
Ultrasound Assisted Synthesis and In Silico Modelling of 1,2,4-Triazole Coupled Acetamide Derivatives of 2-(4-Isobutyl phenyl)propanoic acid as Potential Anticancer Agents
 , and
, and
Abstract
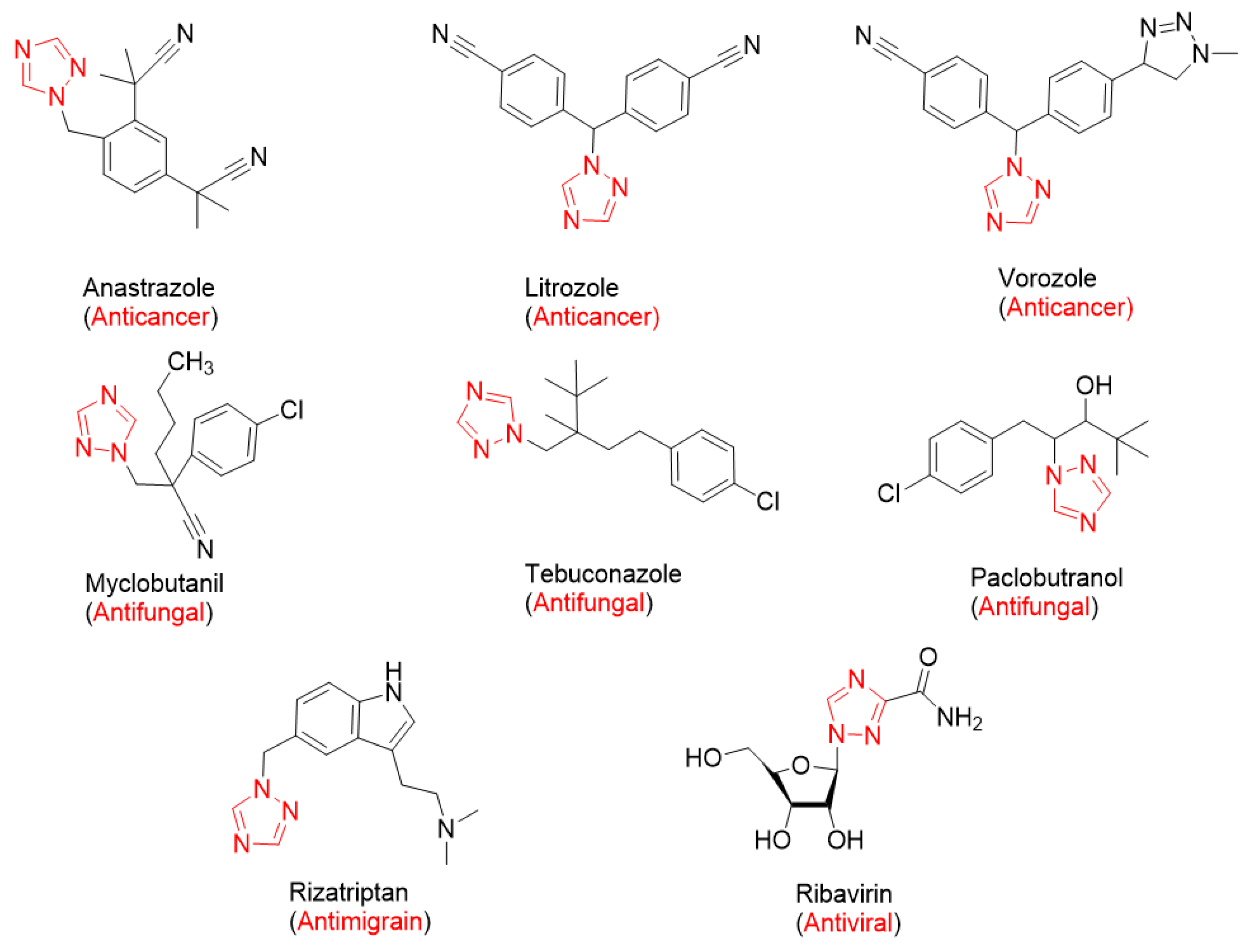
:1. Introduction
2. Results
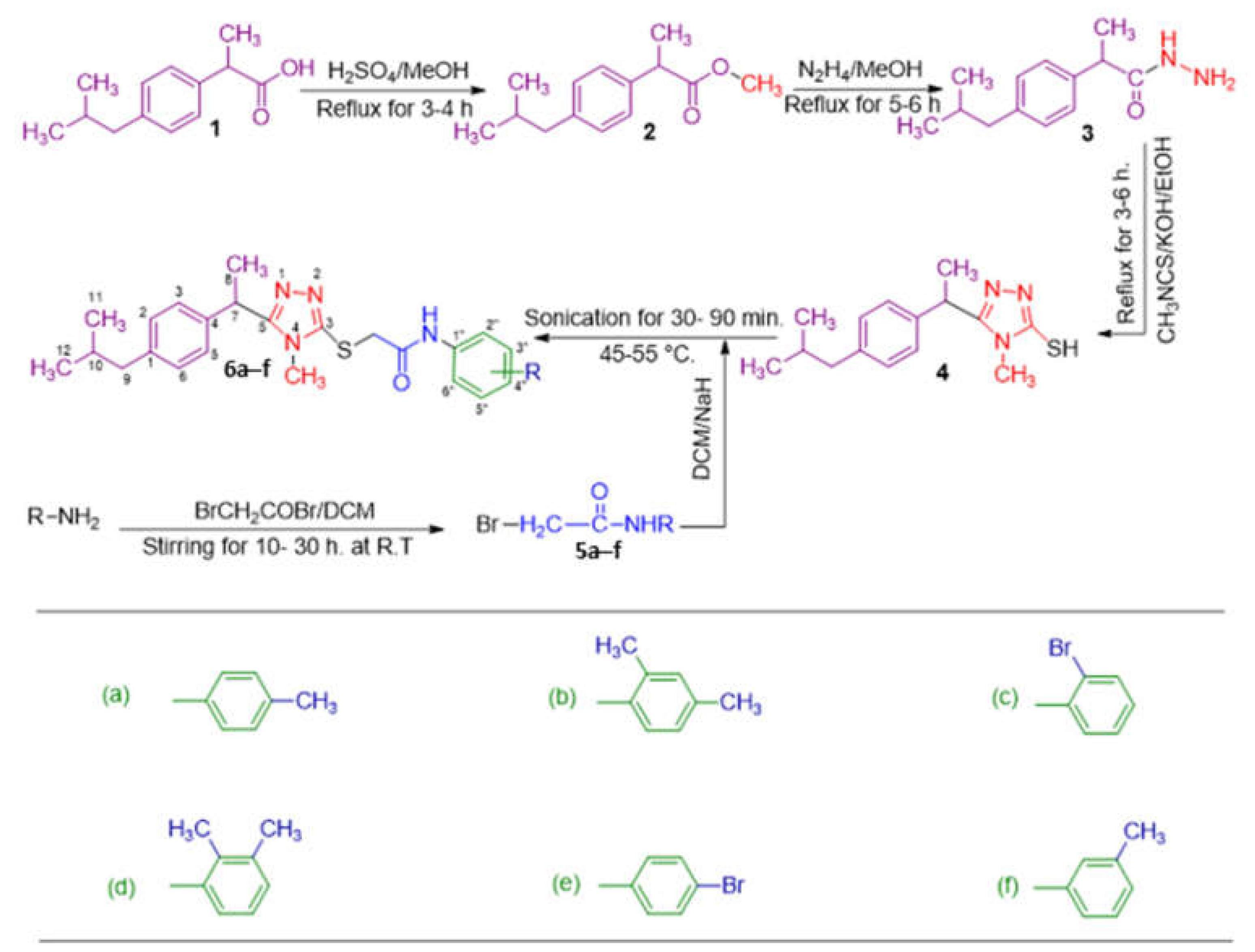
2.1. Chemistry
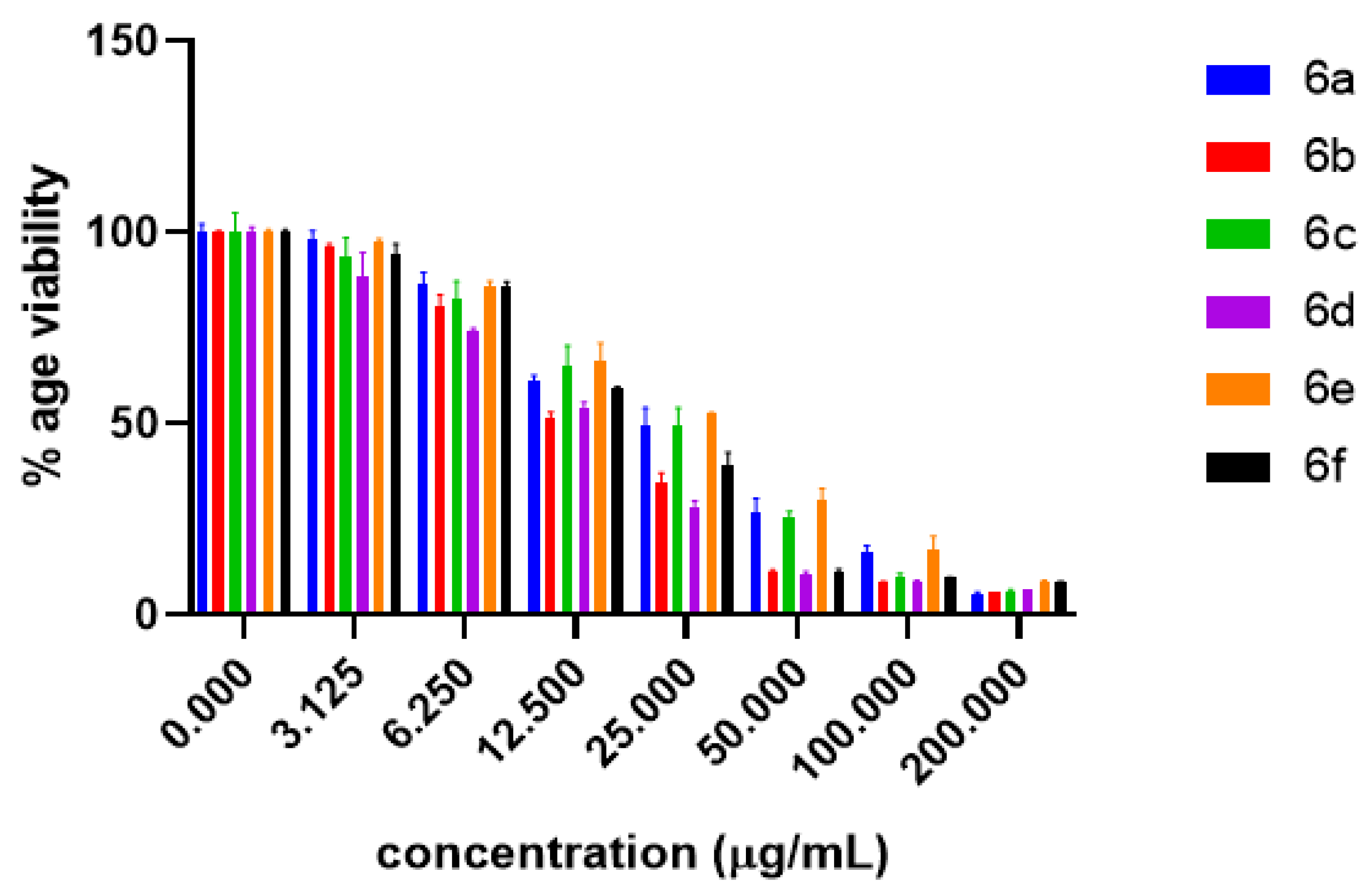
2.2. Anti-Proliferative Potential
2.3. Structure–Activity Relationship (SAR) Study
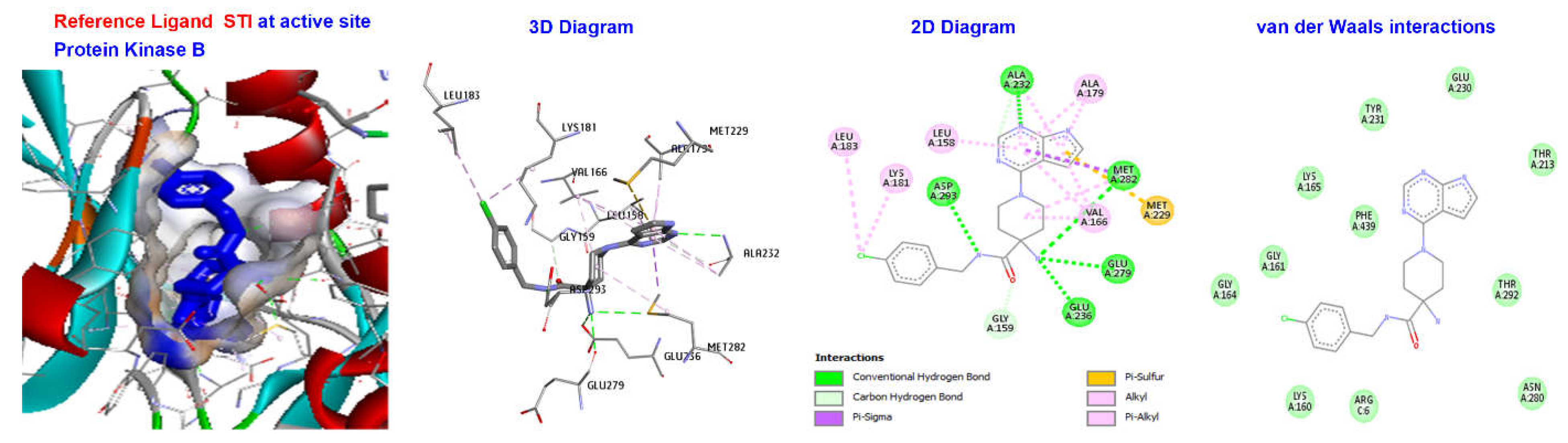
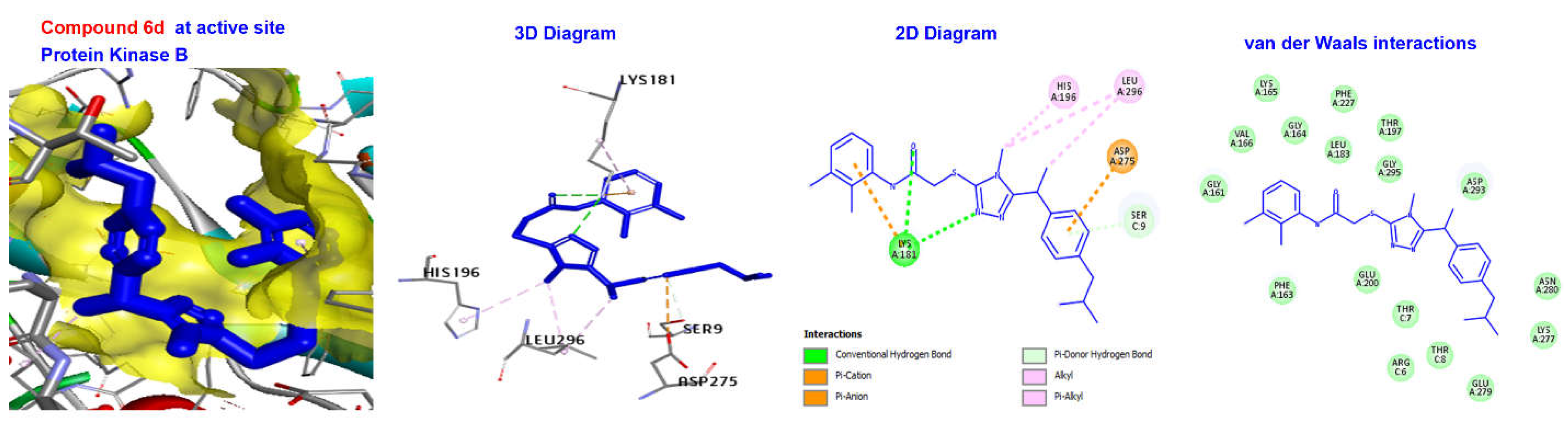
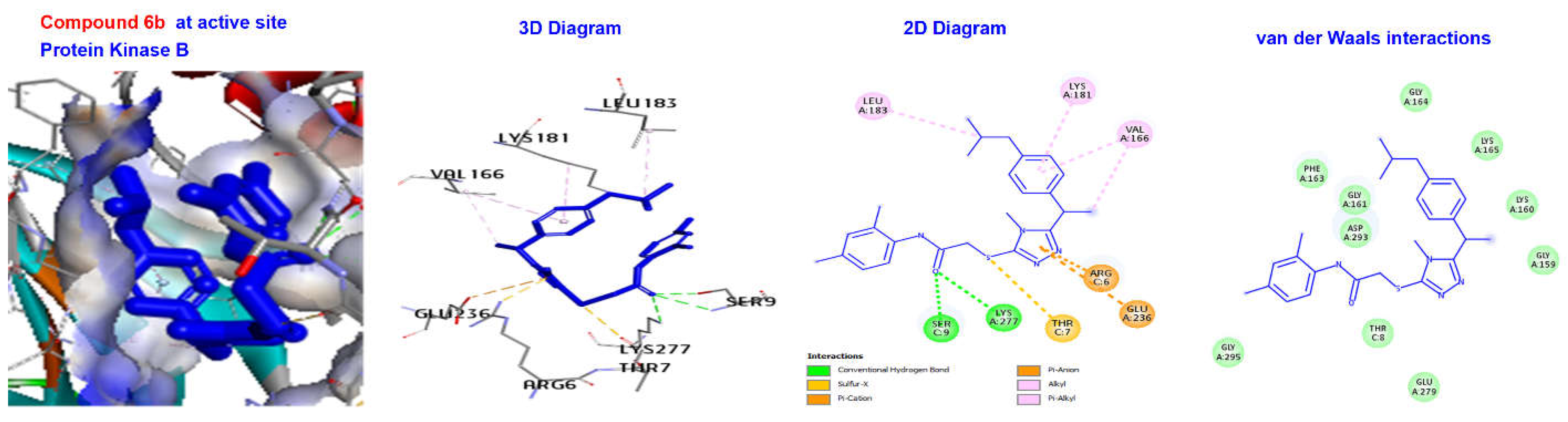
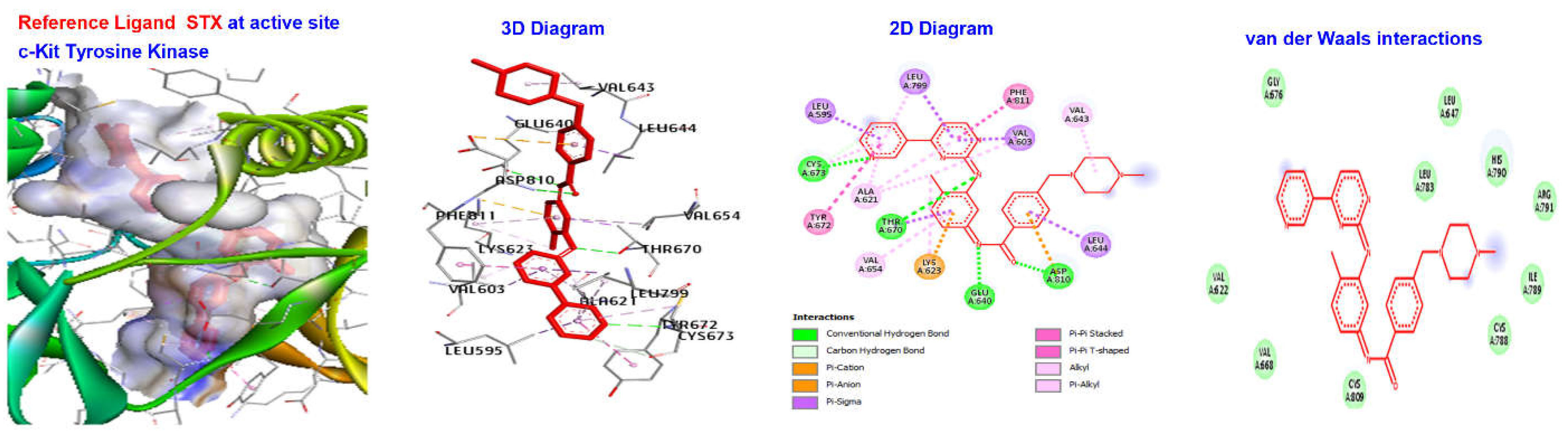
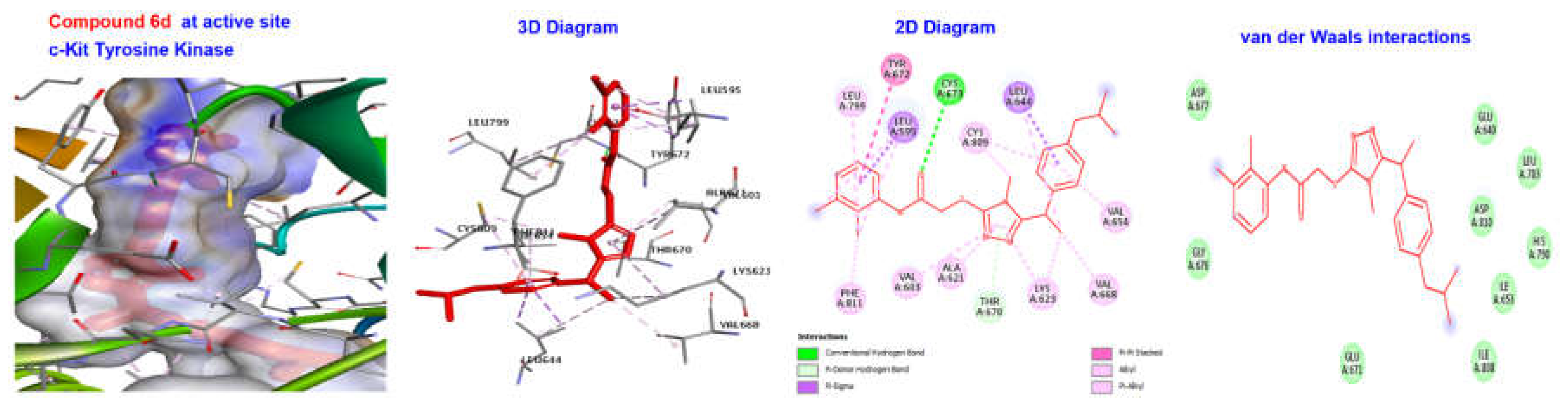
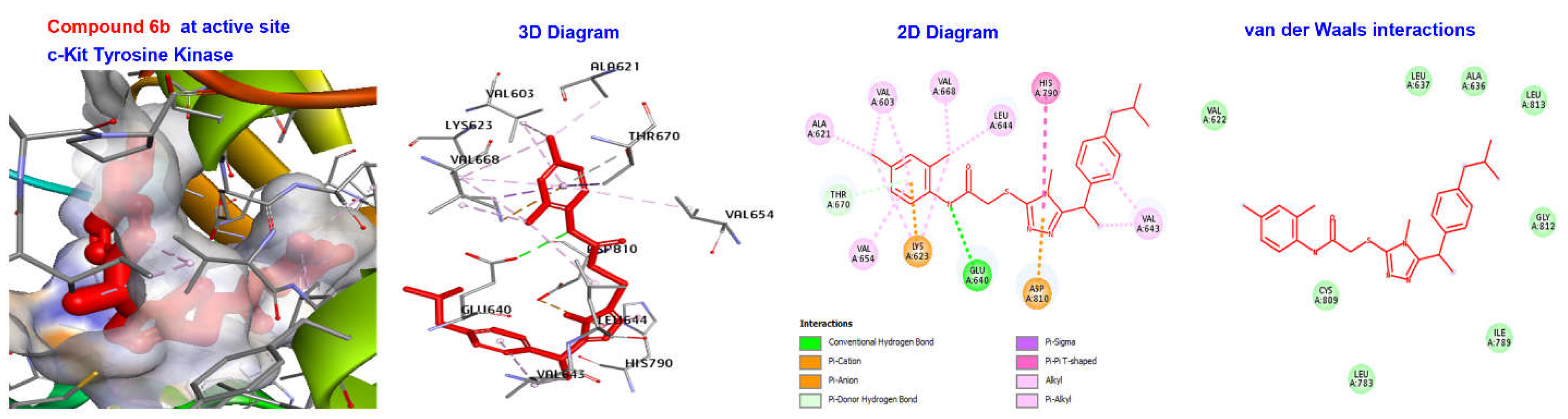
2.4. Molecular Docking
3. Materials and Methods
3.1. General
3.2. General Procedure for the Synthesis of Synthesized N-substituted 5-aryl-1,2,4-triazole-3-acetamide derivatives 6a–6f
3.2.1. Synthesis of methyl 2-(4-isobutylphenyl)propanoate (2)
3.2.2. Synthesis of 2-(4-isobutylphenyl)propane hydrazide (3)
3.2.3. Synthesis of 5-(1-(4-isobutylphenyl) ethyl)-1,2,4-triazole-2-thiol (4)
3.2.4. Synthesis of N-substituted aryl/alkyl 2-chloroacetamides 5a–5f
3.2.5. Synthesis of N-substituted 5-(1-(4-isobutylphenyl)ethyl)-1,2,4-triazole-2-yl- 2-sulfanyl acetamide derivatives 6a–6f
N-(4-Methylphenyl)-2-((5-(1-(4-isobutylphenyl)ethyl)-4-methyl-4H-1,2,4-triazol-3-yl)thio)acetamide (6a)
N-(2,4-Dimethylphenyl)-2-((5-(1-(4-isobutylphenyl)ethyl)-4-methyl-4H-1,2,4-triazol-3-yl)thio)acetamide (6b)
N-(2-Bromophenyl)-2-((5-(1-(4-isobutylphenyl)ethyl)-4-methyl-4H-1,2,4-triazol-3-yl)thio)acetamide (6c)
N-(2,3-Dimethylphenyl)-2-((5-(1-(4-isobutylphenyl)ethyl)-4-methyl-4H-1,2,4-triazol-3-yl)thio)acetamide (6d)
N-(4-Bromophenyl)-2-((5-(1-(4-isobutylphenyl)ethyl)-4-methyl-4H-1,2,4-triazol-3-yl)thio)acetamide (6e)
N-(3-Methylphenyl)-2-((5-(1-(4-isobutylphenyl)ethyl)-4-methyl-4H-1,2,4-triazol-3-yl)thio)acetamide (6f)
3.3. Experimental Procedures for Biological Activities
3.3.1. Cell Culture and Treatment
3.3.2. Determination of Cell Viability
