.; Tislerova, I.; Hajduch, M. Triterpenoid pyrazines and benzopyrazines with cytotoxic activity. J. Nat. Prod. 2007, 70, 526–532. [Google Scholar] [CrossRef]
- Haavikko, R.; Nasereddin, A.; Sacerdoti-Sierra, N.; Kopelyanskiy, D.; Alakurtti, S.; Tikka, M.; Jaffe, C.L.; Yli-Kauhaluoma, J. Heterocycle-fused lupane triterpenoids inhibit Leishmania donovani amastigotes. MedChemComm 2014, 5, 445–451. [Google Scholar] [CrossRef]
- Soural, M.; Hodon, J.; Dickinson, N.J.; Sidova, V.; Gurska, S.; Dzubak, P.; Hajduch, M.; Sarek, J.; Urban, M. Preparation of Conjugates of Cytotoxic Lupane Triterpenes with Biotin. Bioconjug. Chem. 2015, 26, 2563–2570. [Google Scholar] [CrossRef]
- Hodoň, J.; Frydrych, I.; Trhlíková, Z.; Pokorný, J.; Borková, L.; Benická, S.; Vlk, M.; Lišková, B.; Kubíčková, A.; Medvedíková, M.; et al. Triterpenoid pyrazines and pyridines—Synthesis, cytotoxicity, mechanism of action, preparation of prodrugs. Eur. J. Med. Chem. 2022, 243, 114777. [Google Scholar] [CrossRef] [PubMed]
- Suman, P.; Patel, A.; Solano, L.; Jampana, G.; Gardner, Z.S.; Holt, C.M.; Jonnalagadda, S.C. Synthesis and cytotoxicity of Baylis-Hillman template derived betulinic acid-triazole conjugates. Tetrahedron 2017, 73, 4214–4226. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, Y.; Zhu, P.; Liu, J.; Xu, S.; Yao, H.; Jiang, J.; Ye, W.; Wu, X.; Xu, J. Design, synthesis and antitumor activity of triterpenoid pyrazine derivatives from 23-hydroxybetulinic acid. Eur. J. Med. Chem. 2015, 97, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Devendra Rao, S.; Nageswara Rao, B.; Uma Devi, P.; Karteek Rao, A. Isolation of Lupeol, Design and Synthesis of Lupeol Derivatives and their Biological Activity. Orient. J. Chem. 2017, 33, 173–180. [Google Scholar] [CrossRef]
- Zorina, A.D.; Nikiforova, N.S.; Zarubaev, V.V.; Marchenko, S.A.; Selivanov, S.I.; Starova, G.L.; Mehtiev, A.R.; Rodionov, E.I.; Rodionova, A.A.; Trifonov, R.E. Synthesis, structure and in vitro biological evaluation of new lupane and dammarane triterpenoids fused with pyrazine heterocycle. Mendeleev Commun. 2019, 29, 500–502. [Google Scholar] [CrossRef]
- Bhandari, P.; Patel, N.K.; Bhutani, K.K. Synthesis of new heterocyclic lupeol derivatives as nitric oxide and pro-inflammatory cytokine inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 3596–3599. [Google Scholar] [CrossRef]
- Guo, W.B.; Zhang, H.; Yan, W.Q.; Liu, Y.M.; Zhou, F.; Cai, D.S.; Zhang, W.X.; Huang, X.M.; Jia, X.H.; Chen, H.S.; et al. Design, synthesis, and biological evaluation of ligustrazine—Betulin amino-acid/dipeptide derivatives as anti-tumor agents. Eur. J. Med. Chem. 2020, 185, 111839. [Google Scholar] [CrossRef] [PubMed]
- Chu, F.; Xu, X.; Li, G.; Gu, S.; Xu, K.; Gong, Y.; Xu, B.; Wang, M.; Zhang, H.; Zhang, Y.; et al. Amino acid derivatives of ligustrazine-oleanolic acid as new cytotoxic agents. Molecules 2014, 19, 18215–18231. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhang, Y.; Xu, K.; Li, Q.; Zhang, H.; Guo, J.; Pang, D.; Cheng, Y.; Lei, H. A new ligustrazine derivative—Pharmacokinetic evaluation and antitumor activity by suppression of NF-κB/p65 and COX-2 expression in S180 mice. Pharmazie 2013, 68, 782–789. [Google Scholar]
- Qiu, W.W.; Shen, Q.; Yang, F.; Wang, B.; Zou, H.; Li, J.Y.; Li, J.; Tang, J. Synthesis and biological evaluation of heterocyclic ring-substituted maslinic acid derivatives as novel inhibitors of protein tyrosine phosphatase 1B. Bioorg. Med. Chem. Lett. 2009, 19, 6618–6622. [Google Scholar] [CrossRef]
- Li, J.F.; Zhao, Y.; Cai, M.M.; Li, X.F.; Li, J.X. Synthesis and evaluation of a novel series of heterocyclic oleanolic acid derivatives with anti-osteoclast formation activity. Eur. J. Med. Chem. 2009, 44, 2796–2806. [Google Scholar] [CrossRef]
- Zhang, Y.C.; Shen, Q.; Zhu, M.W.; Wang, J.; Du, Y.; Wu, J.; Li, J.X. Modified Quinoxaline-Fused Oleanolic Acid Derivatives as Inhibitors of Osteoclastogenesis and Potential Agent in Anti-Osteoporosis. ChemistrySelect 2020, 5, 1526–1533. [Google Scholar] [CrossRef]
- Zhong, Y.Y.; Chen, H.S.; Wu, P.P.; Zhang, B.J.; Yang, Y.; Zhu, Q.Y.; Zhang, C.G.; Zhao, S.Q. Synthesis and biological evaluation of novel oleanolic acid analogues as potential α-glucosidase inhibitors. Eur. J. Med. Chem. 2019, 164, 706–716. [Google Scholar] [CrossRef]
- Yang, Y.; Guan, D.; Lei, L.; Lu, J.; Liu, J.Q.; Yang, G.; Yan, C.; Zhai, R.; Tian, J.; Bi, Y.; et al. H6, a novel hederagenin derivative, reverses multidrug resistance in vitro and in vivo. Toxicol. Appl. Pharmacol. 2018, 341, 98–105. [Google Scholar] [CrossRef]
- Huang, W.; Wang, Y.; Xu, S.; Qiao, H.; Cheng, H.; Wang, L.; Liu, S.; Tian, Q.; Wang, R.; Wang, H.; et al. Design, synthesis, and tumor drug resistance reversal activity of novel hederagenin derivatives modified by nitrogen-containing heterocycles. Eur. J. Med. Chem. 2022, 232, 114207. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ren, Q.W.; Liu, X.X.; Yang, Y.T.; Wang, B.H.; Zhai, R.; Qi, J.G.; Tian, J.W.; Wang, H.B.; Bi, Y. Synthesis and biological evaluation of novel H6 analogues as drug resistance reversal agents. Eur. J. Med. Chem. 2019, 161, 364–377. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Liu, S.; Huang, W.; Ma, M.; Chen, X.; Zeng, W.; Liang, K.; Wang, H.; Bi, Y.; Li, X. Design, synthesis, and biological evaluation of hederagenin derivatives with improved aqueous solubility and tumor resistance reversal activity. Eur. J. Med. Chem. 2021, 211, 113107. [Google Scholar] [CrossRef]
- Fang, K.; Zhang, X.H.; Han, Y.T.; Wu, G.R.; Cai, D.S.; Xue, N.N.; Guo, W.B.; Yang, Y.Q.; Chen, M.; Zhang, X.Y.; et al. Design, synthesis, and cytotoxic analysis of novel hederagenin-pyrazine derivatives based on partial least squares discriminant analysis. Int. J. Mol. Sci. 2018, 19, 2994. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.P.; Zhang, B.J.; Cui, X.P.; Yang, Y.; Jiang, Z.Y.; Zhou, Z.H.; Zhong, Y.Y.; Mai, Y.Y.; Ouyang, Z.; Chen, H.S.; et al. Synthesis and biological evaluation of novel ursolic acid analogues as potential α-glucosidase inhibitors. Sci. Rep. 2017, 7, 45578. [Google Scholar] [CrossRef]
