

Alvespimycin Inhibits Heat Shock Protein 90 and Overcomes Imatinib Resistance in Chronic Myeloid Leukemia Cell Lines
 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Results
2.1. Alvespimycin Decreases the Metabolic Activity of CML Cell Models
2.2. Apoptosis as the Cell Death Mechanism Triggered by Alvespimycin
2.3. Alvespimycin Promotes Cell Cycle Arrest in G0/G1 Phase
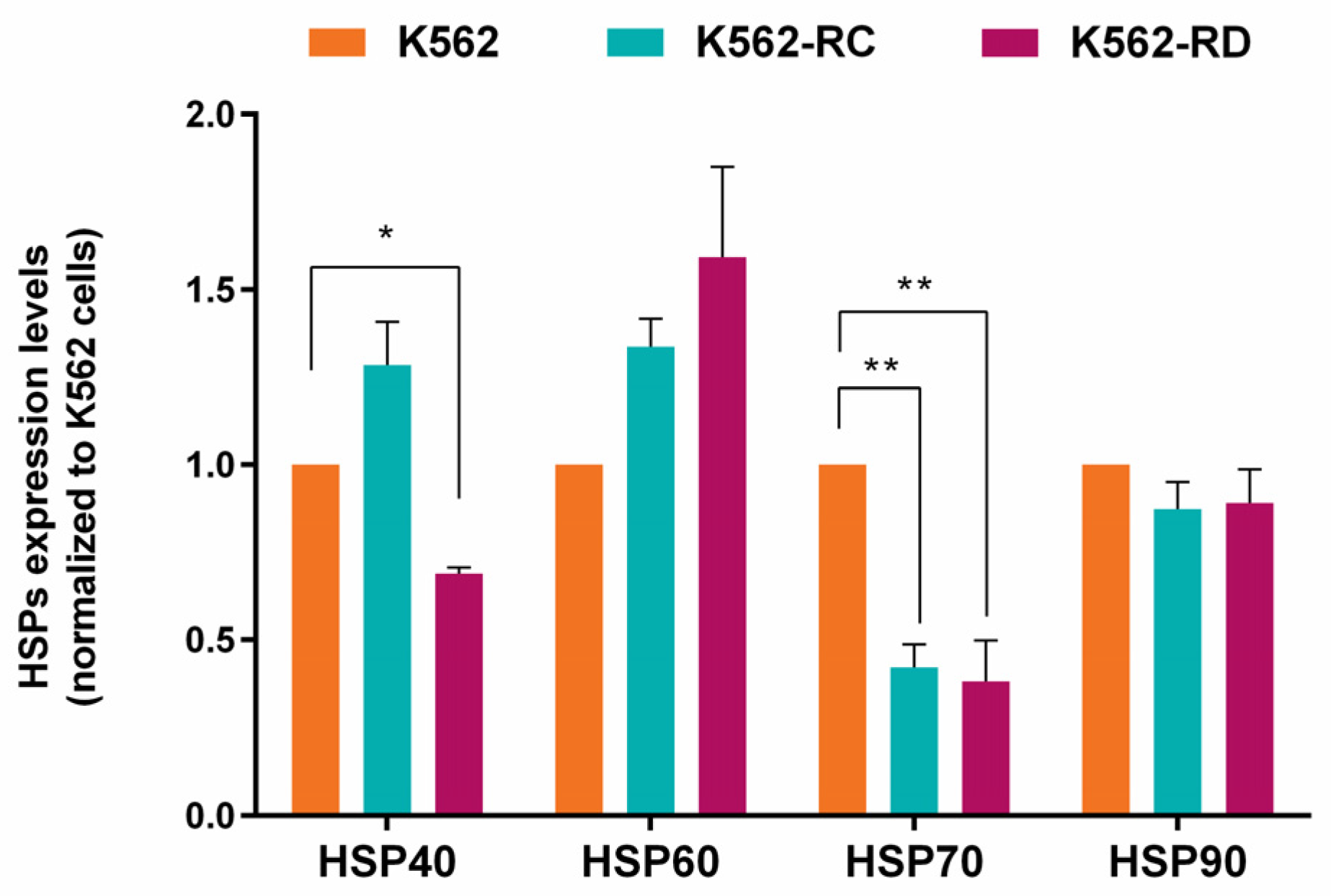
2.4. Heat Shock Proteins Expression
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Culture Conditions
4.2. Metabolic Activity Assay
4.3. Assessment of Cell Death
4.4. Evaluation of Caspases Activity
4.5. Cell Cycle Analysis
4.6. Mitochondrial Membrane Potential Assessment
4.7. Heat Shock Protein Expression Analysis
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ali, M.A.M. Chronic Myeloid Leukemia in the Era of Tyrosine Kinase Inhibitors: An Evolving Paradigm of Molecularly Targeted Therapy. Mol. Diagn. Ther. 2016, 20, 315–333. [Google Scholar] [CrossRef] [PubMed]
- Baccarani, M.; Castagnetti, F.; Gugliotta, G.; Rosti, G. A review of the European LeukemiaNet recommendations for the management of CML. Ann. Hematol. 2015, 94, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Hochhaus, A.; Baccarani, M.; Silver, R.T.; Schiffer, C.; Apperley, J.F.; Cervantes, F.; Clark, R.E.; Cortes, J.E.; Deininger, M.W.; Guilhot, F.; et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia 2020, 34, 966–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alves, R.; Gonçalves, A.C.; Rutella, S.; Almeida, A.M.; De Las Rivas, J.; Trougakos, I.P.; Sarmento Ribeiro, A.B. Resistance to Tyrosine Kinase Inhibitors in Chronic Myeloid Leukemia—From Molecular Mechanisms to Clinical Relevance. Cancers 2021, 13, 4820. [Google Scholar] [CrossRef] [PubMed]
- Khajapeer, K.V.; Baskaran, R. Hsp90 Inhibitors for the Treatment of Chronic Myeloid Leukemia. Leuk. Res. Treat. 2015, 2015, 757694. [Google Scholar] [CrossRef]
- Lianos, G.D.; Alexiou, G.A.; Mangano, A.; Mangano, A.; Rausei, S.; Boni, L.; Dionigi, G.; Roukos, D.H. The role of heat shock proteins in cancer. Cancer Lett 2015, 360, 114–118. [Google Scholar] [CrossRef]
- Birbo, B.; Madu, E.E.; Madu, C.O.; Jain, A.; Lu, Y. Role of HSP90 in Cancer. Int. J. Mol. Sci. 2021, 22, 10317. [Google Scholar] [CrossRef]
- Ren, X.; Li, T.; Zhang, W.; Yang, X. Targeting Heat-Shock Protein 90 in Cancer: An Update on Combination Therapy. Cells 2022, 11, 2556. [Google Scholar] [CrossRef]
- Li, L.; Wang, L.; You, Q.-D.; Xu, X.-L. Heat Shock Protein 90 Inhibitors: An Update on Achievements, Challenges, and Future Directions. J. Med. Chem. 2020, 63, 1798–1822. [Google Scholar] [CrossRef]
