
Critical Evaluation of Two Qualitative Analytical Approaches for Multiclass Determination of Veterinary Drugs in Bovine Muscle Using UHPLC-Q-Orbitrap: The Wind of Change in Brazilian Monitoring

 ,
,
Abstract
:
1. Introduction
2. Results
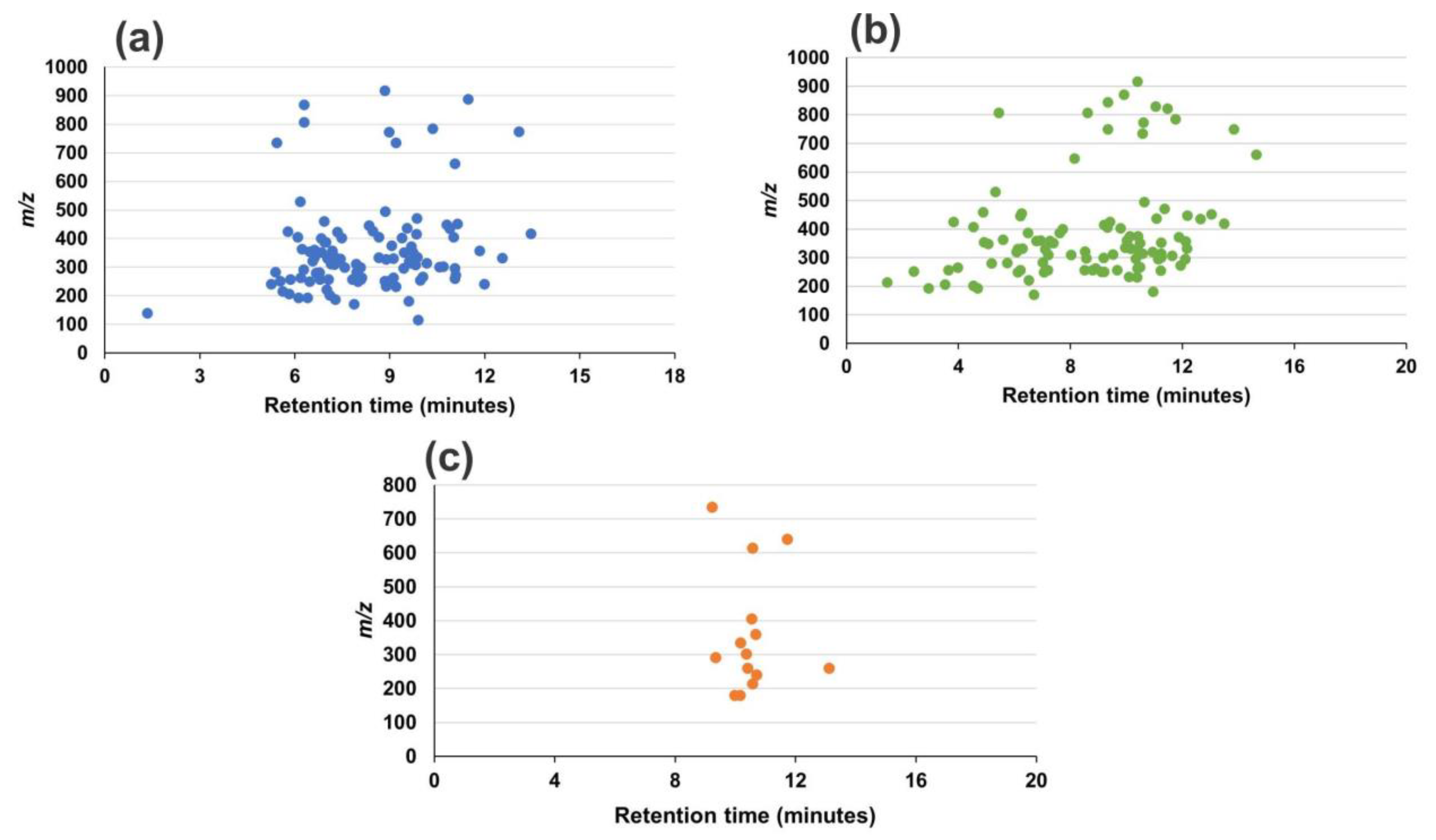
2.1. UHPLC-Q-Orbitrap Analysis Optimization
2.2. Extraction and Cleanup Procedures
2.3. Method Validation
2.3.1. Selectivity/Specificity
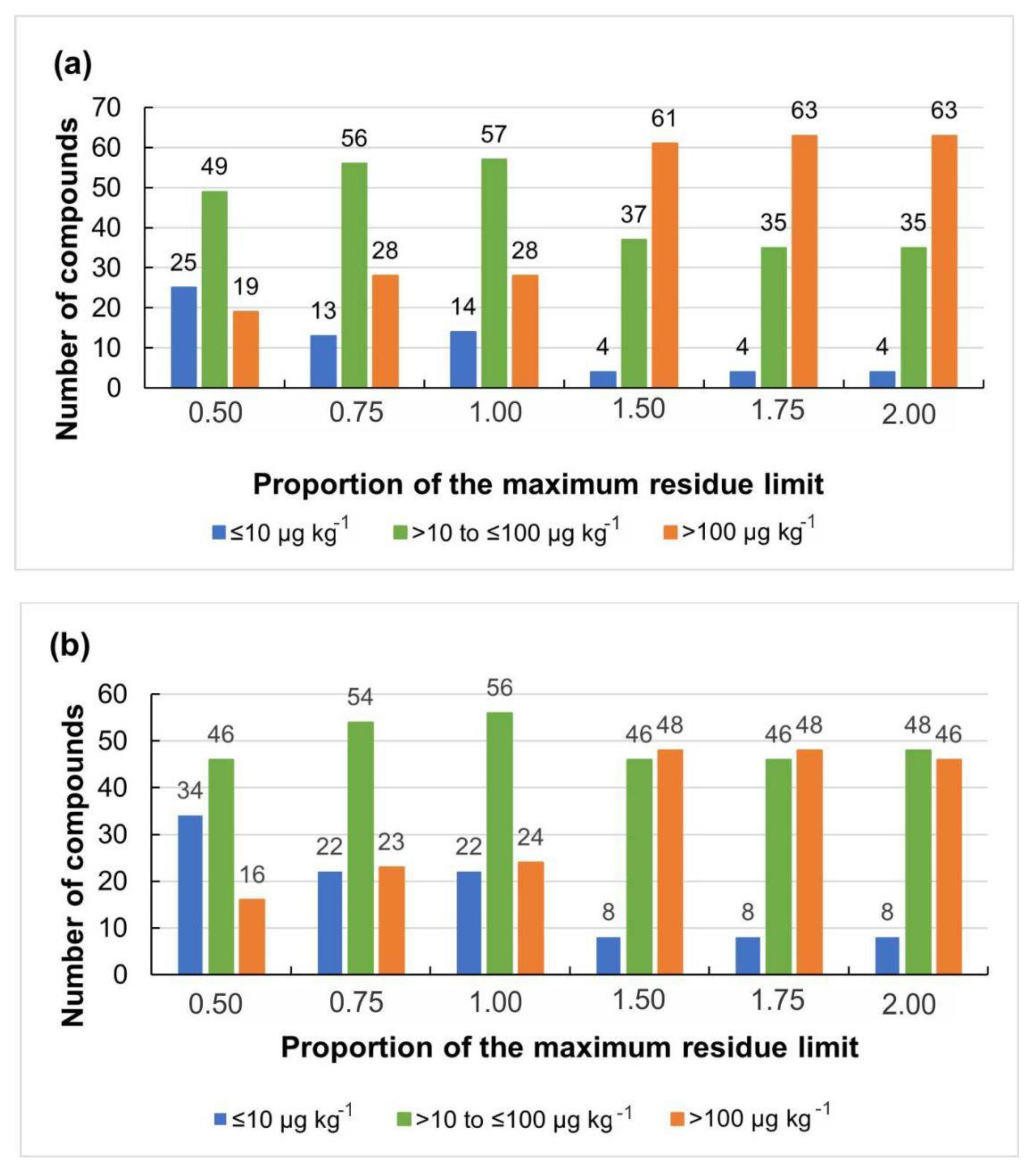
2.3.2. Detection Capability (CCβ)
3. Discussion
3.1. UHPLC-Q-Orbitrap Analysis Optimization
3.2. Extraction and Cleanup Procedures
3.2.1. Selectivity/Specificity
3.2.2. Detection Capability for Screening (CCβ)
4. Materials and Methods
4.1. Scope of Veterinary Drugs
4.2. Standard and Standard Solutions
4.3. Reagents
4.4. Sample Preparation
4.4.1. Method A
4.4.2. Method B
4.5. Instrumentation
4.5.1. Chromatographic Conditions
4.5.2. Q-Orbitrap High-Resolution Mass Spectrometry Parameters
4.6. Qualitative Screening Validation Study
4.6.1. Selectivity/Specificity
4.6.2. Detection Capacity (CCβ)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Confederação da Agricultura e Pecuária do Brasil. PIB do Agronegócio Cresceu Abaixo das pr#ojeções; CEPEA: São Paulo, Brazil, 2022; Available online: https://www.cepea.esalq.usp.br/upload/kceditor/files/Cepea_CNA_PIB_JAn_Dez_2021_Mar%C3%A7o2022.pdf (accessed on 14 September 2022).