- Fu, H.J.; Zhou, Y.R.; Bao, B.H.; Jia, M.X.; Zhao, Y.; Zhang, L.; Li, J.X.; He, H.L.; Zhou, X.M. Tryptophan Hydroxylase 1 (Tph-1)-Targeted Bone Anabolic Agents for Osteoporosis. J. Med. Chem. 2014, 57, 4692–4709. [Google Scholar] [CrossRef] [PubMed]
- Shamraiz, U.; Hussain, H.; Ur Rehman, N.; Al-Shidhani, S.; Saeed, A.; Khan, H.Y.; Khan, A.; Fischer, L.; Csuk, R.; Badshah, A.; et al. Synthesis of new boswellic acid derivatives as potential antiproliferative agents. Nat. Prod. Res. 2020, 34, 1845–1852. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Tu, B.; Liang, J.; Guo, S.; Cao, N.; Chen, S.; Luo, Z.; Li, J.; Zheng, W.; Tang, X.; et al. Synthesis and biological evaluation of pentacyclic triterpenoid derivatives as potential novel antibacterial agents. Bioorg. Chem. 2021, 109, 104692. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhu, Q.; Zhong, Y.; Cui, X.; Jiang, Z.; Wu, P.; Zheng, X.; Zhang, K.; Zhao, S. Synthesis, anti-microbial and anti-inflammatory activities of 18β-glycyrrhetinic acid derivatives. Bioorg. Chem. 2020, 101, 103985. [Google Scholar] [CrossRef]
- Xu, H.; Tang, H.; Feng, H.; Li, Y. Design, synthesis and anticancer activity evaluation of novel C14 heterocycle substituted epi-triptolide. Eur. J. Med. Chem. 2014, 73, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.F.; Tang, Y.B.; Hua, H.M.; Ohkoshi, E.; Goto, M.; Wang, L.T.; Lee, K.H.; Xiao, Z.Y. Discovery of novel andrographolide derivatives as cytotoxic agents. Bioorg. Med. Chem. Lett. 2013, 23, 4056–4060. [Google Scholar] [CrossRef] [PubMed]
- Grigoropoulou, S.; Manou, D.; Antoniou, A.I.; Tsirogianni, A.; Siciliano, C.; Theocharis, A.D.; Athanassopoulos, C.M. Synthesis and Antiproliferative Activity of Novel Dehydroabietic Acid-Chalcone Hybrids. Molecules 2022, 27, 3623. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Sun, X.; Lu, W.; Xu, L.; Shi, J.; Yang, S.; Zhou, M.; Su, F.; Lin, F.; Cao, F. Synthesis of novel, DNA binding heterocyclic dehydroabietylamine derivatives as potential antiproliferative and apoptosis-inducing agents. Drug Deliv. 2020, 27, 216–227. [Google Scholar] [CrossRef]
- De Wang, X.; Li, T.; Li, Y.; Yuan, W.H.; Zhao, Y.Q. 2-Pyrazine-PPD, a novel dammarane derivative, showed anticancer activity by reactive oxygen species-mediate apoptosis and endoplasmic reticulum stress in gastric cancer cells. Eur. J. Pharmacol. 2020, 881, 173211. [Google Scholar] [CrossRef]
- Xu, S.; Li, D.; Pei, L.; Yao, H.; Wang, C.; Cai, H.; Yao, H.; Wu, X.; Xu, J. Design, synthesis and antimycobacterial activity evaluation of natural oridonin derivatives. Bioorg. Med. Chem. Lett. 2014, 24, 2811–2814. [Google Scholar] [CrossRef]
- Xu, S.; Pei, L.; Li, D.; Yao, H.; Cai, H.; Yao, H.; Wu, X.; Xu, J. Synthesis and antimycobacterial evaluation of natural oridonin and its enmein-type derivatives. Fitoterapia 2014, 99, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Zhang, D.; Li, M.; Wu, Q.; Lam, Y.P.Y.; Guo, Y.; Chen, C.; Bai, N.; Malhotra, S.; Li, W.; et al. Discovery of novel, potent, isosteviol-based antithrombotic agents. Eur. J. Med. Chem. 2019, 183, 111722. [Google Scholar] [CrossRef]
- Bruno, M.; Rosselli, S.; Pibiri, I.; Kilgore, N.; Lee, K.H. Anti-HIV Agents Derived from the ent-Kaurane Diterpenoid Linearol. J. Nat. Prod. 2002, 65, 1594–1597. [Google Scholar] [CrossRef] [PubMed]
- Khusnutdinova, E.F.; Smirnova, I.E.; Kazakova, O.B.; Petrova, A.V.; Ha, N.T.T.; Viet, D.Q. Synthesis and evaluation of 2,3-indolotriterpenoids as new α-glucosidase inhibitors. Med. Chem. Res. 2017, 26, 2737–2742. [Google Scholar] [CrossRef]
- Nishiyama, H.; Ono, M.; Sugimoto, T.; Sasai, T.; Asakawa, N.; Ueno, S.; Tominaga, Y.; Yaegashi, T.; Nagaoka, M.; Matsuzaki, T.; et al. 4-Chlorocolchicine derivatives bearing a thiourea side chain at the C-7 position as potent anticancer agents. MedChemComm 2014, 5, 452–458. [Google Scholar] [CrossRef]
- Li, Q.; Zu, Y.; Shi, R.; Yao, L.; Fu, Y.; Yang, Z.; Li, L. Synthesis and antitumor activity of novel 10-substituted camptothecin analogues. Bioorg. Med. Chem. 2006, 14, 7175–7182. [Google Scholar] [CrossRef]
- Zhou, T.; Hou, J.; Wang, M.; Ma, L.; Wu, L.; Wang, S.; Sun, B.; Yao, Z.J. Regio-controlled synthesis of unsymmetrical pyrazine-fused sinomenine derivatives and discriminate substitution effects on TNF-α inhibitory activity. Tetrahedron 2014, 70, 5475–5482. [Google Scholar] [CrossRef]
- Lou, Y.T.; Zhou, H.B.; Zou, J.; Yan, L.C.; Bi, E.G.; Sun, B.; Yao, Z.J. Modification of poorly bioactive sinomenine into more potent immunosuppressive agents by embedding of drug-like fragments. Tetrahedron Lett. 2010, 51, 485–488. [Google Scholar] [CrossRef]
- Watanabe, Y.; Hayashida, K.; Saito, D.; Takahashi, T.; Sakai, J.; Nakata, E.; Kanda, T.; Iwai, T.; Hirayama, S.; Fujii, H.; et al. Design and synthesis of novel δ opioid receptor agonists with an azatricyclodecane skeleton for improving blood-brain barrier penetration. Bioorg. Med. Chem. Lett. 2017, 27, 3495–3498. [Google Scholar] [CrossRef]
- Yekkirala, A.S.; Lunzer, M.M.; McCurdy, C.R.; Powers, M.D.; Kalyuzhny, A.E.; Roerig, S.C.; Portoghese, P.S. N-naphthoyl-beta-naltrexamine (NNTA), a highly selective and potent activator of μ/kappa-opioid heteromers. Proc. Natl. Acad. Sci. USA 2011, 108, 5098–5103. [Google Scholar] [CrossRef]
- Le Naour, M.; Lunzer, M.M.; Powers, M.D.; Portoghese, P.S. Opioid Activity of Spinally Selective Analogues of N-Naphthoyl-β-naltrexamine in HEK-293 Cells and Mice. J. Med. Chem. 2012, 55, 670–677. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Deng, H.; Shen, Q.K.; Quan, Z.S. Tanshinone IIA: Pharmacology, Total Synthesis, and Progress in Structure-modifications. Curr. Med. Chem. 2022, 29, 1959–1989. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, T.; Reker, D.; Schneider, P.; Schneider, G. Counting on natural products for drug design. Nat. Chem. 2016, 8, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Deng, H.; Li, X.T.; Quan, Z.S. Application of Amino Acids in the Structural Modification of Natural Products: A Review. Front. Chem. 2021, 9, 650569. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Structure and pharmacological activity of pyrazine.