- Mellatyar, H.; Talaei, S.; Pilehvar-Soltanahmadi, Y.; Barzegar, A.; Akbarzadeh, A.; Shahabi, A.; Barekati-Mowahed, M.; Zarghami, N. Targeted cancer therapy through 17-DMAG as an Hsp90 inhibitor: Overview and current state of the art. Biomed. Pharmacother. = Biomed. Pharmacother. 2018, 102, 608–617. [Google Scholar] [CrossRef]
- Zhang, Z.; Jing, J.; Ye, Y.; Chen, Z.; Jing, Y.; Li, S.; Hong, W.; Ruan, H.; Liu, Y.; Hu, Q.; et al. Characterization of the dual functional effects of heat shock proteins (HSPs) in cancer hallmarks to aid development of HSP inhibitors. Genome Med. 2020, 12, 101. [Google Scholar] [CrossRef]
- Kamal, A.; Thao, L.; Sensintaffar, J.; Zhang, L.; Boehm, M.F.; Fritz, L.C.; Burrows, F.J. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 2003, 425, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Hertlein, E.; Wagner, A.J.; Jones, J.; Lin, T.S.; Maddocks, K.J.; Towns, W.H., III; Goettl, V.M.; Zhang, X.; Jarjoura, D.; Raymond, C.A.; et al. 17-DMAG targets the nuclear factor-κB family of proteins to induce apoptosis in chronic lymphocytic leukemia: Clinical implications of HSP90 inhibition. Blood 2010, 116, 45–53. [Google Scholar] [CrossRef] [Green Version]
- Ikebe, E.; Kawaguchi, A.; Tezuka, K.; Taguchi, S.; Hirose, S.; Matsumoto, T.; Mitsui, T.; Senba, K.; Nishizono, A.; Hori, M.; et al. Oral administration of an HSP90 inhibitor, 17-DMAG, intervenes tumor-cell infiltration into multiple organs and improves survival period for ATL model mice. Blood Cancer J. 2013, 3, e132. [Google Scholar] [CrossRef] [Green Version]
- Záčková, M.; Moučková, D.; Lopotová, T.; Ondráčková, Z.; Klamová, H.; Moravcová, J. Hsp90—A potential prognostic marker in CML. Blood Cells Mol. Dis. 2013, 50, 184–189. [Google Scholar] [CrossRef]
- Alves, R.; Fonseca, A.R.; Goncalves, A.C.; Ferreira-Teixeira, M.; Lima, J.; Abrantes, A.M.; Alves, V.; Rodrigues-Santos, P.; Jorge, L.; Matoso, E.; et al. Drug transporters play a key role in the complex process of Imatinib resistance in vitro. Leuk. Res. 2015, 39, 355–360. [Google Scholar] [CrossRef]
- Alves, R.; Gonçalves, A.C.; Jorge, J.; Alves, J.; Alves da Silva, A.; Freitas-Tavares, P.; Nascimento Costa, J.M.; Almeida, A.M.; Sarmento-Ribeiro, A.B. Everolimus in combination with Imatinib overcomes resistance in Chronic myeloid leukaemia. Med. Oncol. 2019, 36, 30. [Google Scholar] [CrossRef]
- Gorre, M.E.; Ellwood-Yen, K.; Chiosis, G.; Rosen, N.; Sawyers, C.L. BCR-ABL point mutants isolated from patients with imatinib mesylate-resistant chronic myeloid leukemia remain sensitive to inhibitors of the BCR-ABL chaperone heat shock protein 90. Blood 2002, 100, 3041–3044. [Google Scholar] [CrossRef]
- Ghadban, T.; Dibbern, J.L.; Reeh, M.; Miro, J.T.; Tsui, T.Y.; Wellner, U.; Izbicki, J.R.; Güngör, C.; Vashist, Y.K. HSP90 is a promising target in gemcitabine and 5-fluorouracil resistant pancreatic cancer. Apoptosis Int. J. Program. Cell Death 2017, 22, 369–380. [Google Scholar] [CrossRef]