- Brasil. Ministério da Agricultura, Pecuária e Abastecimento. Indicadores Gerais AGROSTAT; MAPA: Brasília, Brazil, 2022; Available online: https://indicadores.agricultura.gov.br/agrostat/ index.htm (accessed on 14 September 2022).
- United States of America. Department of Agriculture. Graphical Query: Stats by Country; USDA: Washington, DC, USA, 2022. Available online: https://apps.fas.usda.gov/psdonline/app/index.html#/app/topCountriesByCommodity#table28 (accessed on 14 September 2022).
- Dohlman, E.; Hansen, J.; Boussios, D. USDA Agricultural Projections to 2031; USDA: Washington, DC, USA, 2022. Available online: https://www.ers.usda.gov/publications/pub-details/?pubid=103309 (accessed on 14 September 2022).
- Mulchandani, R.; Wang, Y.; Gilbert, M.; Van Boeckel, T.P. Global trends in antimicrobial use in food animals from 2017 to 2030. PLOS Glob. Public Health 2023, 3, e0001305. Available online: https://journals.plos.org/globalpublichealth/article?id=10.1371/journal.pgph.0001305 (accessed on 28 February 2023). [CrossRef]
- U.S. Department of Agriculture. Beef, Short Loin, Porterhouse Steak, Separable Lean Only, Trimmed to 1/8" Fat, Select, Raw; USDA: Washington, DC, USA, 2019. Available online: https://fdc.nal.usda.gov/fdc-app.html#/food-details/746762/nutrients (accessed on 14 September 2022).
- European Comission Regulation. (EC) No 470/2009 of the European Parliament and of the Council of 6 May 2009 laying down Community procedures for the establishment of residue limits of pharmacologically active substances in foodstuffs of animal origin, repealing Council Regulation (EEC) No 2377/90 and amending Directive 2001/82/EC of the European Parliament and of the Council and Regulation (EC) No 726/2004 of the European Parliament and of the Council. Off. J. Eur. Union 2009, L152, 11–22. Available online: https://fdc.nal.usda.gov/fdc-app.html#/food-details/746762/nutrientshttps://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32009R0470&from=EN (accessed on 14 September 2022).
- Food and Agriculture Organization. Codex Alimentarius Commission: Procedural Manual, 27th ed.; FAO: Rome, Italy, 2019; Available online: https://www.fao.org/3/ca2329en/CA2329EN.pdf (accessed on 14 September 2022).
- Kleinubing, N.R.; Ramires, T.; Würfel, S.F.R.; Haubert, L.; Scheik, L.K.; Kremer, F.S.; Lopes, G.V.; Silva, W.P. Antimicrobial resistance genes and plasmids in Campylobacter jejuni from broiler production chain in Southern Brazil. LWT 2021, 144, 111202. [Google Scholar] [CrossRef]
- Martins, B.T.F.; Meirelles, J.L.; Omori, W.P.; Oliveira, R.R.; Yamatogi, R.S.; Call, D.R.; Nero, L.A. Comparative genomics and antibiotic resistance of Yersinia enterocolitica obtained from a pork production chain and human clinical cases in Brazil. Food Res. Int. 2022, 152, 110917. [Google Scholar] [CrossRef]
- Dewdney, J.M.; Maes, L.; Raynaud, J.P.; Blanc, F.; Scheid, J.P.; Jackson, T.; Lens, S.; Verschueren, C. Risk assessment of antibiotic residues of β-lactams and macrolides in food products with regard to their immuno-allergic potential. Food Chem. Toxicol. 1991, 29, 477–483. [Google Scholar] [CrossRef]
- Molina, J.D.A.; Vargas, R.H.L.; Gutierrez, J.A.B.; Gallo-Ortiz, A.; Duarte-Correa, Y. Residues of veterinary drugs and heavy metals in bovine meat from Urabá (Antioquia, Colombia), a promising step forward towards international commercialization. Vet. Anim. Sci. 2021, 13, 100192. [Google Scholar] [CrossRef]
- Saraithong, W. Trade restriction rationale for food safety implementation: Evidence from Southeast Asian Countries. Cogent Econ. Finan. 2018, 6, 1553278. [Google Scholar] [CrossRef]
- Wei, G.-X.; Yuang, J.-K.; Yang, J. Honey safety standards and its impacts on China’s honey export. J. Integr. Agric. 2012, 11, 684–693. [Google Scholar] [CrossRef]
- Barrios, R.E.; Khuntia, H.K.; Bartelt-Hunt, S.L.; Gilley, J.E.; Schmidt, A.M.; Snow, D.D.; Li, X. Fate and transport of antibiotics and antibiotic resistance genes in runoff and soil as affected by the timing of swine manure slurry application. Sci. Total Environ. 2020, 712, 136505. [Google Scholar] [CrossRef]
- Brasil, Ministério da Agricultura. Portaria nº 86, de 26 de Janeiro de 1979. Aprova o Programa Nacional de Controle de Resíduos Biológicos em Carnes, a Ser Executado no Âmbito da Secretaria Nacional de Defesa Agropecuária. In Diário Oficial da União; 7 January 1979. Available online: https://www.ibama.gov.br/component/legislacao/?view=legislacao&legislacao=91640 (accessed on 14 September 2022).
- Brasil, Ministério da Agricultura, Pecuária e Abastecimento. Resultados do Plano Nacional de Controle de Resíduos e Contaminantes—PNCRC 2021; MAPA: Brasília, Brazil, 2021. Available online: https://www.gov.br/agricultura/pt-br/assuntos/inspecao/produtos-animal/plano-de-nacional-de-controle-de-residuos-e-contaminantes/consolidado-resultados-pncrc-2021.pdf (accessed on 14 September 2022).