3.4. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Park, S.K.; Cho, L.Y.; Yang, J.J.; Park, B.; Chang, S.H.; Lee, K.-S.; Kim, H.; Yoo, K.-Y.; Lee, C.-T.; The Scientific Committee, Korean Academy of Tuberculosis and Respiratory Diseases. Lung cancer risk and cigarette smoking, lung tuberculosis according to histologic type and gender in a population based case–Control study. Lung Cancer 2010, 68, 20–26. [Google Scholar] [CrossRef]
- Clayton, P.E.; Banerjee, I.; Murray, P.G.; Renehan, A.G. Growth hormone, the insulin-like growth factor axis, insulin and cancer risk. Nat. Rev. Endocrinol. 2011, 7, 11–24. [Google Scholar] [CrossRef]
- Porta, C.; Riboldi, E.; Sica, A. Mechanisms linking pathogens-associated inflammation and cancer. Cancer Lett. 2011, 305, 250–262. [Google Scholar] [CrossRef]
- Rosa, R.; Monteleone, F.; Zambrano, N.; Bianco, R. In vitro and in vivo models for analysis of resistance to anticancer molecular therapies. Curr. Med. Chem. 2014, 21, 1595–1606. [Google Scholar] [CrossRef]
- Antunes, M.M.; Amarante, T.R.; Valente, A.A.; Almeida Paz, F.A.; Gonçalves, I.S.; Pillinger, M. A Linear Trinuclear Oxidodiperoxido-molybdenum (VI) Complex with Single Triazole Bridges: Catalytic Activity in Epoxidation, Alcoholysis, and Acetalization Reactions. ChemCatChem 2018, 10, 2782–2791. [Google Scholar] [CrossRef]
- Gomha, S.M.; Abdel-aziz, H.M.; El-Reedy, A.A. Facile Synthesis of Pyrazolo [3, 4-c] pyrazoles Bearing Coumarine Ring as Anticancer Agents. J. Heterocycl. Chem. 2018, 55, 1960–1965. [Google Scholar] [CrossRef]
- Henary, M.; Kananda, C.; Rotolo, L.; Savino, B.; Owens, E.A.; Cravotto, G. Benefits and applications of microwave-assisted synthesis of nitrogen containing heterocycles in medicinal chemistry. RSC Adv. 2020, 10, 14170–14197. [Google Scholar] [CrossRef] [PubMed]
- Goss, P.E. Pre-clinical and clinical review of vorozole, a new third generation aromatase inhibitor. Breast Cancer Res. Treat. 1998, 49, S59–S65. [Google Scholar] [CrossRef] [PubMed]
- Schiller, D.S.; Fung, H.B. Posaconazole: An extended-spectrum triazole antifungal agent. Clin. Ther. 2007, 29, 1862–1886. [Google Scholar] [CrossRef] [PubMed]