- Lee, H.J.; Shin, S.; Kang, J.; Han, K.C.; Kim, Y.H.; Bae, J.W.; Park, K.H. HSP90 Inhibitor, 17-DMAG, Alone and in Combination with Lapatinib Attenuates Acquired Lapatinib-Resistance in ER-positive, HER2-Overexpressing Breast Cancer Cell Line. Cancers 2020, 12, 2630. [Google Scholar] [CrossRef]
- Li, J.J.; Zhang, J.J.; Wang, X.; Sun, Z.M. Effects of 17-DMAG on diffuse large B-cell lymphoma cell apoptosis. Exp. Ther. Med. 2017, 14, 3727–3731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lancet, J.E.; Gojo, I.; Burton, M.; Quinn, M.; Tighe, S.M.; Kersey, K.; Zhong, Z.; Albitar, M.X.; Bhalla, K.; Hannah, A.L.; et al. Phase I study of the heat shock protein 90 inhibitor alvespimycin (KOS-1022, 17-DMAG) administered intravenously twice weekly to patients with acute myeloid leukemia. Leukemia 2010, 24, 699–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maddocks, K.; Hertlein, E.; Chen, T.L.; Wagner, A.J.; Ling, Y.; Flynn, J.; Phelps, M.; Johnson, A.J.; Byrd, J.C.; Jones, J.A. A phase I trial of the intravenous Hsp90 inhibitor alvespimycin (17-DMAG) in patients with relapsed chronic lymphocytic leukemia/small lymphocytic lymphoma. Leuk. Lymphoma 2016, 57, 2212–2215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatia, S.; Diedrich, D.; Frieg, B.; Ahlert, H.; Stein, S.; Bopp, B.; Lang, F.; Zang, T.; Kröger, T.; Ernst, T.; et al. Targeting HSP90 dimerization via the C terminus is effective in imatinib-resistant CML and lacks the heat shock response. Blood 2018, 132, 307–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, W.; Ye, X.; Huang, X.; Lel, W.; You, L.; Wang, L.; Chen, X.; Qian, W. Hsp90 inhibitor, BIIB021, induces apoptosis and autophagy by regulating mTOR-Ulk1 pathway in imatinib-sensitive and -resistant chronic myeloid leukemia cells. Int. J. Oncol. 2016, 48, 1710–1720. [Google Scholar] [CrossRef] [Green Version]
- Radujkovic, A.; Schad, M.; Topaly, J.; Veldwijk, M.R.; Laufs, S.; Schultheis, B.S.; Jauch, A.; Melo, J.V.; Fruehauf, S.; Zeller, W.J. Synergistic activity of imatinib and 17-AAG in imatinib-resistant CML cells overexpressing BCR-ABL—Inhibition of P-glycoprotein function by 17-AAG. Leukemia 2005, 19, 1198–1206. [Google Scholar] [CrossRef]
- Rao, R.; Nalluri, S.; Fiskus, W.; Balusu, R.; Joshi, A.; Mudunuru, U.; Buckley, K.M.; Robbins, K.; Ustun, C.; Reuther, G.W.; et al. Heat shock protein 90 inhibition depletes TrkA levels and signaling in human acute leukemia cells. Mol. Cancer Ther. 2010, 9, 2232–2242. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, S.N.; Leng, X.; Perazzona, B.; Sun, X.; Lin, Y.H.; Arlinghaus, R.B. Combination of JAK2 and HSP90 inhibitors: An effective therapeutic option in drug-resistant chronic myelogenous leukemia. Genes Cancer 2016, 7, 201–208. [Google Scholar] [CrossRef] [Green Version]