- Mauricio, A.Q.; Lins, E.S.; Alvarenga, M.B. A national residue control plan from the analytical perspective: The Brazilian case. Anal. Chim. Acta 2009, 637, 333–336. [Google Scholar] [CrossRef]
- Barreto, F.; Ribeiro, C.; Hoff, R.B.; Costa, T.D. Determination of chloramphenicol, thiamphenicol, florfenicol and florfenicol amine in poultry, swine, bovine and fish by liquidchromatography-tandem mass spectrometry. J. Chromatogr. A 2016, 1449, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Jank, L.; Martins, M.T.; Arsand, J.B.; Motta, T.M.C.; Hoff, R.B.; Barreto, F.; Pizzolato, T.M. High-throughput method for macrolides and lincosamides antibiotics residues analysis in milk and muscle using a simple liquid–liquid extraction technique and liquid chromatography–electrospray–tandem mass spectrometry analysis (LC–MS/MS). Talanta 2015, 144, 686–695. [Google Scholar] [CrossRef] [PubMed]
- Pastore, V.A.A.; Santos, F.A.; Lana, M.A.G.; Silva, G.R.; Figueiredo, T.C.; Assis, D.C.S.; Cançado, S.V. Development and validation of a multiresidue method for the determination of macrocyclic lactones, monensin, and fipronil in bovine liver by UHPLC-MS/MS using a QuEChERS extraction. Food Anal. Methods 2022, 15, 3177–3188. [Google Scholar] [CrossRef]
- Silva, G.R.; Lima, J.A.; Souza, L.F.; Santos, F.A.; Lana, M.A.G.; Assis, D.C.S.; Cançado, S.V. Multiresidue method for identification and quantification of avermectins, benzimidazoles and nitroimidazoles residues in bovine muscle tissue by ultra-high performance liquid chromatography tandem mass spectrometry (UHPLC-MS/MS) using a QuEChERS approach. Talanta 2017, 171, 307–320. [Google Scholar] [CrossRef]
- Martins, M.T.; Melo, J.; Barreto, F.; Hoff, R.B.; Jank, L.; Bittencourt, M.S.; Arsand, J.B.; Schapoval, E.E.S. A simple, fast and cheap non-SPE screening method for antibacterial residue analysis in milk and liver using liquid chromatography–tandem mass spectrometry. Talanta 2014, 129, 374–383. [Google Scholar] [CrossRef]
- Kaufmann, A. The current role of high-resolution mass spectrometry in food analysis. Anal. Bioanal. Chem. 2012, 403, 1233–1249. [Google Scholar] [CrossRef]
- Kaufmann, A.; Arrizabalaga-Larrañaga, A.; Blokland, M.H.; Sterk, S.S. Potential and limitation of retrospective HRMS based data analysis: “Have meat-producing animals been exposed to illegal growth promotors such as SARMs? Food Control 2023, 147, 109611. [Google Scholar] [CrossRef]
- You, Y.; Proctor, R.M.; Guo, K.; Li, X.; Xue, E.; Guan, F.; Robinson, M.A. Use of high resolution/accurate mass full scan/data-dependent acquisition for targeted/non-targeted screening in equine doping control. Anal. Methods 2021, 13, 1565–1575. [Google Scholar] [CrossRef]
- Jongedijk, E.; Fifeik, M.; Arrizabalaga-Larrañaga, A.; Polzer, J.; Blokland, M.; Sterk, S. Use of high-resolution mass spectrometry for veterinary drug multi-residue analysis. Food Control 2023, 145, 109488. [Google Scholar] [CrossRef]
- Spisso, B.F.; Pereira, M.U.; Ferreira, R.G. Methods of analysis for the determination of veterinary drugs in food: Focus on antimicrobials. In Encyclopedia of Analytical Chemistry: Applications, Theory and Instrumentation; Meyers, R.A., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2022. [Google Scholar] [CrossRef]
- Hepperle, J.; Dörk, D.; Barth, A.; Taşdelen, B.; Anastassiades, M. Studies to improve the extraction yields of incurred pesticide residues from crops using the QuEChERS method. J. AOAC Int. 2015, 98, 450–463. [Google Scholar] [CrossRef]
- Dasenaki, M.E.; Michali, C.S.; Thomaidis, N.S. Analysis of 76 veterinary pharmaceuticals from 13 classes including aminoglycosides in bovine muscle by hydrophilic interaction liquid chromatography–tandem mass spectrometry. J. Chromatogr. A 2016, 1452, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Gosetti, F.; Mazzucco, E.; Zampieri, D.; Gennaro, M.C. Signal suppression/enhancement in high-performance liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2010, 1217, 3929–3937. [Google Scholar] [CrossRef]
- Saluti, G.; Diamanti, I.; Giusepponi, D.; Pucciarini, L.; Rossi, R.; Moretti, S.; Sardella, R.; Galarinia, R. Simultaneous determination of aminoglycosides and colistins in food. Food Chem. 2018, 266, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Tian, H.; Lian-feng Ai, L.-F.; Liang, S.-X. Screening and quantification of 146 veterinary drug residues in beef and chicken using QuEChERS combined with high performance liquid chromatography-quadrupole orbitrap mass spectrometry. Food Chem. 2023, 408, 135207. [Google Scholar] [CrossRef] [PubMed]
- Sardela, V.F.; Martucci, M.E.P.; Araújo, A.L.D.; Leal, E.C.; Oliveira, D.S.; Carneiro, G.R.A.; Deventer, K.; Van Eenoo, P.; Pereira, H.M.G.; Aquino Neto, F.R. Comprehensive analysis by liquid chromatography Q-Orbitrap mass spectrometry: Fast screening of peptides and organic molecules. J. Mass Spectrom. 2018, 53, 476–503. [Google Scholar] [CrossRef] [PubMed]
- Moretti, S.; Cavanna, D.; Lambertini, F.; Catellani, D.; Sammarco, G.; Barola, C.; Paoletti, F.; Saluti, G.; Galarini, R.; Suman, M. Practical approach to develop a multi-group screening method for detection of mycotoxins, pesticides and veterinary drugs in food. J. Mass. Spectrom. 2020, 55, 4618. [Google Scholar] [CrossRef] [PubMed]
- Pugajeva, I.; Ikkere, L.E.; Judjallo, E.; Bartkevics, V. Determination of residues and metabolites of more than 140 pharmacologically active substances in meat by liquid chromatography coupled to high resolution Orbitrap mass spectrometry. J. Pharm. Biomed. Anal. 2019, 166, 252–263. [Google Scholar] [CrossRef]
- Uczay, F.; Bandeira, N.M.G.; Floriano, L.; Prestes, O.D.; Adaime, M.B.; Zanella, R. Determination of avermectins residues in soybean, bean, and maize using a QuEChERS-based method and ultra-high-performance liquid chromatography coupled to tandem mass spectrometry. Separations 2021, 8, 214. [Google Scholar] [CrossRef]
- Moscou, I.C.; Dasenaki, M.E.; Thomaidis, N.S. Ionization study and simultaneous determination of avermectins and milbemycines in fish tissue by LC-ESI-MS/MS. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2019, 1104, 134–140. [Google Scholar] [CrossRef]
- Sismotto, M.; Paschoal, J.A.R.; Teles, J.A.; Rezende, R.A.E.; Reyes, F.G.R. A simple liquid chromatography coupled to quadrupole time of flight mass spectrometry method for macrolide determination in tilapia fillets. J. Food Compos. Anal. 2014, 34, 153–162. [Google Scholar] [CrossRef]
- Grunwald, L.; Petz, M. Food processing effects on residues: Penicillins in milk and yoghurt. Anal. Chim. Acta 2003, 483, 73–79. [Google Scholar] [CrossRef]
- Qi, P.; Zhou, Q.-Q.; Lin, Z.-H.; Liu, J.; Cai, W.-Y.; Mao, X.-W.; Jiang, J.J. Qualitative screening and quantitative determination of multiclass water-soluble synthetic dyes in foodstuffs by liquid chromatography coupled to quadrupole Orbitrap mass spectrometry. Food Chem. 2021, 360, 129948. [Google Scholar] [CrossRef] [PubMed]
- Campanholi, K.S.; Ana Beatriz Zanqui, A.B.; Morais, F.A.P.; Jaski, J.M.; Gonçalves, R.S.; Silva Junior, R.C.; Cardozo-Filho, L.; Caetano, W. Obtaining phytotherapeutic chlorophyll extracts using pressurized liquid technology. J. Supercrit. Fluids 2022, 180, 105457. [Google Scholar] [CrossRef]
- Gałuszka, A.; Migaszewski, Z.; Namieśnik, J. The 12 principles of green analytical chemistry and the SIGNIFICANCE mnemonic of green analytical practices. TrAC 2013, 50, 78–84. [Google Scholar] [CrossRef]
- Cifuentes, A. Food analysis: Present, future, and foodomics. Int. Sch. Res. Netw. 2012, 2012, 801607. [Google Scholar] [CrossRef]