- Bektaş, H.; Ceylan, Ş.; Demirbaş, N.; Alpay-Karaoğlu, Ş.; Sökmen, B.B. Antimicrobial and antiurease activities of newly synthesized morpholine derivatives containing an azole nucleus. Med. Chem. Res. 2013, 22, 3629–3639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Serwy, W.S.; Mohamed, N.A.; Abbas, E.M.; Abdel-Rahman, R.F. Synthesis and anti-inflammatory properties of novel 1, 2, 4-triazole derivatives. Res. Chem. Intermed. 2013, 39, 2543–2554. [Google Scholar] [CrossRef]
- Sancak, K.; Ünver, Y.; Ünlüer, D.; Düğdü, E.; Kör, G.; Celik, F.; Birinci, E. Synthesis, characterization, and antioxidant activities of new trisubstituted triazoles. Turk. J. Chem. 2012, 36, 457–466. [Google Scholar] [CrossRef]
- Plech, T.; Kaproń, B.; Łuszczki, J.J.; Wujec, M.; Paneth, A.; Siwek, A.; Kołaczkowski, M.; Żołnierek, M.; Nowak, G. Studies on the anticonvulsant activity and influence on GABA-ergic neurotransmission of 1, 2, 4-triazole-3-thione-based compounds. Molecules 2014, 19, 11279–11299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alanazi, M.M.; Elwan, A.; Alsaif, N.A.; Obaidullah, A.J.; Alkahtani, H.M.; Al-Mehizia, A.A.; Alsubaie, S.M.; Taghour, M.S.; Eissa, I.H. Discovery of new 3-methylquinoxalines as potential anti-cancer agents and apoptosis inducers targeting VEGFR-2: Design, synthesis, and in silico studies. J. Enzym. Inhib. Med. Chem. 2021, 36, 1732–1750. [Google Scholar] [CrossRef]
- Kaur, N. Synthesis of six-and seven-membered heterocycles under ultrasound irradiation. Synth. Commun. 2018, 48, 1235–1258. [Google Scholar] [CrossRef]
- Ammar, H.B.; Chtourou, M.; Frikha, M.H.; Trabelsi, M. Green condensation reaction of aromatic aldehydes with active methylene compounds catalyzed by anion-exchange resin under ultrasound irradiation. Ultrason. Sonochem. 2015, 22, 559–564. [Google Scholar] [CrossRef]
- Bai, G.-Y.; Lan, X.-W.; Chen, G.-F.; Liu, X.-F.; Li, T.-Y.; Shi, L.-J. A novel sodium iodide and ammonium molybdate co-catalytic system for the efficient synthesis of 2-benzimidazoles using hydrogen peroxide under ultrasound irradiation. Ultrason. Sonochem. 2014, 21, 520–526. [Google Scholar] [CrossRef]
- Suslick, K.S. Sonochemistry. Science 1990, 247, 1439–1445. [Google Scholar] [CrossRef]