- Zeng, D.; Gao, M.; Zheng, R.; Qin, R.; He, W.; Liu, S.; Wei, W.; Huang, Z. The HSP90 inhibitor KW-2478 depletes the malignancy of BCR/ABL and overcomes the imatinib-resistance caused by BCR/ABL amplification. Exp. Hematol. Oncol. 2022, 11, 33. [Google Scholar] [CrossRef]
- Georgakis, G.V.; Li, Y.; Younes, A. The heat shock protein 90 inhibitor 17-AAG induces cell cycle arrest and apoptosis in mantle cell lymphoma cell lines by depleting cyclin D1, Akt, Bid and activating caspase 9. Br. J. Haematol. 2006, 135, 68–71. [Google Scholar] [CrossRef]
- Fukuyo, Y.; Hunt, C.R.; Horikoshi, N. Geldanamycin and its anti-cancer activities. Cancer Lett. 2010, 290, 24–35. [Google Scholar] [CrossRef]
- Kim, J.G.; Lee, S.C.; Kim, O.H.; Kim, K.H.; Song, K.Y.; Lee, S.K.; Choi, B.J.; Jeong, W.; Kim, S.J. HSP90 inhibitor 17-DMAG exerts anticancer effects against gastric cancer cells principally by altering oxidant-antioxidant balance. Oncotarget 2017, 8, 56473–56489. [Google Scholar] [CrossRef] [Green Version]
- Burrows, F.; Zhang, H.; Kamal, A. Hsp90 activation and cell cycle regulation. Cell Cycle 2004, 3, 1530–1536. [Google Scholar] [CrossRef] [Green Version]
- Karkoulis, P.K.; Stravopodis, D.J.; Voutsinas, G.E. 17-DMAG induces heat shock protein 90 functional impairment in human bladder cancer cells: Knocking down the hallmark traits of malignancy. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2016, 37, 6861–6873. [Google Scholar] [CrossRef]
- George, P.; Bali, P.; Annavarapu, S.; Scuto, A.; Fiskus, W.; Guo, F.; Sigua, C.; Sondarva, G.; Moscinski, L.; Atadja, P.; et al. Combination of the histone deacetylase inhibitor LBH589 and the hsp90 inhibitor 17-AAG is highly active against human CML-BC cells and AML cells with activating mutation of FLT-3. Blood 2005, 105, 1768–1776. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.M.; Bae, J.H.; Kim, M.J.; Lee, H.S.; Lee, M.K.; Chung, B.S.; Kim, D.W.; Kang, C.D.; Kim, S.H. Bcr-Abl-independent imatinib-resistant K562 cells show aberrant protein acetylation and increased sensitivity to histone deacetylase inhibitors. J. Pharmacol. Exp. Ther. 2007, 322, 1084–1092. [Google Scholar] [CrossRef] [Green Version]
- Pocaly, M.; Lagarde, V.; Etienne, G.; Ribeil, J.A.; Claverol, S.; Bonneu, M.; Moreau-Gaudry, F.; Guyonnet-Duperat, V.; Hermine, O.; Melo, J.V.; et al. Overexpression of the heat-shock protein 70 is associated to imatinib resistance in chronic myeloid leukemia. Leukemia 2007, 21, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Hong, D.S.; Banerji, U.; Tavana, B.; George, G.C.; Aaron, J.; Kurzrock, R. Targeting the molecular chaperone heat shock protein 90 (HSP90): Lessons learned and future directions. Cancer Treat. Rev. 2013, 39, 375–387. [Google Scholar] [CrossRef]