- García-Cañas, V.; Simó, C.; Herrero, M.; Ibáñez, E.; Cifuentes, A. Present and future challenges in food analysis: Foodomics. Anal. Chem. 2012, 84, 10150–10159. [Google Scholar] [CrossRef]
- Kaufmann, A.; Butcher, P.; Maden, K.; Walker, S.; Widmer, M. Using in silico fragmentation to improve routine residue screening in complex matrices. J. Am. Soc. Mass Spectrom. 2017, 28, 2705–2715. [Google Scholar] [CrossRef]
- Berendsen, B.J.A.; Stolker, L.A.M.; Nielen, M.W.F. Selectivity in the sample preparation for the analysis of drug residues in products of animal origin using LC-MS. TrAC 2013, 43, 229–239. [Google Scholar] [CrossRef]
- Dasenaki, M.E.; Bletsou, A.A.; Koulis, G.A.; Thomaidis, N.S. Qualitative multiresidue screening method for 143 veterinary drugs and pharmaceuticals in milk and fish tissue using liquid chromatography quadrupole-time-of-flight mass spectrometry. J. Agric. Food Chem. 2015, 63, 4493–4508. [Google Scholar] [CrossRef]
- Turnipseed, S.B.; Storey, J.M.; Lohne, J.J.; Andersen, W.C.; Burger, R.; Johnson, A.S.; Madson, M.R. Wide-scope screening method for multiclass veterinary drug residues in fish, shrimp, and eel using liquid chromatography–quadrupole high-resolution mass spectrometry. J. Agric. Food Chem. 2017, 65, 7252–7267. [Google Scholar] [CrossRef]
- Boix, C.; Ibáñez, M.; Sancho, J.V.; León, N.; Yusá, V.; Hernández, F. Qualitative screening of 116 veterinary drugs in feed by liquid chromatography–high resolution mass spectrometry: Potential application to quantitative analysis. Food Chem. 2014, 160, 313–332. [Google Scholar] [CrossRef] [PubMed]
- Stolker, A.A.M.; Rutgers, P.; Oosterink, E.; Lasaroms, J.J.P.; Peters, R.J.B.; van Rhijn, J.A.; Nielen, M.W.F. Comprehensive screening and quantification of veterinary drugs in milk using UPLC-ToF-MS. Anal. Bioanal. Chem. 2008, 391, 2309–2322. [Google Scholar] [CrossRef] [PubMed]
- Turnipseed, S.B.; Lohne, J.J.; Storey, J.M.; Andersen, W.C.; Young, S.L.; Carr, J.R.; Madson, M.R. Challenges in implementing a screening method for veterinary drugs in milk using liquid chromatography quadrupole time-of-flight mass spectrometry. J. Agric. Food Chem. 2014, 62, 3660–3674. [Google Scholar] [CrossRef] [PubMed]
- Kaklamanos, G.; Vincent, U.; Holst, C. Analysis of antimicrobial agents in pig feed by liquid chromatography coupled to orbitrap mass spectrometry. J. Chromatogr. A 2013, 1293, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, A.; Butcher, P.; Maden, K.; Widmer, M. Quantitative multiresidue method for about 100 veterinary drugs in different meat matrices by sub 2-μm particulate high-performance liquid chromatography coupled to time of flight mass spectrometry. J. Chromatogr A 2008, 1194, 66–79. [Google Scholar] [CrossRef]
- Kang, J.; Fan, C.-L.; Cao, Y.-F.; Wang, H.-J.; Peng, X.; Wang, Z.-B.; Chang, Q.-Y.; Hub, X.-Y.; Pang, G.-F. Multi-residue screening of 100 multi-class veterinary drugs in milk powder by liquid chromatography coupled to quadrupole time-of-flight mass spectrometry. Anal. Methods 2014, 6, 8337–8349. [Google Scholar] [CrossRef]
- Kaufmann, A.; Butcher, P.; Maden, K.; Walker, S.; Widmer, M. Quantification of anthelmintic drug residues in milk and muscle tissues by liquid chromatography coupled to Orbitrap and liquid chromatography coupled to tandem mass spectrometry. Talanta 2011, 85, 991–1000. [Google Scholar] [CrossRef]
- Dasenaki, M.E.; Thomaidis, N.S. Multi-residue determination of 115 veterinary drugs and pharmaceutical residues in milk powder, butter, fish tissue and eggs using liquid chromatography–tandem mass spectrometry. Anal. Chim. Acta 2015, 880, 103–121. [Google Scholar] [CrossRef]
- Mastovska, K.; Lightfield, A.R. Streamlining methodology for the multiresidue analysis of beta-lactam antibiotics in bovine kidney using liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2008, 1202, 118–123. [Google Scholar] [CrossRef]
- Freitas, A.; Barbosa, J.; Ramos, F. Multi-residue and multi-class method for the determination of antibiotics in bovine muscle by ultra-high-performance liquid chromatography tandem mass spectrometry. Meat Sci. 2014, 98, 58–64. [Google Scholar] [CrossRef]
- Lopes, R.P.; Reyes, R.C.; Romero-González, R.; Frenich, A.G.; Vidal, J.L.M. Development and validation of a multiclass method for the determination of veterinary drug residues in chicken by ultra high performance liquid chromatography–tandem mass spectrometry. Talanta 2012, 89, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Gros, M.; Rodríguez-Mozaz, S.; Barceló, D. Rapid analysis of multiclass antibiotic residues and some of their metabolites in hospital, urban wastewater and river water by ultra-high-performance liquid chromatography coupled to quadrupole-linear ion trap tandem mass spectrometry. J. Chromatogr. A 2013, 1292, 173–188. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Xu, X.; Han, M.; Shiting Qiu, S.; Hou, X. Development of a modified QuEChERS method based on magnetic multiwalled carbon nanotubes for the simultaneous determination of veterinary drugs, pesticides and mycotoxins in eggs by UPLC-MS/MS. Food Chem. 2019, 276, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Mitscher, L.A. (Ed.) The Chemistry of the Tetracycline Antibiotics; Marcel Dekker: New York, NY, USA, 1978. [Google Scholar]
- Castro, M.D.L.; Silva, M.P. Strategies for solid sample treatment. Trends Anal. Chem. 1997, 16, 16–24. [Google Scholar] [CrossRef]
- Clark, S.B.; Turnipseed, S.B.; Nandre, G.J.; Madson, M.R.; Hurlbut, J.A.; Sofos, J.N. Confirmation of phenylbutazone residues in bovine kidney by liquid chromatography/mass spectrometry. J. AOAC Int. 2002, 85, 1009–1014. [Google Scholar] [CrossRef]
- Martines, M.A.U.; Davolos, M.R.; Jafelicci Junior, M. O efeito do ultra-som em reações químicas. Quím. Nova 2000, 23, 251–256. Available online: https://www.scielo.br/j/qn/a/gTBJfM66ZcYw9LjnhRwqzPy/?format=pdf&lang=pt (accessed on 24 April 2022). [CrossRef]
- Bittencourt, M.S.; Martins, M.T.; Albuquerque, F.G.S.; Barreto, F.; Hoff, R. High-throughput multiclass screening method for antibiotic residue analysis in meat using liquid chromatography-tandem mass spectrometry: A novel minimum sample preparation procedure. Food Addit. Contam. Part A 2012, 29, 508–516. [Google Scholar] [CrossRef]
- Kemmerich, M.; Demarco, M.; Bernardi, G.; Prestes, O.D.; Adaime, M.B.; Zanella, R. Balls-in-tube matrix solid phase dispersion (BiT-MSPD): An innovative and simplified technique for multiresidue determination of pesticides in fruit samples. J. Chromatogr. A 2020, 1612, 460640. [Google Scholar] [CrossRef]
- Rizetti, T.M.; Souza, M.P.; Prestes, O.D.; Adaime, M.B.; Zanella, R. Optimization of sample preparation by central composite design for multi-class determination of veterinary drugs in bovine muscle, kidney and liver by ultra-high-performance liquid chromatographic-tandem mass spectrometry. Food Chem. 2018, 246, 404–413. [Google Scholar] [CrossRef]
- Goulart, S.M.; Queiroz, M.E.L.R.; Neves, A.A.; Queiroz, J.H. Low-temperature clean-up method for the determination of pyrethroids in milk using gas chromatography with electron capture detection. Talanta 2008, 75, 1320–1323. [Google Scholar] [CrossRef]
- Vieira, H.P.; Neves, A.A.; Queiroz, M.L.R. Otimização e validação da técnica de extração líquido-líquido com partição em baixa temperatura (ELL-PBT) para piretróides em água e análise por CG. Quím. Nova 2007, 30, 535–540. Available online: https://www.scielo.br/j/qn/a/YCy6kCDZHpTMkktNvhkvqqQ/?format=pdf&lang=pt (accessed on 24 April 2022). [CrossRef]