- Shabir, G.; Shafique, I.; Saeed, A. Ultrasound assisted synthesis of 5–7 membered heterocyclic rings in organic molecules. J. Heterocycl. Chem. 2022, 59, 1669–1702. [Google Scholar] [CrossRef]
- Wu, X.; Joyce, E.M.; Mason, T.J. Evaluation of the mechanisms of the effect of ultrasound on Microcystis aeruginosa at different ultrasonic frequencies. Water Res. 2012, 46, 2851–2858. [Google Scholar] [CrossRef] [PubMed]
- Savun-Hekimoğlu, B.; Ince, N.H. Decomposition of PPCPs by ultrasound-assisted advanced Fenton reaction: A case study with salicylic acid. Ultrason. Sonochem. 2017, 39, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Gul, S.; Abbasi, M.A.; Khan, K.M.; Nafeesa, K.; Siddiqa, A.; Akhtar, M.N.; Shahid, M.; Subhani, Z. Synthesis, antimicrobial evaluation and hemolytic activity of 2-[[5-alkyl/aralkyl substituted-1, 3, 4-oxadiazol-2-yl] thio]-N-[4-(4-morpholinyl) phenyl] acetamide derivatives. J. Saudi Chem. Soc. 2017, 21, S425–S433. [Google Scholar] [CrossRef]
- Khan, S.G.; Bokhari, T.H.; Anjum, F.; Akhter, N.; Rasool, S.; Shah, S.A.A.; Shahid, M.; Arshad, A. Synthesis, characterization, antibacterial, hemolytic and thrombolytic activity evaluation of 5-(3-chlorophenyl)-2-((N-(substituted)-2-acetamoyl) sulfanyl)-1, 3, 4-oxadiazole derivatives. Pak. J. Pharm. Sci. 2020, 33, 871–876. [Google Scholar]
- Aziz-ur-Rehman; Khan, S.G.; Naqvi, S.A.R.; Ahmad, M.; Akhtar, N.; Bokhari, T.H.; Irfan, M.; Usman, A.; Batool, S.; Rasool, S.; et al. Synthesis, spectral analysis and biological evaluation of 2-{[(morpholin-4-yl) ethyl] thio}-5-phenyl/aryl-1, 3, 4-oxadiazole derivatives. Pak. J. Pharm. Sci. 2021, 34, 441–446. [Google Scholar]
- Zappaterra, F.; Rodriguez, M.E.M.; Summa, D.; Semeraro, B.; Costa, S.; Tamburini, E. Biocatalytic approach for direct esterification of ibuprofen with sorbitol in biphasic media. Int. J. Mol. Sci. 2021, 22, 3066. [Google Scholar] [CrossRef]
- Iqbal, K.; Jamal, Q.; Iqbal, J.; Afreen, M.S.; Sandhu, M.Z.A.; Dar, E.; Farooq, U.; Mushtaq, M.F.; Arshad, N.; Iqbal, M.M. Synthesis of N-substituted acetamide derivatives of azinane-bearing 1, 3, 4-oxadiazole nucleus and screening for antibacterial activity. Trop. J. Pharm. Res. 2017, 16, 429–437. [Google Scholar] [CrossRef] [Green Version]