- Huang, Y.-H.; Yeh, C.-T. Functional Compartmentalization of HSP60-Survivin Interaction between Mitochondria and Cytosol in Cancer Cells. Cells 2020, 9, 23. [Google Scholar] [CrossRef] [Green Version]
- Scaltriti, M.; Dawood, S.; Cortes, J. Molecular Pathways: Targeting Hsp90—Who Benefits and Who Does Not. Clin. Cancer Res. 2012, 18, 4508–4513. [Google Scholar] [CrossRef]
- Mendes, J.; Gonçalves, A.C.; Alves, R.; Jorge, J.; Pires, A.; Ribeiro, A.; Sarmento-Ribeiro, A.B. L744,832 and Everolimus Induce Cytotoxic and Cytostatic Effects in Non-Hodgkin Lymphoma Cells. Pathol. Oncol. Res. 2016, 22, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, A.C.; Cortesão, E.; Oliveiros, B.; Alves, V.; Espadana, A.I.; Rito, L.; Magalhães, E.; Lobão, M.J.; Pereira, A.; Nascimento Costa, J.M.; et al. Oxidative stress and mitochondrial dysfunction play a role in myelodysplastic syndrome development, diagnosis, and prognosis: A pilot study. Free Radic. Res. 2015, 49, 1081–1094. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| sub-G1 (%) | G0/G1 (%) | S (%) | G2/M (%) | |
|---|---|---|---|---|
| K562 Cells | ||||
| Control | 1.4 ± 0.2 | 40.0 ± 2.4 | 44.2 ± 3.9 | 15.8 ± 1.8 |
| Alvespimycin 10 nM | 5.6 ± 2.9 | 40.1 ± 0.7 | 46.4 ± 1.2 | 13.5 ± 0.9 |
| Alvespimycin 100 nM | 9.8 ± 2.0 ** | 60.3 ± 5.4 ** | 21.3 ± 7.3 | 18.4 ± 2.0 |
| K562-RC Cells | ||||
| Control | 0.2 ± 0.2 | 52.4 ± 3.6 | 33.6 ± 3.0 | 14.0 ± 0.8 |
| Alvespimycin 10 nM | 0.6 ± 0.4 $ | 52.6 ± 2.5 | 33.6 ± 2.6 | 13.8 ± 0.2 |
| Alvespimycin 100 nM | 17.4 ± 5.2 ** | 62.0 ± 5.8 | 28.5 ± 3.2 | 9.5 ± 3.4 |
| K562-RD Cells | ||||
| Control | 0.2 ± 0.2 | 44.4 ± 1.5 | 36.6 ± 2.6 | 19.0 ± 2.2 |
| Alvespimycin 10 nM | 0.8 ± 0.4 $ | 45.0 ± 2.2 | 36.4 ± 2.5 | 18.6 ± 1.7 |
| Alvespimycin 100 nM | 3.0 ± 0.8 ** | 41.3 ± 1.4 | 32.7 ± 3.8 | 26.0 ± 4.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alves, R.; Santos, D.; Jorge, J.; Gonçalves, A.C.; Catarino, S.; Girão, H.; Melo, J.B.; Sarmento-Ribeiro, A.B. Alvespimycin Inhibits Heat Shock Protein 90 and Overcomes Imatinib Resistance in Chronic Myeloid Leukemia Cell Lines. Molecules 2023, 28, 1210. https://doi.org/10.3390/molecules28031210
Alves R, Santos D, Jorge J, Gonçalves AC, Catarino S, Girão H, Melo JB, Sarmento-Ribeiro AB. Alvespimycin Inhibits Heat Shock Protein 90 and Overcomes Imatinib Resistance in Chronic Myeloid Leukemia Cell Lines. Molecules. 2023; 28(3):1210. https://doi.org/10.3390/molecules28031210
Chicago/Turabian StyleAlves, Raquel, Diogo Santos, Joana Jorge, Ana Cristina Gonçalves, Steve Catarino, Henrique Girão, Joana Barbosa Melo, and Ana Bela Sarmento-Ribeiro. 2023. "Alvespimycin Inhibits Heat Shock Protein 90 and Overcomes Imatinib Resistance in Chronic Myeloid Leukemia Cell Lines" Molecules 28, no. 3: 1210. https://doi.org/10.3390/molecules28031210