- Molognoni, L.; Daguer, H.; Ploêncio, L.A.S.; Lindner, J.D. A multi-purpose tool for food inspection: Simultaneous determination of various classes of preservatives and biogenic amines in meat and fish products by LC-MS. Talanta 2018, 178, 1053–1066. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, L.G.; Ramkumar, A.; Moloney, M.; Kurz, M.H.S.; Gonçalves, F.F.; Prestes, O.D.; Danaher, M. Vibrational extraction QuEChERS for analysis of antiparasitic agents in fish by liquid chromatography coupled with tandem mass spectrometry. Anal. Bioanal. Chem. 2019, 411, 6913–6929. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, X.; Liu, X.; Cao, Y.; Shi, Z.; Sun, H. Multi-class, multi-residue analysis of trace veterinary drugs in milk by rapid screening and quantification using ultra-performance liquid chromatography–quadrupole time-of-flight mass spectrometry. J. Dairy Sci. 2015, 98, 8433–8444. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Chu, X.; Yun Ling, Y.; Huang, J.; Chang, J. High-throughput screening of pesticide and veterinary drug residues in baby food by liquid chromatography coupled to quadrupole Orbitrap mass spectrometry. J. Chromatogr. A 2014, 1347, 122–128. [Google Scholar] [CrossRef]
- Zhao, F.; Gao, X.; Tang, Z.; Luo, X.; Wu, M.; Xu, J.; Fu, X. Development of a simple multi-residue determination method of 80 veterinary drugs in Oplegnathus punctatus by liquid chromatography coupled to quadrupole Orbitrap mass spectrometry. J. Chromatogr. B 2017, 1065–1066, 20–28. [Google Scholar] [CrossRef]
- Andersson, A.; Pålsheden, H. Comparison of the efficiency of different GLC multi-residue methods on crops containing pesticide residues. Fresenius J. Anal. Chem. 1991, 339, 365–367. [Google Scholar] [CrossRef]
- Hiemstra, M.; Kok, A. Comprehensive multi-residue method for the target analysis of pesticides in crops using liquid chromatography–tandem mass spectrometry. J. Chromatogr. A 2007, 1154, 3–25. [Google Scholar] [CrossRef]
- Rejczak, T.; Tuzimski, T. A review of recent developments and trends in the QuEChERS sample preparation approach. Open Chem. 2015, 13, 980–1010. Available online: https://www.degruyter.com/document/doi/10.1515/chem-2015-0109/html (accessed on 24 April 2022). [CrossRef]
- González-Curbelo, M.A.; Socas-Rodríguez, B.; Herrera-Herrera, A.V.; González-Sálamo, J.; Hernández-Borges, J.; Rodríguez-Delgado, M.A. Evolution and applications of the QuEChERS method. TrAC 2015, 71, 169–185. [Google Scholar] [CrossRef]
- Desmarchelier, A.; Fan, K.; Tien, M.M.; Savoy, M.-C.; Tarres, A.; Fuger, D.; Goyon, A.; Bessaire, T.; Mottier, P. Determination of 105 antibiotic, anti-inflammatory, antiparasitic agents and tranquilizers by LC-MS/MS based on an acidic QuEChERS-like extraction. Food Addit. Contam. Part A 2018, 35, 647–661. [Google Scholar] [CrossRef] [PubMed]
- Perestrelo, R.; Silva, P.; Porto-Figueira, P.; Pereira, J.A.M.; Silva, C.; Medina, S.; Câmara, J.S. QuEChERS: Fundamentals, relevant improvements, applications and future trends. Anal. Chim. Acta 2019, 1070, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Lehotay, S.J.; Matovská, K.; Yun, S.J. Evaluation of two fast and easy methods for pesticide residue analysis in fatty food matrixes. J. AOAC Int. 2005, 88, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Lehotay, S.; Son, K.A.; Kwon, H.; Koesukwiwat, U.; Fu, W.; Mastovska, K.; Hoh, E.; Leepipatpiboon, N. Comparison of QuEChERS sample preparation methods for the analysis of pesticide residues in fruits and vegetables. J. Chromatogr. A 2010, 1217, 2548–2560. [Google Scholar] [CrossRef] [PubMed]
- Moretti, S.; Dusi, G.; Giusepponi, D.; Pellicciotti, S.; Rossi, R.; Saluti, G.; Cruciani, G.; Galarini, R. Screening and confirmatory method for multiclass determination of 62 antibiotics in meat. J. Chromatograp. A 2016, 1429, 175–199. [Google Scholar] [CrossRef]
- Borràs, S.; Kaufmann, A.; Companyó, R. Correlation of precursor and product ions in single-stage high resolution mass spectrometry. A tool for detecting diagnostic ions and improving the precursor elemental composition elucidation. Anal. Chim. Acta 2013, 772, 47–58. [Google Scholar] [CrossRef]
- Kaufmann, A. High-resolution mass spectrometry for bioanalytical applications: Is this the new gold standard? J. Mass Spectrom. 2020, 44, e4533. [Google Scholar] [CrossRef]
- Vanini, G.; Barra, T.A.; Souza, L.M.; Madeira, N.C.L.; Gomes, A.O.; Romão, W.; Azevedo, D.A. Characterization of nonvolatile polar compounds from Brazilian oils by electrospray ionization with FT-ICR MS and Orbitrap-MS. Fuel 2020, 282, 118790. [Google Scholar] [CrossRef]
- Kaufmann, A.; Butcher, P.; Maden, K.; Walker, S.; Widmer, M. Reliability of veterinary drug residue confirmation: High resolution mass spectrometry versus tandem mass spectrometry. Anal. Chim. Acta 2015, 856, 54–67. [Google Scholar] [CrossRef]
- Kaufmann, A.; Mol, W.G. Product ion isotopologue pattern: A tool to improve the reliability of elemental composition elucidations of unknown compounds in complex matrices. Rapid Commun. Mass Spectrom. 2016, 30, 791–799. [Google Scholar] [CrossRef]
- Lehotay, S.J.; Sapozhnikova, Y.; Mol, H.G. Current issues involving screening and identification of chemical contaminants in foods by mass spectrometry. TrAC 2015, 69, 62–75. [Google Scholar] [CrossRef]
- Moretti, S.; Lega, F.; Rigoni, L.; Saluti, G.; Giusepponi, D.; Gioiello, A.; Manuali, E.; Rossi, R.; Galarini, R. Multiclass screening method to detect more than fifty banned substances in bovine bile and urine. Anal. Chim. Acta 2018, 1032, 56–57. [Google Scholar] [CrossRef] [PubMed]
- Varenina, I.; Bilandžić, N.; Luburić, Đ.B.; Kolanović, B.S.; Varga, I. High resolution mass spectrometry method for the determination of 13 antibiotic groups in bovine, swine, poultry and fish meat: An effective screening and confirmation analysis approach for routine laboratories. Food Control 2022, 133, 108576. [Google Scholar] [CrossRef]
- Brasil. Ministério da Agricultura, Pecuária e Abastecimento. Instrução Normativa nº 5, de 23 de Abril de 2019. Aprova o Plano de Amostragem e os Limites de Referência para o Plano Nacional de Controle de Resíduos e Contaminantes em Produtos de Origem Animal—PNCRC de 2019 para as Cadeias de Carnes Bovina, Suína, Caprina, Ovina, Equina, de Coelho, de aves e de Avestruz, de Leite, Pescado, mel e Ovos. In Diário Oficial da União; 25 April 2019; p. 4. Available online: https://pesquisa.in.gov.br/imprensa/jsp/visualiza/index.jsp?data=23/04/2019&jornal=515&pagina=4 (accessed on 14 October 2022).
- Food and Agriculture Organization. Maximum Residue Limits (MRLs) and Risk Management Recommendations (RMRs) for Residues of Veterinary Drugs in Foods; FAO: Rome, Italy, 2021; Available online: https://www.fao.org/fao-who-codexalimentarius/sh-proxy/en/?lnk=1&url=https%253A%252F%252Fworkspace.fao.org%252Fsites%252Fcodex%252FStandards%252FCXM%2B2%252FMRL2e.pdf (accessed on 14 September 2022).
- European Union. Commission Regulation (EU) No 37/2010 of 22 December 2009 on Pharmacologically Active Substances and Their Classification Regarding Maximum Residue Limits in Foodstuffs of Animal Origin (Text with EEA Relevance). Off. J. Eur. Union. 2010, p. 15. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32010R0037&from=EN (accessed on 14 September 2022).
- Paula, R.A.O. Resíduos de Antimicrobianos em Produtos de Origem Animal: Análise Crítica das Bulas e Avaliação Histórica dos Programas de Monitoramento no Brasil. Master’s Thesis, Universidade Federal de Minas Gerais, Belo Horizonte, Brazil, 2017. Available online: https://repositorio.ufmg.br/handle/1843/BUOS-B56P9R (accessed on 14 September 2022).