- Thongnest, S.; Chawengrum, P.; Keeratichamroen, S.; Lirdprapamongkol, K.; Eurtivong, C.; Boonsombat, J.; Kittakoop, P.; Svasti, J.; Ruchirawat, S. Vernodalidimer L, a sesquiterpene lactone dimer from Vernonia extensa and anti-tumor effects of vernodalin, vernolepin, and vernolide on HepG2 liver cancer cells. Bioorg. Chem. 2019, 92, 103197. [Google Scholar] [CrossRef]
- Johnston Paul, A.; Grandis Jennifer, R. STAT3 signaling: Anticancer strategies and challenges. Mol. Interv. 2011, 11, 18–26. [Google Scholar] [CrossRef] [Green Version]
- Dong, C.; Liu, S.; Cui, Y.; Guo, Q. 12-Lipoxygenase as a key pharmacological target in the pathogenesis of diabetic nephropathy. Eur. J. Pharmacol. 2020, 879, 173122. [Google Scholar] [CrossRef]
- Vardanyan, S.; Avagyan, A.; Aghekyan, A.; Sargsyan, A.; Harutyunyan, S.; Gasparyan, H. Synthesis and Some Transformations of 5-(1, 4-Benzodioxan-2-yl)-4-phenyl-4H-1, 2, 4-triazole-3-thiol. Russ. J. Org. Chem. 2020, 56, 436–439. [Google Scholar] [CrossRef]
- Washburn, K.; Neary, J. P2 purinergic receptors signal to STAT3 in astrocytes: Difference in STAT3 responses to P2Y and P2X receptor activation. Neuroscience 2006, 142, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Sylvester, R.; Tighe, A.P.; Chen, S.; Gudas, L.J. Transcriptional activation of the suppressor of cytokine signaling-3 (SOCS-3) gene via STAT3 is increased in F9 REX1 (ZFP-42) knockout teratocarcinoma stem cells relative to wild-type cells. J. Mol. Biol. 2008, 377, 28–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.F.; Kader, F.B.; Arman, M.; Ahmed, S.; Lyzu, C.; Sakib, S.A.; Tanzil, S.M.; Zim, A.I.U.; Imran, M.A.S.; Venneri, T. Pharmacological insights and prediction of lead bioactive isolates of Dita bark through experimental and computer-aided mechanism. Biomed. Pharmacother. 2020, 131, 110774. [Google Scholar] [CrossRef] [PubMed]
- McHardy, T.; Caldwell, J.J.; Cheung, K.-M.; Hunter, L.J.; Taylor, K.; Rowlands, M.; Ruddle, R.; Henley, A.; de Haven Brandon, A.; Valenti, M. Discovery of 4-amino-1-(7 H-pyrrolo [2, 3-d] pyrimidin-4-yl) piperidine-4-carboxamides as selective, orally active inhibitors of protein kinase B (Akt). J. Med. Chem. 2010, 53, 2239–2249. [Google Scholar] [CrossRef]