Figure 2.
Cinnamic acid–pyrazine derivatives 1–19.

Figure 3.
Cinnamic acid–pyrazine derivatives 20–33.

Figure 4.
Cinnamic acid–pyrazine derivatives 34–45.

Figure 5.
Chalcone–pyrazine derivatives 46–67.

Figure 6.
Pyrazine-based resveratrol derivatives, hetero analogs 68–75, and combretastatin A-4-pyrazine derivative 76–77.
Figure 6.
Pyrazine-based resveratrol derivatives, hetero analogs 68–75, and combretastatin A-4-pyrazine derivative 76–77.

Figure 7.
Curcumin–pyrazine derivatives 78–87.

Figure 8.
Flavono–pyrazine derivatives 88–95.

Figure 9.
Coumarin–pyrazine derivatives 96–105.

Figure 10.
Coumarin–pyrazine derivatives 106–116.

Figure 11.
Anthraquinone–pyrazine derivatives 117–126.

Figure 12.
Anthraquinone–pyrazine derivatives 127–135.

Figure 13.
Anthraquinone–pyrazine derivatives 136–147.

Figure 14.
Anthraquinone–pyrazine derivatives 148–160.

Figure 15.
Anthraquinone–pyrazine derivatives 161–183.

Figure 16.
Anthraquinone–pyrazine derivatives 184–188.

Figure 17.
Anthraquinone–pyrazine derivatives 189–210.

Figure 18.
Structure of FA-AMA-DOX (211), O-naphthoquinone 212, and xanthone derivatives 213–214.

Figure 19.
Lignin–pyrazine derivatives 215–226.

Figure 20.
Lignin–pyrazine derivatives 227–239.

Figure 21.
Steroidal–pyrazine derivatives 240–247.

Figure 22.
Steroidal–pyrazine derivatives 248–256.

Figure 23.
Steroidal–pyrazine derivatives 257–264.

Figure 24.
Steroidal–pyrazine derivatives 265–269.

Figure 25.
Betulinic acid–pyrazine derivatives 270–274.

Figure 26.
Betulinic acid–pyrazine derivatives 275–286.

Figure 27.
A 23-hydroxyl betulinic acid–pyrazine derivative 287.

Figure 28.
Lupeol–pyrazine derivatives 288–289 and betulin–pyrazine derivatives 290–291.

Figure 29.
Oleanolic acid–pyrazine derivatives 292–296.

Figure 30.
Oleanolic acid–pyrazine derivatives 297–309.

Figure 31.
Hederagenin–pyrazine derivatives 310–314.

Figure 32.
Other pentacyclic triterpenes–pyrazine derivatives 315–320.

Figure 33.
Other terpenes–pyrazine derivatives 321–328.

Figure 34.
Other terpenes–pyrazine derivatives 329–335.

Figure 35.
Alkaloid–pyrazine derivatives 336–344.