- European Commission. Commission Implementing Regulation (EU) 2021/808 of 22 March 2021 on the performance of analytical methods for residues of pharmacologically active substances used in food-producing animals and on the interpretation of results as well as on the methods to be used for sampling and repealing Decisions 2002/657/EC and 98/179/EC. Off. J. Eur. Union 2021, 180, 84–109. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32021R0808&from=EN (accessed on 14 September 2022).
- European Comission. Community Reference Laboratories Residues. Guidelines for the Validation of Screening Methods for Residues of Veterinary Medicines: Initial Validation and Transfer; CRLs: Berlin, Germany, 2010; Available online: https://food.ec.europa.eu/system/files/2016-10/cs_vet-med-residues_guideline_validation_screening_en.pdf (accessed on 14 October 2022).
- Gondim, C.S.; Coelho, O.A.M.; Alvarenga, R.L.; Junqueira, R.G.; Souza, S.V.C. An appropriate and systematized procedure for validating qualitative methods: Its application in the detection of sulfonamide residues in raw milk. Anal. Chim. Acta. 2014, 830, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Agência Nacional de Vigilância Sanitária. Instrução Normativa nº 162, de 1 de julho de 2022. Estabelece a Ingestão Diária Aceitável (IDA), a Dose de Referência Aguda (DRfA) e os Limites Máximos de Resíduos (LMR) para Insumos Farmacêuticos Ativos (IFA) de Medicamentos Veterinários em Alimentos de Origem Animal. In Diário Oficial da União; 6 July 2022; p. 238. Available online: https://pesquisa.in.gov.br/imprensa/jsp/visualiza/index.jsp?data=06/07/2022&jornal=515&pagina=238 (accessed on 14 October 2022).
- US Food & Drug Administration. CFR—Code of Federal Regulations Title 21: Food and Drug; FDA: Silver Spring, MD, USA, 2022. Available online: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=556 (accessed on 14 October 2022).
- The Japan Food Chemical Research Foundation. Maximum Residue Limits (MRLs) List of Agricultural Chemicals in Foods; JFCRF: Toyonaka, Japan, 2022; Available online: https://db.ffcr.or.jp/front/ (accessed on 14 October 2022).
- Parker, C. Agricultural and Veterinary Chemicals Code (MRL Standard) Instrument 2019; Australian Pesticides and Veterinary Medicines Authority: Sydney, Australia, 2019. Available online: https://www.legislation.gov.au/Details/F2019L01105/871ed0cc-ebbe-4268-b6ca-174e8dafc46e (accessed on 14 October 2022).
- Huet, A.-C.; Bienenmann-Ploum, M.; Vincent, U.; Delahaut, P. Screening methods and recent developments in the detection of anticoccidials. Anal. Bioanal. Chem. 2013, 405, 7733–7751. [Google Scholar] [CrossRef]
- Wode, F.; van Baar, P.; Dünnbier, U.; Hecht, F.; Taute, T.; Jekel, M.; Reemtsma, T. Search for over 2000 current and legacy micropollutants on a wastewater infiltration site with a UPLC-high resolution MS target screening method. Water Res. 2015, 69, 274–283. [Google Scholar] [CrossRef]
- Chemaxon. logP Plugin; Chemaxon: Budapst, Hungary, 2022. Available online: https://docs.chemaxon.com/display/docs/logp-plugin.md#src-1806637-logpplugin-fig-4 (accessed on 6 December 2022).
- Chemaxon. Pka Plugin; Chemaxon: Budapst, Hungary, 2022. Available online: https://docs.chemaxon.com/display/docs/pka-plugin.md (accessed on 6 December 2022).
- Chemaxon. Solubility prediction; Chemaxon: Budapst, Hungary, 2022. Available online: https://docs.chemaxon.com/display/docs/solubility-predictor.md (accessed on 6 December 2022).
- Perkin Elmer Informatics. Chemdraw Vesion 18.1: User Guide; Perkin Elmer: Waltham, MA, USA, 2019. Available online: https://chem.beloit.edu/classes/programs/ChemDraw_18_manual.pdf (accessed on 6 December 2022).
- Chemspider. Search ChemSpider; Royal Society of Chemistry: London, UK, 2022. Available online: http://www.chemspider.com (accessed on 6 December 2022).
- National Library of Medicine. PubChem: Explore Chemistry; National Library of Medicine; Rockville Pike: Bethesda, MA, USA, 2022. Available online: https://pubchem.ncbi.nlm.nih.gov (accessed on 6 December 2022).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Result | Criterion | Assessment | ||
|---|---|---|---|---|---|
| Method A | Method B | Method A | Method B | ||
| Description | Solid-liquid extraction using a mixture of solvents, assisted by ultrasound; cleanup in low temperature and hexane; UHPLC-Q-Orbitrap analysis | QuEChERS method -Acetonitrile extraction using a mixture of salts (d-SPE) and ultrasound-assisted; cleanup and centrifugal separation; UHPLC-Q-Orbitrap analysis | Less complexity possible | Less Steps | More steps |
| Technique complexity/time consumption | More time | Less time | Shortest possible time per sample | ||
| Sample weight | 1 g | 5 g | Smallest possible | Lower consumption | Higher consumption |
| Solvent consumption | 2 mL ACN; 2 mL MeOH; 1 mL hexane | 10 mL of ACN acidified with 2% v/v of acetic acid | Less possible | Lower consumption | Higher consumption |
| Consumption of solutions | 2 mL 0.1% (w/v) EDTA in water with 0.1% (v/v) formic acid; 0.5 mL of 0.01% aqueous formic acid/MeOH solution (75:25 v/v) | Aqueous extract: 1 mL mobile phase A; Organic extract: 1 mL mobile phase B | Smallest possible | Higher consumption | Lower consumption |
| Amount of solvents | None | Na2SO4 (4 g); NaCl (2 g); MgSO4 (250 mg); C18 (250 mg) | Smallest possible | No consumption | Higher consumption |
| Group/Class— Pharmacologically Active Substance | MRL in Bovine Muscle (µg kg−1) | CCβ (0.50 MRL) (µg kg−1) | Method A | Method B | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Validation Parameter | Validation Parameter | |||||||||||||||
| Selectivity | CCβ | Selectivity | CCβ | |||||||||||||
| Ratio of the Maximum Limit (MRL) | Ratio of the Maximum Limit (MRL) | |||||||||||||||
| 0.00 (Blank Samples) | 0.50 | 0.75 | 1.00 | 1.50 | 1.75 | 2.00 | 0.00 (Blank Samples) | 0.50 | 0.75 | 1.00 | 1.50 | 1.75 | 2.00 | |||
| Antimicrobials/ Aminoglycosides | ||||||||||||||||
| Amikacin | 500 | 250 | ||||||||||||||
| Apramycin | 1000 | 500 | ||||||||||||||
| Dihydrostreptomycin | 600 | 300 | ||||||||||||||
| Spectinomycin | 500 | 250 | ||||||||||||||
| Streptomycin | 500 | 250 | ||||||||||||||
| Gentamicin (C1, C1a, C2-C2a) | 100 | 50 | ||||||||||||||
| Hygromycin B | 500 | 250 | ||||||||||||||
| Kanamycin A | 100 | 50 | ||||||||||||||
| Neomycin B | 500 | 250 | ||||||||||||||
| Tobramycin | 500 | 250 | ||||||||||||||
| Antimicrobials/ Amphenicols | ||||||||||||||||
| Chloramphenicol | 0.3 | 0.15 | 20 | 8 | 9 | 11 | 13 | 18 | 20 | |||||||
| Florfenicol | 200 | 100 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Thiamphenicol | 50 | 25 | 20 | 12 | 15 | 16 | 17 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Antimicrobials/ Betalactams | ||||||||||||||||
| Amoxicillin | 50 | 25 | ||||||||||||||
| Ampicillin | 50 | 25 | 20 | 2 | 14 | 16 | 20 | 20 | 20 | 20 | 0 | 5 | 11 | 13 | 17 | 20 |
| Cephalexin | 200 | 100 | 20 | 9 | 15 | 18 | 20 | 20 | 20 | 20 | 0 | 0 | 7 | 15 | 17 | 20 |
| Cefalonium | 10 | 5 | ||||||||||||||