- Thomsen, R.; Christensen, M.H. MolDock: A new technique for high-accuracy molecular docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Alkyl/Aryl IC50 | Value (µg/mL) | Conventional Method | Ultrasound Assisted Method | ||
|---|---|---|---|---|---|---|
| Product %age Yield | Time for Reaction | Product %age Yield | Time for Reaction | |||
| 6a | 4-methylphenyl | 22.385 | 65 | 16 h. | 83 | 45 min. |
| 6b | 2,4-dimethylphenyl | 14.133 | 70 | 16 h. | 89 | 39 min. |
| 6c | 2-Bromophenyl | 24.482 | 71 | 26 h. | 77 | 80 min. |
| 6d | 2,3-dimethylphenyl | 13.004 | 60 | 15 h. | 75 | 40 min. |
| 6e | 4-Bromophenyl | 28.399 | 75 | 22 h. | 85 | 75 min. |
| 6f | 3-methylphenyl | 15.451 | 73 | 18 h. | 87 | 50 min. |
| Sorafenib | 05.971 | |||||
| Concentration (µg/mL) | 6a | 6b | 6c | 6d | 6e | 6f |
|---|---|---|---|---|---|---|
| 0 | 100 ± 2.33 | 100 ± 0.47 | 100 ± 5.20 | 100 ± 1.61 | 100 ± 0.85 | 100 ± 0.75 |
| 3.125 | 98.18 ± 2.33 | 96.63 ± 0.47 | 93.51 ± 5.20 | 88.75 ± 6.03 | 97.57 ± 0.85 | 94.39 ± 2.65 |
| 6.25 | 86.65 ± 2.92 | 81.10 ± 2.74 | 82.51 ± 4.74 | 74.18 ± 0.77 | 85.67 ± 1.64 | 86.12 ± 1.01 |
| 12.5 | 61.35 ± 1.30 | 51.63 ± 1.44 | 65.11 ± 5.30 | 54.02 ± 1.61 | 66.47 ± 4.71 | 59.07 ± 0.75 |
| 25 | 49.53 ± 4.65 | 34.77 ± 2.41 | 49.53 ± 4.65 | 36.37 ± 1.39 | 52.67 ± 0.26 | 38.98 ± 3.56 |
| 50 | 26.50 ± 4.05 | 11.58 ± 0.22 | 25.62 ± 1.51 | 10.40 ± 0.88 | 29.81 ± 3.18 | 11.36 ± 0.73 |
| 100 | 16.37 ± 1.76 | 8.74 ± 0.08 | 9.97 ± 1.00 | 8.93 ± 0.17 | 16.80 ± 3.94 | 9.71 ± 0.27 |
| 200 | 5.45 ± 0.55 | 6.04 ± 0.00 | 6.00 + 0.70 | 6.77 + 0.02 | 8.97 + 0.14 | 8.70 + 0.15 |
| DMSO (-ve Control) | 100 ± 0.0 | 100 ± 0.0 | 100 ± 0.0 | 100 ± 0.0 | 100 ± 0.0 | 100 ± 0.0 |
| Targets | Phosphatidylinositol 3-Kinase Alpha (PI3Kalpha) | Protein Kinase B (Akt) (PKB) | Human Aurora B Kinase (AURKB) | c-Kit Tyrosine Kinase (c-Kit) | Signal Transducer and Activator of Transcription 3 (STAT3) |
|---|---|---|---|---|---|
| PDB ID | 4FA6 | 2 × 39 | 4AF3 | 1T46 | 6NJS |