Figure 36.
Alkaloid–pyrazine derivatives 345–348.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
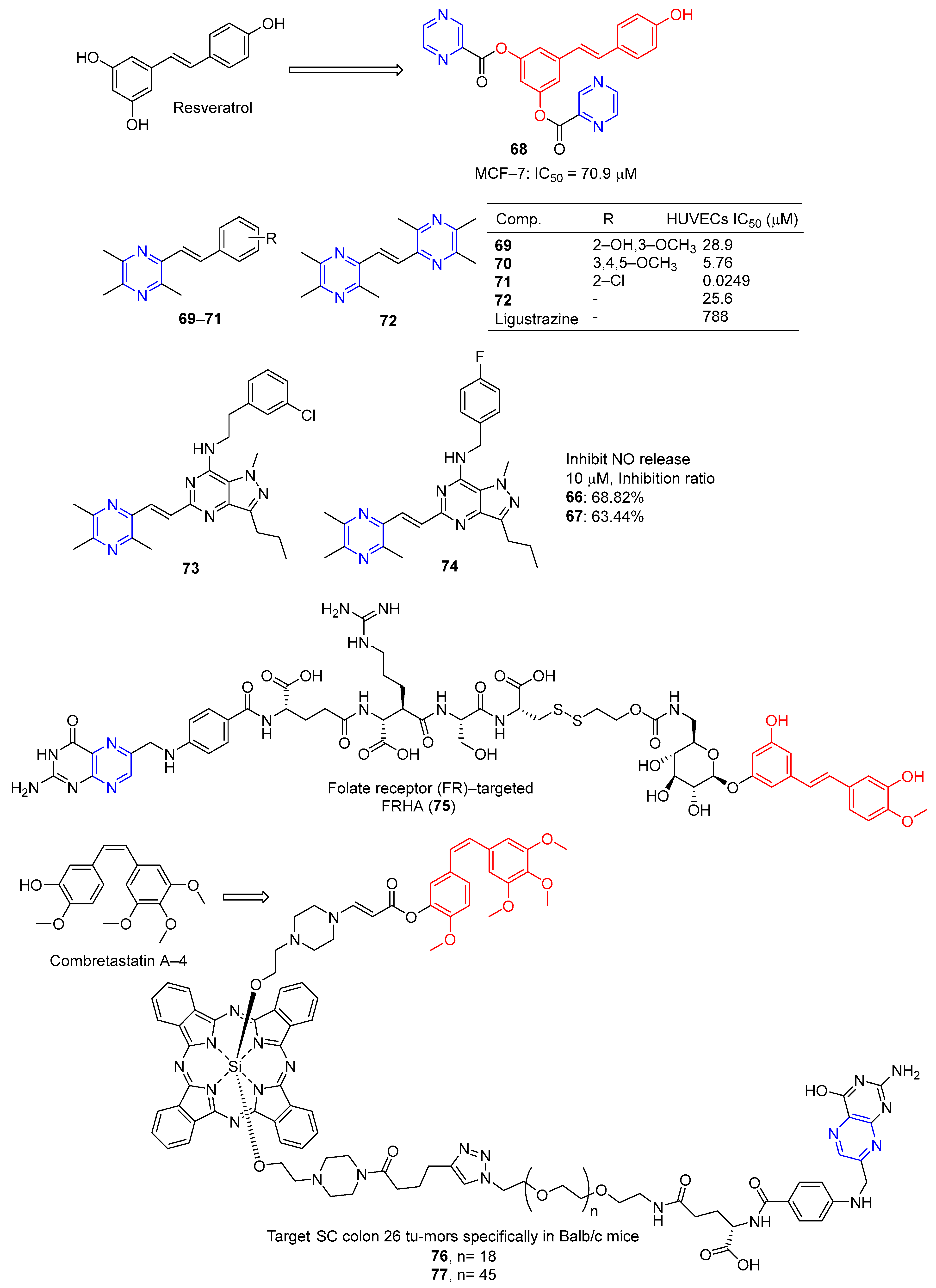{kind=link}
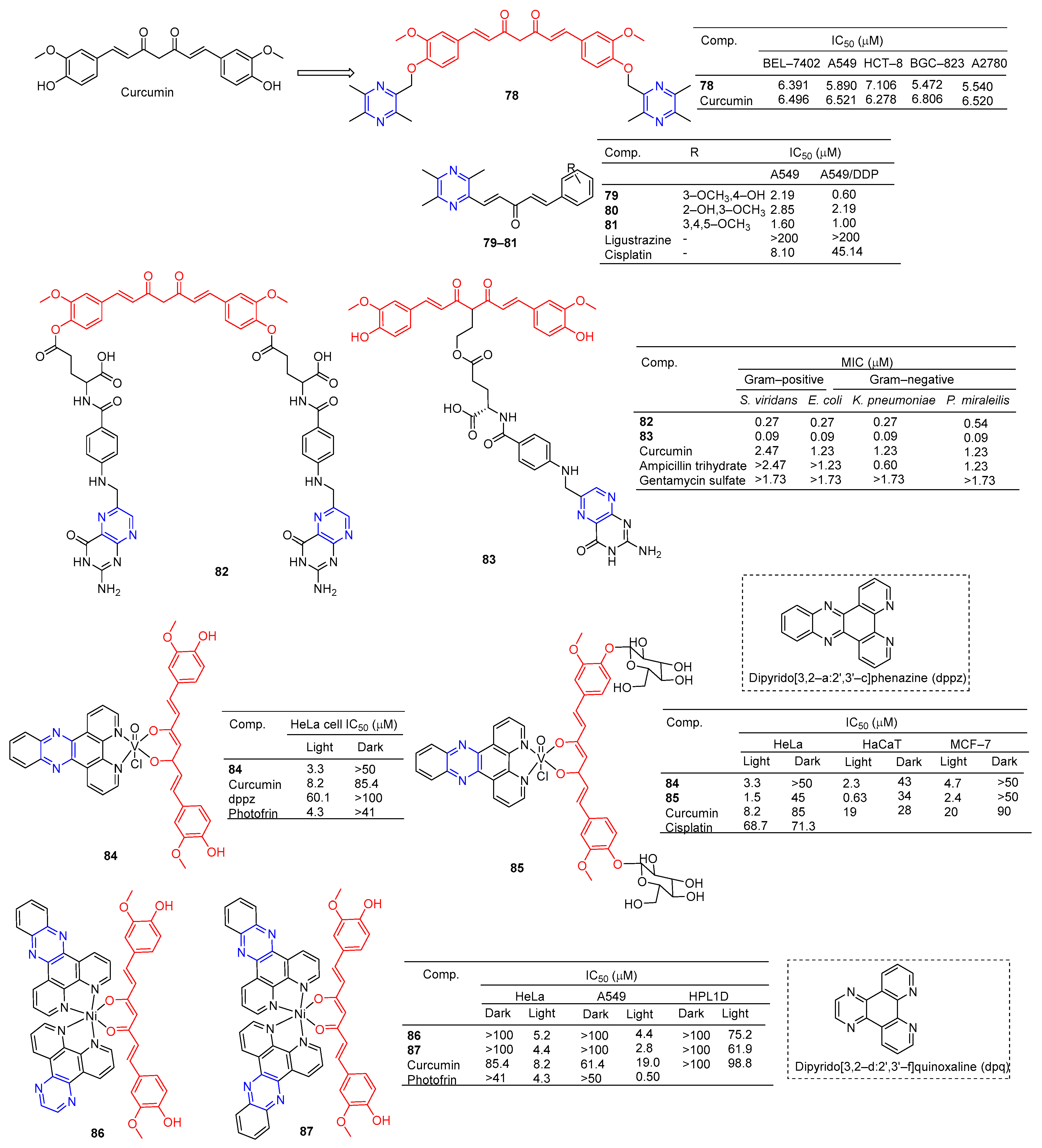{kind=link}
{kind=link}
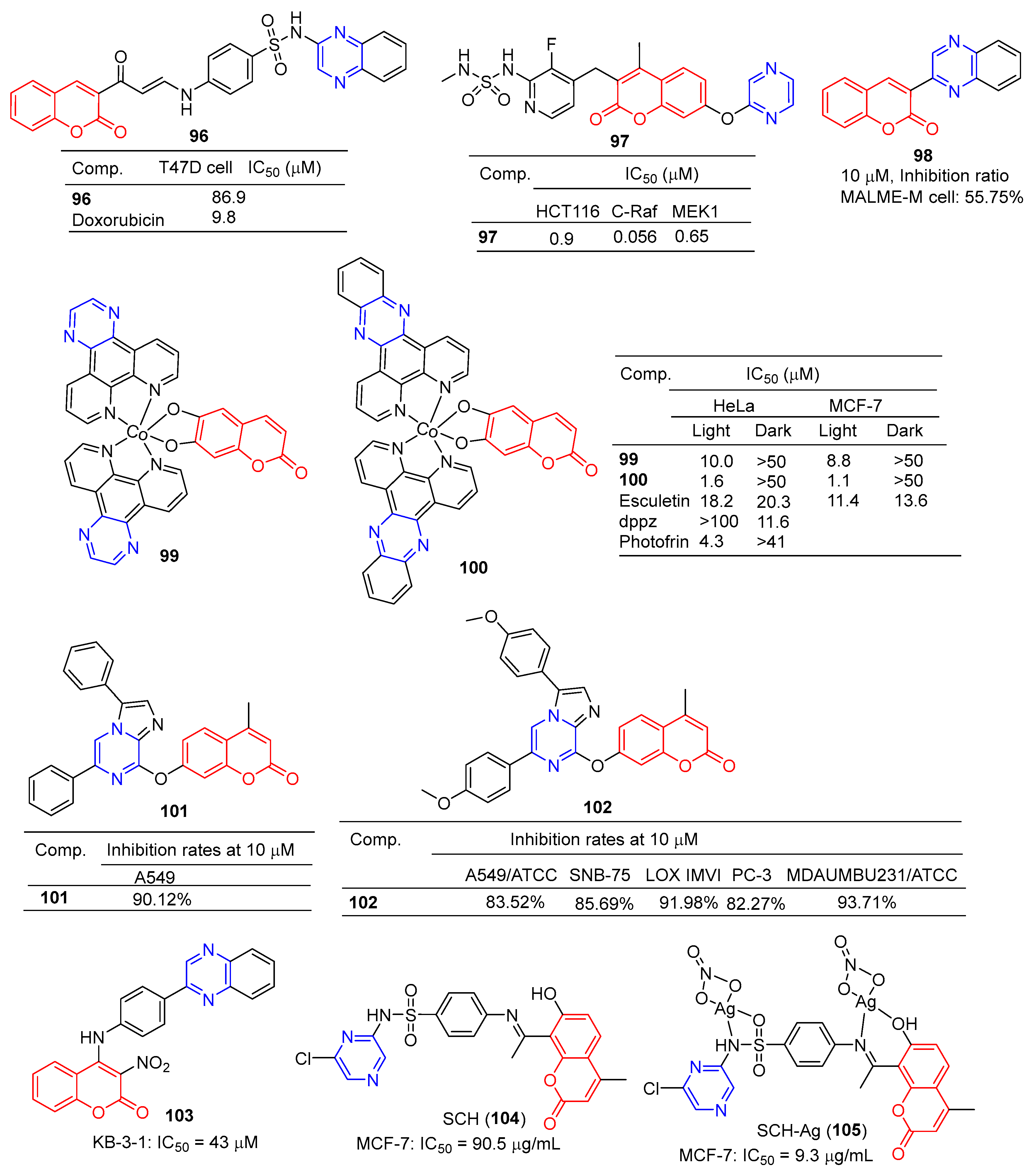{kind=link}
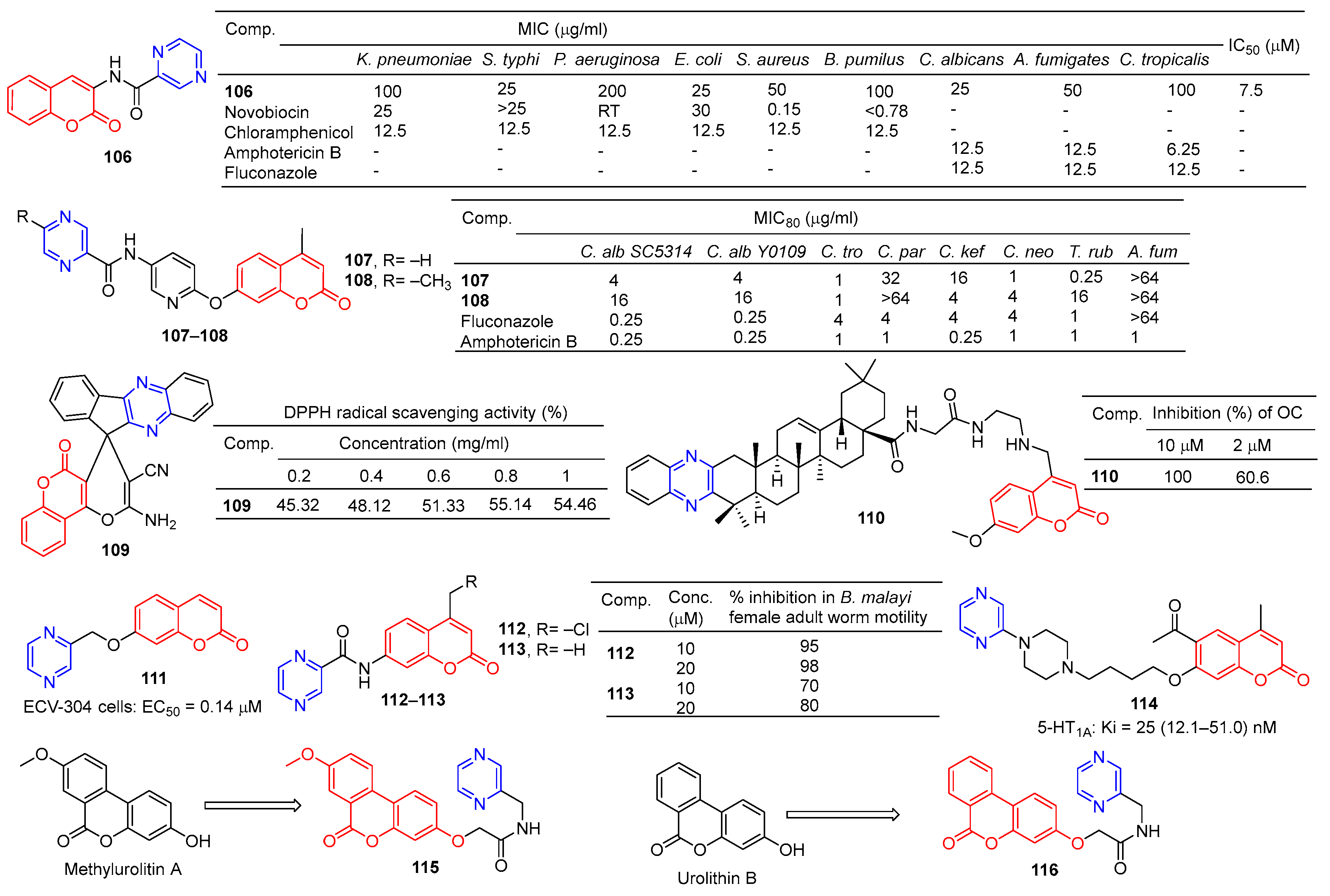{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
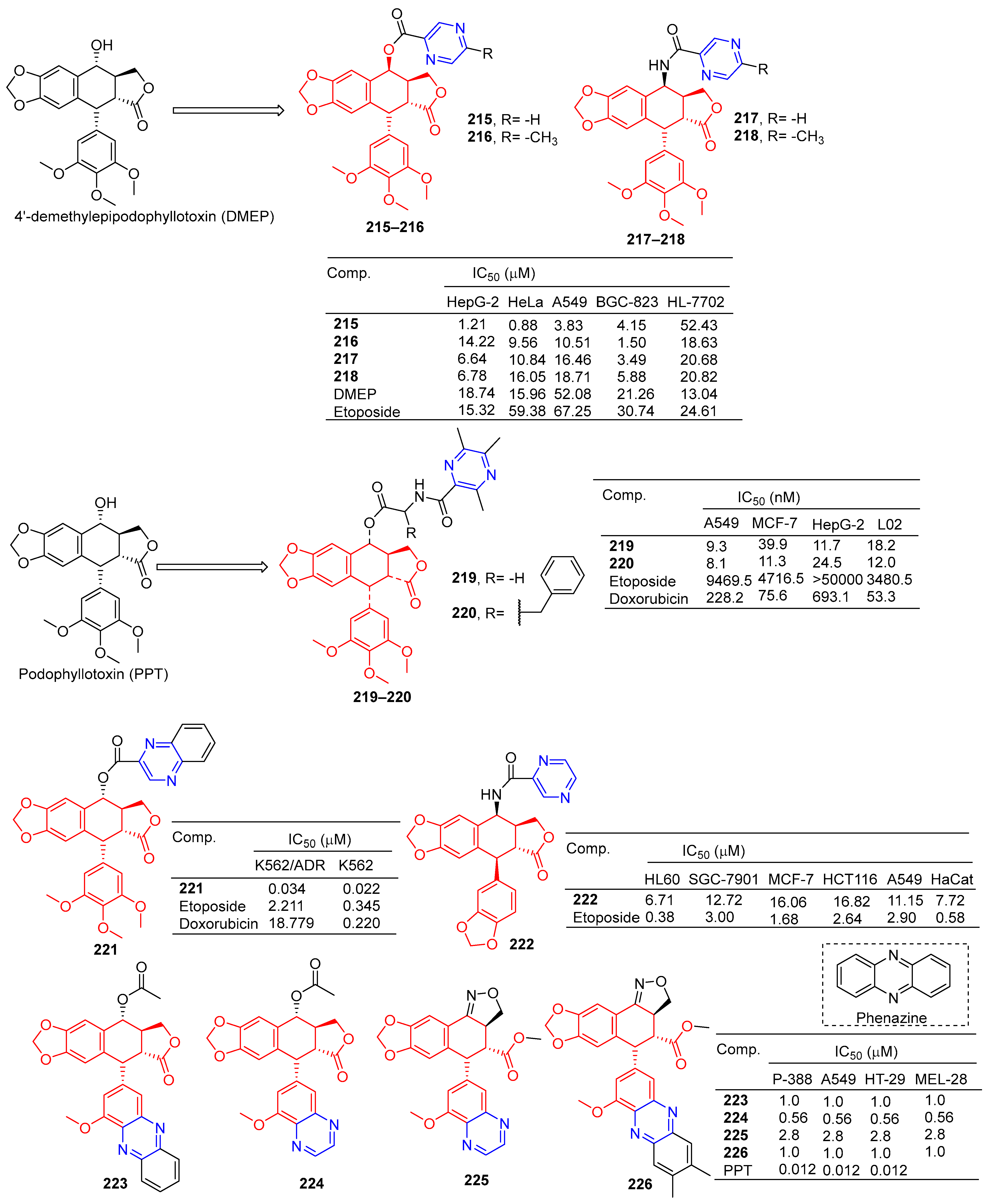{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
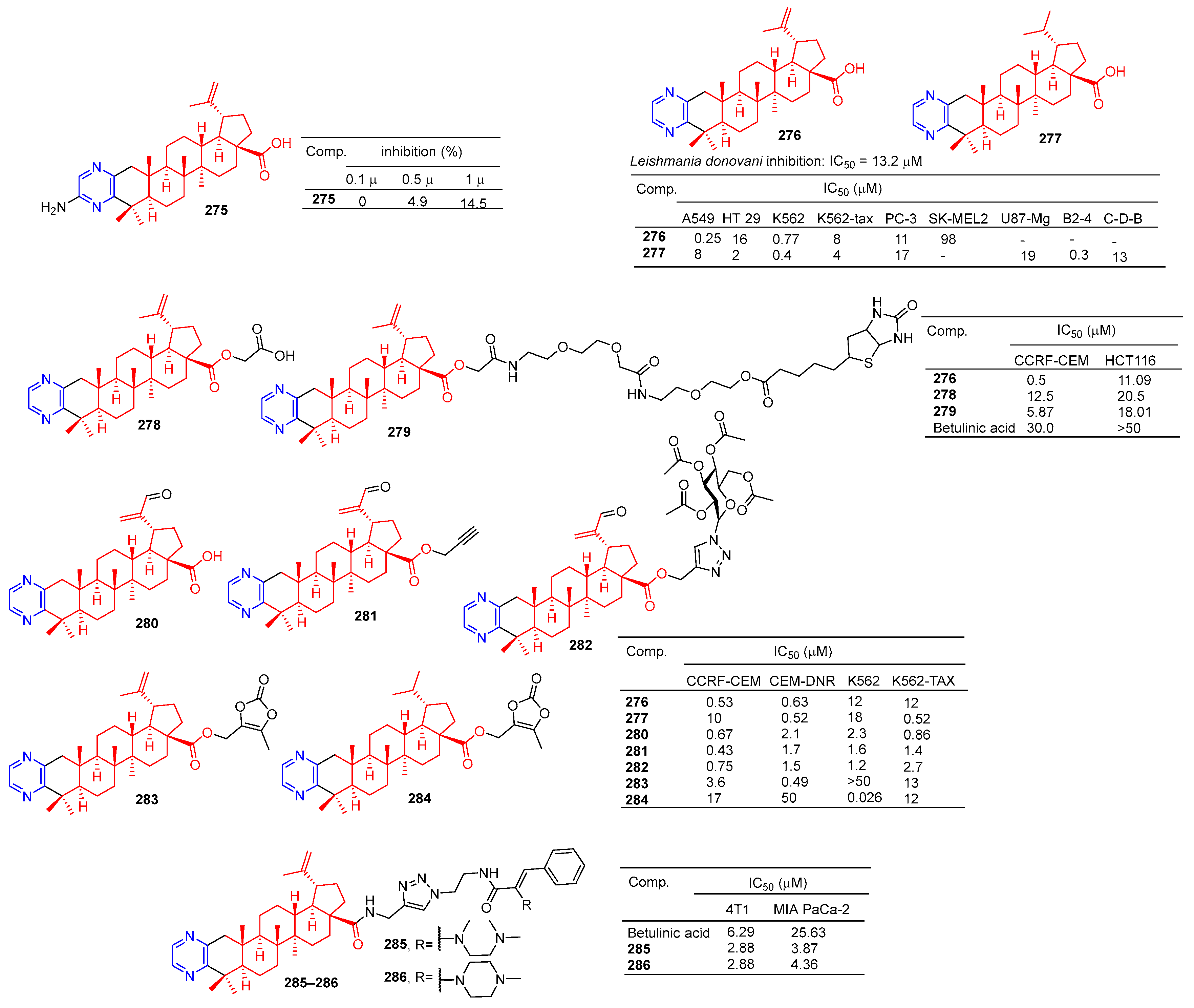{kind=link}
{kind=link}
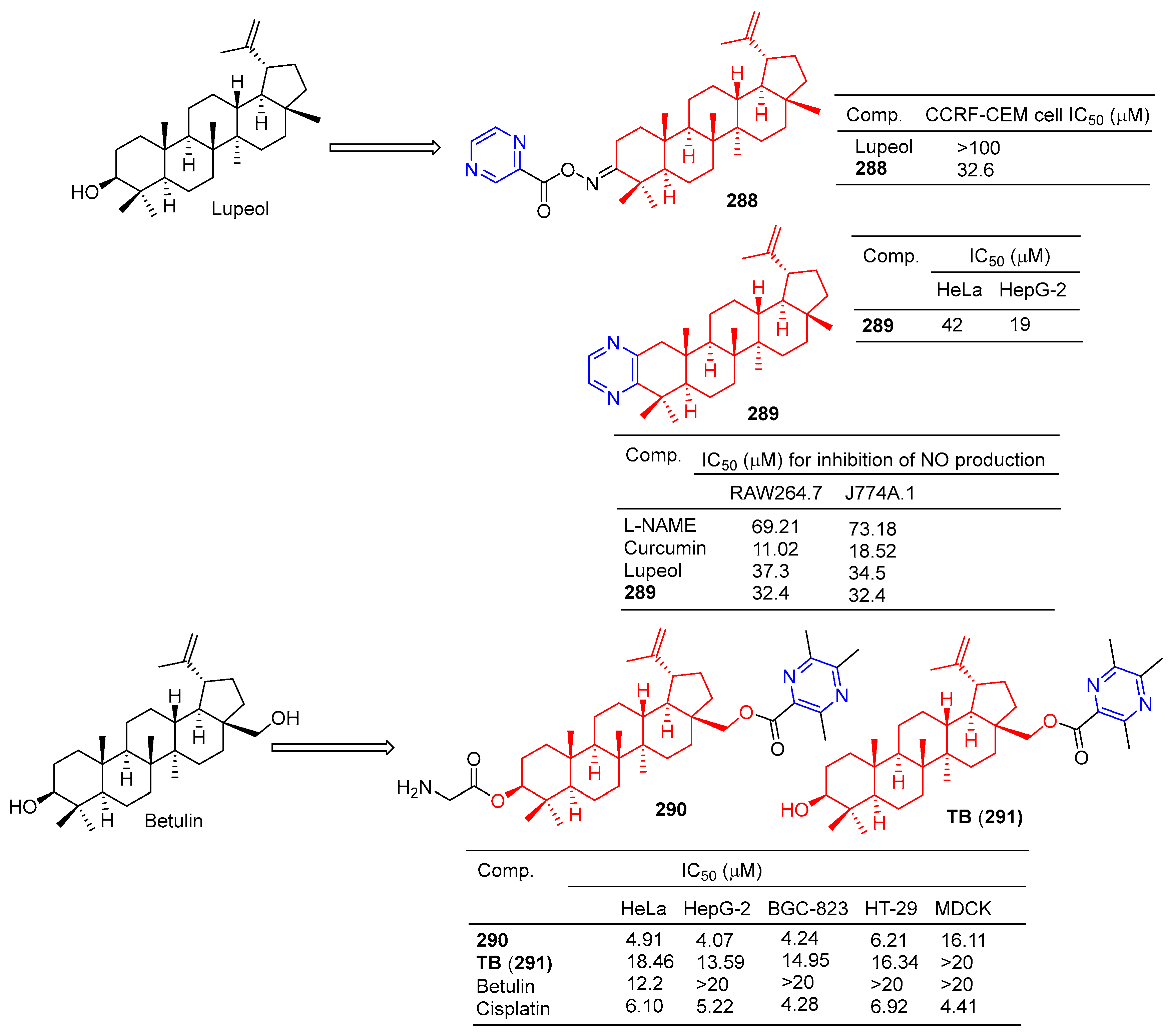{kind=link}
{kind=link}
{kind=link}
{kind=link}
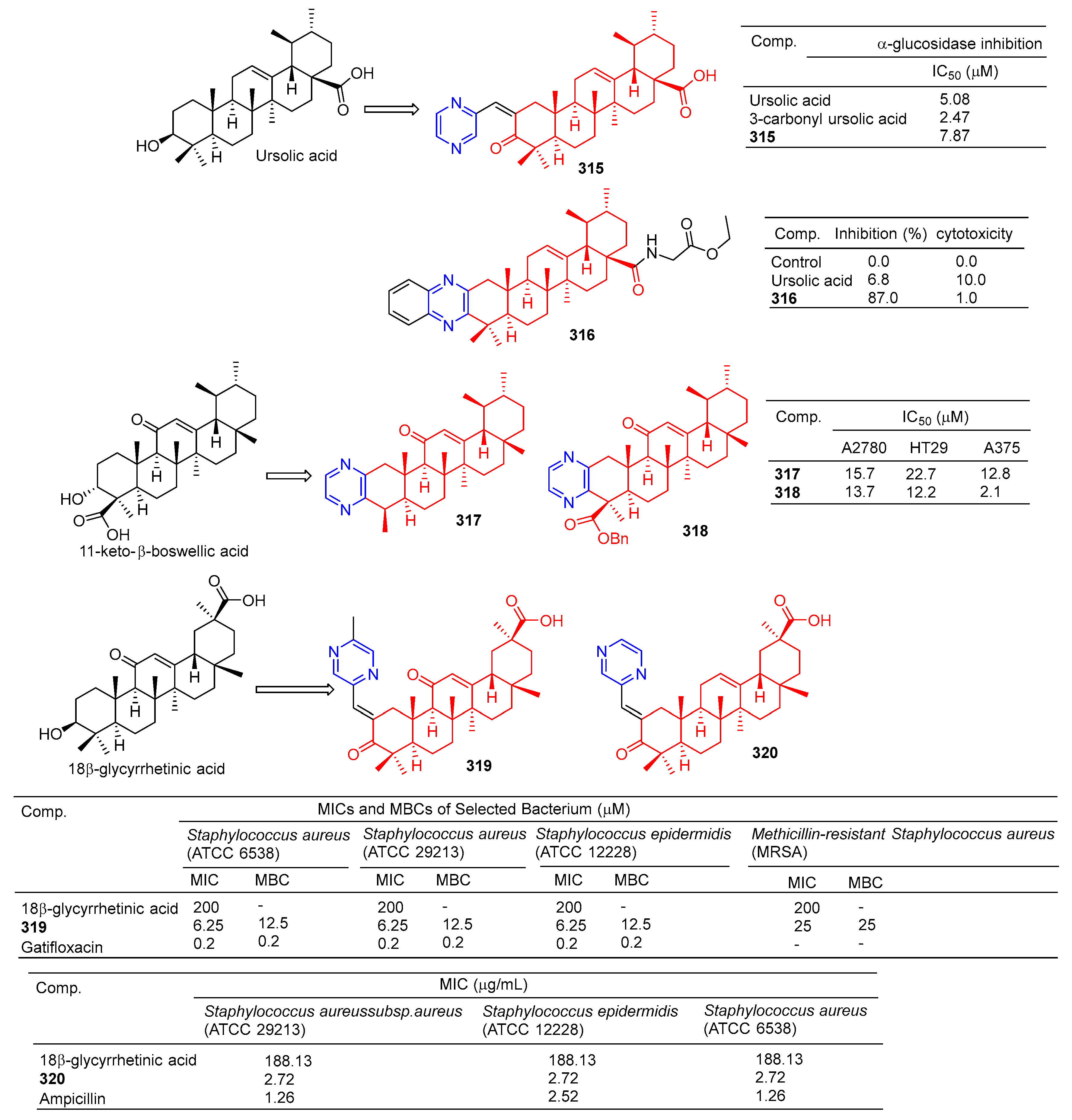{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Examples of pyrazine-containing drugs and their pharmacological applications.
| Drug | Structure | Biological Activity | Refs. |
|---|---|---|---|
| Acipimox |  | Hypolipidemic agent | [8] |