| Cephapirin | 50 | 25 | 20 | 0 | 0 | 0 | 2 | 18 | 16 | 20 | 4 | 13 | 18 | 19 | 20 | 20 |
| Cefazolin | 50 | 25 | 20 | 0 | 0 | 6 | 14 | 18 | 18 | |||||||
| Cefoperazone | 50 | 25 | 20 | 10 | 18 | 20 | 20 | 20 | 20 | |||||||
| Cefquinome | 50 | 25 | 20 | 0 | 0 | 2 | 4 | 8 | 4 | 20 | 7 | 12 | 16 | 19 | 20 | 20 |
| Cloxacillin | 300 | 150 | 20 | 2 | 7 | 13 | 19 | 18 | 18 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Dicloxacillin | 300 | 150 | 20 | 18 | 18 | 16 | 20 | 20 | 20 | 20 | 20 | 18 | 20 | 20 | 20 | 20 |
| Nafcillin | 300 | 150 | 20 | 19 | 20 | 20 | 20 | 20 | 20 | |||||||
| Oxacillin | 300 | 150 | 20 | 18 | 18 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Benzylpenicillin | 50 | 25 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 19 | 20 | 20 | 20 | 20 | 20 |
| Phenoxymethylpenicillin/Phenoxymethyl V penicillin | 25 | 12.5 | 20 | 4 | 10 | 13 | 17 | 20 | 20 | 20 | 5 | 9 | 11 | 17 | 20 | 20 |
| Antimicrobials/ Lincosamides | ||||||||||||||||
| Clindamycin | 50 | 25 | 20 | 13 | 20 | 19 | 20 | 19 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Lincomycin | 100 | 50 | 20 | 0 | 2 | 8 | 20 | 20 | 20 | |||||||
| Antimicrobials/ Macrolides | ||||||||||||||||
| Azithromycin | 50 | 25 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | |||||||
| Erythromycin A | 200 | 100 | 20 | 10 | 16 | 20 | 20 | 20 | 20 | |||||||
| Spiramycin I | 200 | 100 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | |||||||
| Tilmicosin | 100 | 50 | 20 | 6 | 14 | 15 | 16 | 18 | 18 | 20 | 18 | 18 | 19 | 19 | 20 | 20 |
| Tylosin A | 100 | 50 | 20 | 0 | 3 | 4 | 7 | 13 | 14 | 20 | 19 | 19 | 20 | 20 | 20 | 20 |
| Antimicrobials/ Quinolones-Fluoroquinolones | ||||||||||||||||
| Nalidixic acid | 20 | 10 | 20 | 20 | 20 | 18 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Oxolinic acid | 100 | 50 | 20 | 20 | 20 | 18 | 20 | 20 | 20 | |||||||
| Ciprofloxacin | 100 | 50 | 20 | 18 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 18 | 20 | 20 | 20 |
| Danofloxacin | 200 | 100 | 20 | 17 | 18 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Difloxacin | 400 | 200 | 20 | 20 | 16 | 16 | 20 | 18 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Enrofloxacin | 100 | 50 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Flumequine | 500 | 250 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Norfloxacin | 20 | 10 | 20 | 20 | 20 | 20 | 20 | 20 | 18 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Sarafloxacin | 10 | 5 | 20 | 9 | 13 | 15 | 19 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Antimicrobials/ Sulphonamides | ||||||||||||||||
| Sulfachlorpyridazine | 100 | 50 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Sulfadiazine | 100 | 50 | 20 | 17 | 18 | 20 | 20 | 20 | 19 | |||||||
| Sulfadimethoxine | 100 | 50 | 20 | 19 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Sulfadoxine | 100 | 50 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Sulfisoxazole/ Sulfafurazole | 100 | 50 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Sulfamerazine | 100 | 50 | 20 | 19 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Sulfamethazine/Sulfadimidine | 100 | 50 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Sulfathiazole | 100 | 50 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Sulfamethoxazole | 100 | 50 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 0 | 2 | 9 | 12 | 18 | 20 |
| Sulfaquinoxaline | 100 | 50 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Antimicrobials/ Tetracyclines | ||||||||||||||||
| Chlortetracycline | 200 | 100 | 20 | 18 | 20 | 20 | 20 | 19 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Doxycycline | 100 | 50 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | |||||||
| Oxytetracycline | 200 | 100 | 20 | 18 | 19 | 20 | 20 | 19 | 20 | 20 | 7 | 8 | 17 | 20 | 20 | 20 |
| Tetracycline | 200 | 100 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 6 | 13 | 20 | 20 | 20 | 20 |
| Antimicrobials/ Others | ||||||||||||||||
| Bromhexine | 10 | 5 | 20 | 7 | 9 | 16 | 17 | 30 | 20 | 20 | 3 | 9 | 18 | 19 | 20 | 20 |
| Dapsone | 10 | 5 | 20 | 0 | 0 | 2 | 5 | 9 | 6 | |||||||
| Rifampicin | 10 | 5 | 20 | 19 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Tiamulin | 100 | 50 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Antimicrobials Prospect/ Quinolones-Fluoroquinolones | ||||||||||||||||
| Marbofloxacin | 150 | 75 | 20 | 16 | 16 | 20 | 20 | 20 | 20 | 20 | 18 | 19 | 20 | 20 | 20 | 20 |
| Antimicrobials Prospect/ Macrolides | ||||||||||||||||
| Josamycin/Leucomycin A3 | 100 | 50 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | |||||||
| Leucomycin/Leucomycin A5 | 100 | 50 | 20 | 2 | 2 | 2 | 8 | 12 | 14 | |||||||
| Tildipirosin | 400 | 200 | 20 | 19 | 19 | 19 | 20 | 20 | 20 | 20 | 18 | 18 | 19 | 20 | 20 | 20 |
| Tulathromycin A | 300 | 150 | 20 | 15 | 18 | 18 | 19 | 19 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Antimicrobials Prospect/Sulfonamides | ||||||||||||||||
| Phthalylsulfathiazole | 100 | 50 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Sulfacetamide/N-Sulfanilylacetamide | 100 | 50 | 20 | 9 | 16 | 17 | 19 | 20 | 20 | |||||||
| Sulfamethoxypyridazine | 100 | 50 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Antimicrobials Prospect/Others | ||||||||||||||||
| Diminazene | 500 | 250 | 20 | 20 | 16 | 16 | 20 | 18 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Fosfomycin | 500 | 250 | ||||||||||||||
| Isoniazid | 10 | 5 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | |||||||
| Novobiocin | 1000 | 500 | ||||||||||||||
| Rifaximin | 10 | 5 | 20 | 18 | 20 | 20 | 20 | 20 | 20 | |||||||
| Antibiotic Growth Promoters | ||||||||||||||||
| Dichloroisoeverninic acid | 800 | 400 | 20 | 17 | 20 | 20 | 20 | 20 | 20 | 20 | 17 | 20 | 20 | 20 | 20 | 20 |
| Halquinol/Chlorhydroxyquinoline(7-Chloro-8-quinolinol) | 40 | 20 | 20 | 15 | 16 | 15 | 18 | 19 | 20 | |||||||
| Halquinol/Chlorhydroxyquinoline (5,7-Dichloro-8-quinolinol/Chloroxine) | 40 | 20 | 20 | 18 | 19 | 19 | 20 | 20 | 20 | |||||||
| Halquinol/Chlorhydroxyquinoline (5-Chloro-8-hydroxyquinoline) | 40 | 20 | 20 | 14 | 16 | 16 | 18 | 20 | 20 | |||||||
| Virginiamycin M1 | 100 | 50 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 18 | 18 | 18 | 19 | 20 | 20 |
| Anticoccidials | ||||||||||||||||
| Amprolium | 500 | 250 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | |||||||
| Clopidol | 200 | 100 | 20 | 20 | 9 | 17 | 18 | 20 | 20 | |||||||
| Diaveridine | 50 | 25 | 20 | 0 | 5 | 11 | 14 | 20 | 20 | |||||||
| Decoquinate | 1000 | 500 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Diclazuril | 50 | 25 | 20 | 19 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Ethopabate | 500 | 250 | 20 | 18 | 18 | 20 | 20 | 20 | 20 | 20 | 7 | 11 | 12 | 13 | 17 | 18 |
| Lasalocid A | 10 | 5 | 20 | 18 | 18 | 18 | 19 | 20 | 20 | |||||||
| Maduramicin | 30 | 15 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | ||||||
| Monensin A | 10 | 5 | 20 | 20 | 9 | 14 | 15 | 16 | 19 | 20 | ||||||
| Narasin A | 15 | 7.5 | 20 | |||||||||||||
| 4,4′-Dinitrocarbanilide – DNC | 4000 | 2000 | 20 | 12 | 20 | 16 | 20 | 20 | 20 | 20 | ||||||