| Centre of docking | X:44 | X:43 | X:21 | X:28 | X:13 |
| Coordinates | Y:14 | Y:31 | Y:-22 | Y:26 | Y:56 |
| Z:31 | Z:111 | Z:-10 | Z:39 | Z:0.32 | |
| References Ligand | 0TA | X39 | VX6 | STI | KQV |
| Ligands | Mol. Dock Score | Mol. Dock Score | Mol. Dock Score | Mol. Dock Score | Mol. Dock Score |
| 6a | −145.41 | −164.897 | −133.498 | −158.744 | −109.036 |
| 6b | −138.651 | −169.631 | −143.585 | −170.426 | −115.252 |
| 6c | −140.069 | −161.63 | −132.878 | −153.031 | −107.5 |
| 6d | −137.122 | −176.152 | −145.486 | −180.052 | −117.807 |
| 6e | −132.298 | −159.691 | −131.958 | −151.185 | −106.026 |
| 6f | −133.085 | −167.609 | −141.576 | −161.024 | −111.411 |
| Reference Molecules | −112.819 | −130.624 | −144.231 | −181.533 | −197.521 |
| Ligand | (∆G) (kcal/mol) | Category | Types | Interactions Residues |
|---|---|---|---|---|
| 6a | −164.897 | H Bond | Conventional H Bond | ARG6, SER9 |
| Other | Sulfur-X | THR7 | ||
| Hydrophobic | Alkyl | VAL166, LYS181, LYS181, LEU183 | ||
| 6b | −169.631 | H Bond | Conventional H Bond | SER9, LYS277 |
| Other | Sulfur-X Pi- Cation | THR7 ARG6, GLU236 | ||
| Hydrophobic | Alkyl | VAL166, LYS181, LEU183, | ||
| 6c | −167.609 | Hydrophobic | Alkyl | LYS181, LEU296 |
| 6d | −176.152 | H Bond | Conventional H Bond Doner H Bond | LYS181 SERC9 |
| Other | Pi-Anion | ASP275 | ||
| Hydrophobic | Alkyl | HIS196, LEU296 | ||
| 6e | −161.63 | H Bond | Conventional H Bond | THR162, PHE163 |
| Hydrophobic | Alkyl | VAL166 | ||
| 6f | −159.691 | H Bond Other | Conventional H Bond | LYS160, LYS181 |
| C-H Bond | ASP293 | |||
| Hydrophobic | Alkyl | VAL166, ALA179, LEU158, VAL166, MET282 | ||
| Reference Ligand | H Bond | Conventional H Bond | ALA232, ASP293, MET282, GLU279, GLU236, | |
| X39 | −130.624 | Other | C-H Bond | GLY159, MET229 |
| Hydrophobic | Alkyl | VAL166, ALA179, LEU158, LEU183, LYS181 |
| Ligand | ACE (kcal/mol) | Category | Types | Interactions Residues |
|---|---|---|---|---|
| 6a | −158.744 | H Bond | Conventional H Bond | GLU640 |
| Hydrophobic | Alkyl | ALA621, LYS623, VAL643, VAL603, LYS623, VAL603 | ||