| Amiloride |  | Potassium sparing diuretic | [9] |
| Benzamil |  | Potassium sparing diuretic | [10] |
| Bortezomib |  | Proteasome inhibitor | [11,12,13] |
| Glipizide |  | Anti-diabetic agent | [14] |
| Morinamide |  | Anti-tubercular agent | [15] |
| Pyrazinamide |  | Anti-tubercular agent | [16] |
| Oltipraz |  | Schistosomicide and antitumor | [17] |
| Rimonabant (non-aryl derivative) |  | Cannabinoid receptor antagonist | [18] |
| Elpetrigine |  | Antiepileptic | [19] |
| Verenicline |  | Used to treat smoking addiction | [20] |
| Zibotentan |  | Anticancer agent | [21] |
| Amiloride |  | EnaC blocker | [22] |
| Paritaprevir |  | NS3-4A serine protease inhibitor hepatitis C treatment | [23] |
| Eszopiclone |  | Insomnia | [24,25] |
| Zopiclone |  | Treatment of sleep disorders | [26] |
| Cephalostatin 1 |  | Anticancer | [27,28] |
| Favipiravir |  | Antiviral (approved in Japan, influenza; FDA clearance, COVID-19) | [29,30,31] |
| (-)-Barrenazine A |  | Anticancer | [32] |
| (-)-Barrenazine B |  | Anticancer | [32] |
| Alocasin A |  | Anticancer | [33] |
| AKN-028 |  | Acute myeloid leukemia | [34,35] |
| Botryllazine |  | Anticancer | [36,37] |
| Phenazine-1-carboxylic acid |  | Antibacterial | [38] |
| 2-Bromo-1-hydroxy phenazine |  | Antibacterial | [39] |
| Griseolutein A |  | Antibacterial | [40] |
| HP-14 |  | Biofilm-eradicating agent | [41] |
| Iodinin |  | Antibacterial | [42] |
| Myxin |  | Antibacterial | [43] |
| Quinoxidine |  | Antibacterial | [44] |
| Dioxidine |  | Antibacterial | [44] |
| NC-190 |  | Anticancer | [45] |
| NC-182 |  | Anticancer | [46] |
| Erdafitinib |  | Anticancer | [47] |
| Pralatrexate |  | Anticancer | [48] |
| Methotrexate |  | Anticancer | [49] |