| Robenidine | 200 | 100 | ||||||||||||||
| Salinomycin | 20 | 10 | 20 | 0 | 0 | 0 | 4 | 5 | 9 | 20 | 19 | 20 | 20 | 20 | 20 | 20 |
| Toltrazuril | 100 | 50 | 20 | 13 | 15 | 16 | 16 | 17 | 17 | |||||||
| Trimethoprim | 50 | 25 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | |||||||
| Anti-inflammatories/ Steroidal | ||||||||||||||||
| Prednisolone | 4 | 2 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | |||||||
| Prednisone | 4 | 2 | ||||||||||||||
| Anti-inflammatories Prospect/Steroidal | ||||||||||||||||
| Isoflupredone acetate/9-Fluoroprednisolone acetate | 10 | 5 | ||||||||||||||
| Flumetasone | 10 | 5 | 20 | 10 | 17 | 18 | 20 | 20 | 20 | |||||||
| Anti-Inflammatory/ Non-Steroidal | ||||||||||||||||
| Mefenamic acid | 20 | 10 | 20 | 18 | 20 | 20 | 20 | 20 | 20 | 20 | 16 | 18 | 18 | 19 | 20 | 20 |
| Tolfenamic acid | 50 | 25 | 20 | 19 | 20 | 20 | 20 | 20 | 20 | 20 | 9 | 10 | 17 | 18 | 20 | 20 |
| Carprofen | 500 | 250 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | |||||||
| Ketoprofen | 50 | 25 | 20 | 0 | 2 | 2 | 4 | 6 | 12 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Diclofenac | 5 | 2,5 | 20 | 18 | 18 | 19 | 20 | 20 | 20 | |||||||
| Flunixin | 20 | 10 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 10 | 12 | 18 | 17 | 20 | 20 |
| Indomethacin | 20 | 10 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | |||||||
| Meloxicam | 20 | 10 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 0 | 0 | 2 | 4 | 8 | 13 |
| Naproxen | 20 | 10 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | |||||||
| Nimesulide | 20 | 10 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | |||||||
| Piroxycam | 20 | 10 | 20 | 17 | 19 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Propyphenazone | 20 | 10 | 20 | 18 | 19 | 20 | 20 | 20 | 20 | |||||||
| Antiparasitic/ Avermectins | ||||||||||||||||
| Avermectin B1a/Abamectin B1a | 20 | 10 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | |||||||
| Doramectin | 10 | 5 | ||||||||||||||
| Emamectin B1a | 2 | 1 | ||||||||||||||
| Eprinomectin B1a | 100 | 50 | ||||||||||||||
| Ivermectin B1a/22,23-Dihydroavermectin B1 | 30 | 15 | ||||||||||||||
| Moxidectin | 20 | 10 | 20 | 13 | 15 | 16 | 19 | 20 | 20 | |||||||
| Antiparasitic/ Benzimidazoles | ||||||||||||||||
| Albendazole | 100 | 50 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | |||||||
| Albendazole sulfone | 100 | 50 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | |||||||
| Albendazole sulfoxide | 100 | 50 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | |||||||
| Closantel | 1000 | 500 | 20 | 19 | 19 | 19 | 19 | 19 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Febantel | 100 | 50 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Fenbendazole | 100 | 50 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 19 | 20 | 20 | 20 | 20 | 20 |
| Fenbendazole sulfone | 100 | 50 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Flubendazole | 20 | 10 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| 2-Aminoflubendazole | 20 | 10 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Levamisole | 10 | 5 | 20 | 6 | 6 | 16 | 19 | 19 | 19 | 20 | 19 | 19 | 20 | 20 | 20 | 20 |
| Mebendazole | 20 | 10 | 20 | 20 | 19 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Oxibendazole | 100 | 50 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Oxfendazole | 100 | 50 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Thiabendazole | 100 | 50 | 20 | 19 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Triclabendazole | 250 | 125 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | |||||||
| Antiparasitic/ Phenylpyrazoles | ||||||||||||||||
| Fipronil | 500 | 250 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Fipronil sulfone | 500 | 250 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | |||||||
| Antiparasitic/ Nitroimidazoles | ||||||||||||||||
| Dimetridazole | 10 | 5 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | |||||||
| Hydroxydimetridazole/HMMNI | 10 | 5 | 20 | 19 | 19 | 20 | 20 | 20 | 20 | |||||||
| Ipronidazole | 3 | 1.5 | 20 | 0 | 0 | 0 | 0 | 2 | 6 | 20 | 13 | 15 | 16 | 19 | 20 | 20 |
| Hydroxy Ipronidazole | 3 | 1.5 | 20 | 2 | 2 | 10 | 18 | 20 | 18 | |||||||
| Metronidazole | 10 | 5 | ||||||||||||||
| Hydroxymetronidazole | 10 | 5 | 20 | 0 | 4 | 9 | 11 | 12 | 16 | |||||||
| Ronidazole/1-Methyl-2-carbamoyloxymethyl-5-nitroimidazole | 10 | 5 | 20 | 3 | 7 | 15 | 19 | 20 | 20 | |||||||
| Antiparasitic/ Isoquinoline-pyrazines | ||||||||||||||||
| Praziquantel | 300 | 150 | 20 | 18 | 18 | 19 | 20 | 20 | 20 | |||||||
| Beta-Agonists | ||||||||||||||||
| Cimaterol | 10 | 5 | 20 | 19 | 20 | 20 | 20 | 20 | 20 | |||||||
| Clenbuterol | 0.2 | 0.1 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | |||||||
| Ractopamine | 10 | 5 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | |||||||
| Salbutamol | 10 | 5 | 20 | 9 | 13 | 19 | 20 | 20 | 20 | |||||||
| Zilpaterol | 0.5 | 0.25 | 20 | 4 | 8 | 10 | 15 | 17 | 19 | |||||||
| Sedatives | ||||||||||||||||
| Acepromazine | 10 | 5 | 20 | 11 | 13 | 20 | 20 | 20 | 19 | 20 | ||||||
| Azaperol | 60 | 30 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | |||||||
| Azaperone | 60 | 30 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | |||||||
| Carazolol | 5 | 2.5 | 20 | 11 | 15 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Chlorpromazine | 10 | 5 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Prospecting sedatives | ||||||||||||||||
| Xylazine | 20 | 10 | 20 | 11 | 14 | 15 | 20 | 20 | 20 | |||||||
| Corants | ||||||||||||||||
| Gentian violet/Crystal violet | 10 | 5 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | |||||||
| Leucocrystal Violet | 10 | 5 | ||||||||||||||
| Malachite green | 10 | 5 | 20 | 18 | 14 | 14 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Leucomalachite green | 10 | 5 | ||||||||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paula, R.A.d.O.; Gondim, C.d.S.; Schmidt, E.M.; Diniz, M.H.G.M.; Lana, M.A.G.; Oliveira, L.S.d. Critical Evaluation of Two Qualitative Analytical Approaches for Multiclass Determination of Veterinary Drugs in Bovine Muscle Using UHPLC-Q-Orbitrap: The Wind of Change in Brazilian Monitoring. Molecules 2023, 28, 4150. https://doi.org/10.3390/molecules28104150
Paula RAdO, Gondim CdS, Schmidt EM, Diniz MHGM, Lana MAG, Oliveira LSd. Critical Evaluation of Two Qualitative Analytical Approaches for Multiclass Determination of Veterinary Drugs in Bovine Muscle Using UHPLC-Q-Orbitrap: The Wind of Change in Brazilian Monitoring. Molecules. 2023; 28(10):4150. https://doi.org/10.3390/molecules28104150
Chicago/Turabian StylePaula, Ramon Alves de Oliveira, Carina de Souza Gondim, Eduardo Morgado Schmidt, Maria Helena Glicério Marcelina Diniz, Mary Ane Gonçalves Lana, and Leandro Soares de Oliveira. 2023. "Critical Evaluation of Two Qualitative Analytical Approaches for Multiclass Determination of Veterinary Drugs in Bovine Muscle Using UHPLC-Q-Orbitrap: The Wind of Change in Brazilian Monitoring" Molecules 28, no. 10: 4150. https://doi.org/10.3390/molecules28104150