| 6b | −170.426 | H Bond Other | Conventional H Bond Pi- doner H Bond Pi-Cation Pi-Pi T shaped | GLU640 THR670 LYS273, ASP810 HIS790 |
| Hydrophobic | Alkyl | VAL603, ALA621, VAL643, VAL654, LEU644, VAL668 | ||
| 6c | −153.031 | H Bond | Conventional H Bond | CYS673 |
| Hydrophobic | Pi-Alkyl | TYR672, PHE811 | ||
| Hydrophobic | Alkyl | ALA621, LYS623, VAL643 VAL668, EU783, CYS788 VAL603, LYS623, LEU783 | ||
| 6d | −180.052 | H Bond Other | Conventional H Bond Pi-doner H-Bond Pi-Sigma Pi-Pi staked | CYS673 THR670 LEU595, LEU644 TYR672 |
| Hydrophobic | Alkyl | LEU644, VAL654, CYS809, LEU799, LYS623, PHE811, VAL668, VAL603, ALA621 | ||
| 6e | −151.185 | H Bond | Conventional H Bond | ASP810 |
| Other | Pi-Sulfur | PHE811 | ||
| Hydrophobic | Alkyl | LEU595, LEU644, VAL654 EU799, CYS809, LEU647 LEU783, LEU644, VAL654 | ||
| 6f | −161.024 | H Bond | Conventional H Bond | ILE808 |
| Hydrophobic | Alkyl | HIS790 | ||
| Reference Ligand | H Bond | Conventional H Bond | ALA232, ASP293, MET282, GLU279, GLU236, | |
| STI | −181.533 | Other | Carbon H Bond | GLY159, MET229 |
| Hydrophobic | Alkyl | VAL166, ALA179, LEU A158, LEU A183, LYS A181 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahmood, S.; Khan, S.G.; Rasul, A.; Christensen, J.B.; Abourehab, M.A.S. Ultrasound Assisted Synthesis and In Silico Modelling of 1,2,4-Triazole Coupled Acetamide Derivatives of 2-(4-Isobutyl phenyl)propanoic acid as Potential Anticancer Agents. Molecules 2022, 27, 7984. https://doi.org/10.3390/molecules27227984
Mahmood S, Khan SG, Rasul A, Christensen JB, Abourehab MAS. Ultrasound Assisted Synthesis and In Silico Modelling of 1,2,4-Triazole Coupled Acetamide Derivatives of 2-(4-Isobutyl phenyl)propanoic acid as Potential Anticancer Agents. Molecules. 2022; 27(22):7984. https://doi.org/10.3390/molecules27227984
Chicago/Turabian StyleMahmood, Sadaf, Samreen Gul Khan, Azhar Rasul, Jørn Bolstad Christensen, and Mohammed A. S. Abourehab. 2022. "Ultrasound Assisted Synthesis and In Silico Modelling of 1,2,4-Triazole Coupled Acetamide Derivatives of 2-(4-Isobutyl phenyl)propanoic acid as Potential Anticancer Agents" Molecules 27, no. 22: 7984. https://doi.org/10.3390/molecules27227984