| Selinexor |  | Anticancer | [50] |
| Gilteritinib |  | Anticancer | [51] |
| Grazoprevir |  | Anti-hepatitis C virus | [52] |
| Telaprevir |  | Anti-hepatitis C virus | [53] |
| Triamterene |  | Potassium-sparing diuretics | [54] |
| Folic acid |  | Reduction of the neural tube defect risk | [55] |
| Selexipag (NS-304) |  | Pulmonary arterial hypertension | [56] |
| 2-Pyrazinecarboxamide |  | Antituberculosis | [57] |
| Thionazine |  | Insecticide and nematicide | [58] |
| Tetramethylpyrazine |  | Anti-inflammatory | [59] |
| Sulfalen |  | Urinary tract infection | [60] |
| Brimonidine |  | Glaucoma | [61] |
| Echinomycin |  | Antibacterial | [62] |
| Chloroquinoxaline sulfonamide |  | Anticancer | [63] |
| Brimondine |  | Rosacea | [64] |
| Pyrazine-2-diazohydroxide |  | Antitumor | [65] |
| Acipimox |  | Hyperlipidaemia | [66] |
| Sulfalene (sulfamethoxypyrazine) |  | Resistant falciparum malaria and antibacterial | [67] |
| Mirfentanil |  | Selectivity for the µ opioid receptor; analgesic | [68] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Chen, G.-Q.; Guo, H.-Y.; Quan, Z.-S.; Shen, Q.-K.; Li, X.; Luan, T. Natural Products–Pyrazine Hybrids: A Review of Developments in Medicinal Chemistry. Molecules 2023, 28, 7440. https://doi.org/10.3390/molecules28217440
AMA Style
Chen G-Q, Guo H-Y, Quan Z-S, Shen Q-K, Li X, Luan T. Natural Products–Pyrazine Hybrids: A Review of Developments in Medicinal Chemistry. Molecules. 2023; 28(21):7440. https://doi.org/10.3390/molecules28217440
Chicago/Turabian StyleChen, Guo-Qing, Hong-Yan Guo, Zhe-Shan Quan, Qing-Kun Shen, Xiaoting Li, and Tian Luan. 2023. "Natural Products–Pyrazine Hybrids: A Review of Developments in Medicinal Chemistry" Molecules 28, no. 21: 7440. https://doi.org/10.3390/molecules28217440