
Actions of Novel Angiotensin Receptor Blocking Drugs, Bisartans, Relevant for COVID-19 Therapy: Biased Agonism at Angiotensin Receptors and the Beneficial Effects of Neprilysin in the Renin Angiotensin System

 , , and
, , and 
Abstract
:1. Introduction
2. Methods
2.1. Isolated Tissues
2.2. In Silico Studies
3. Results and Discussion
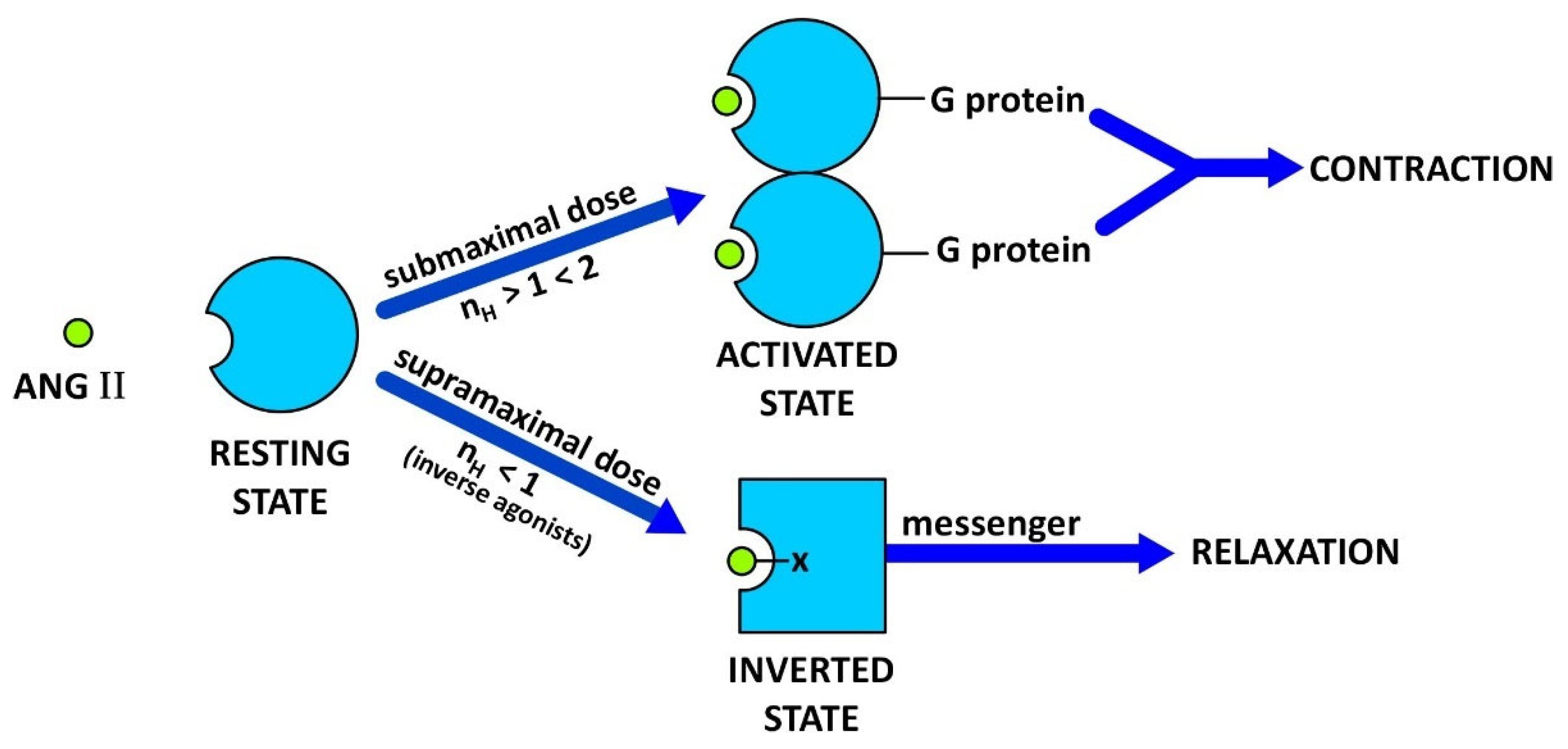
3.1. Two-State Receptor and Biased Agonism
3.1.1. Angiotensin Peptides and Antipeptides—Regulators of RAS
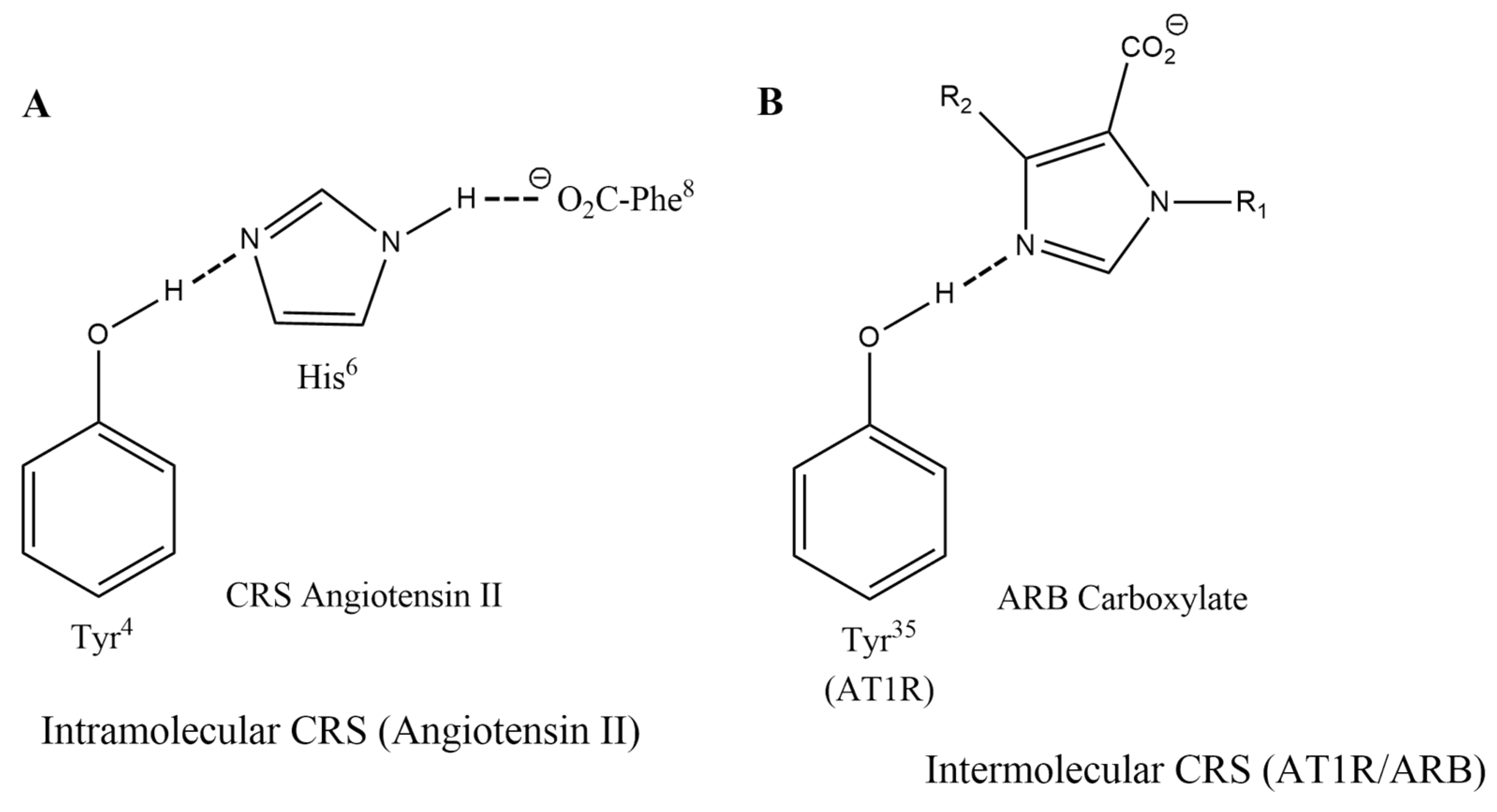
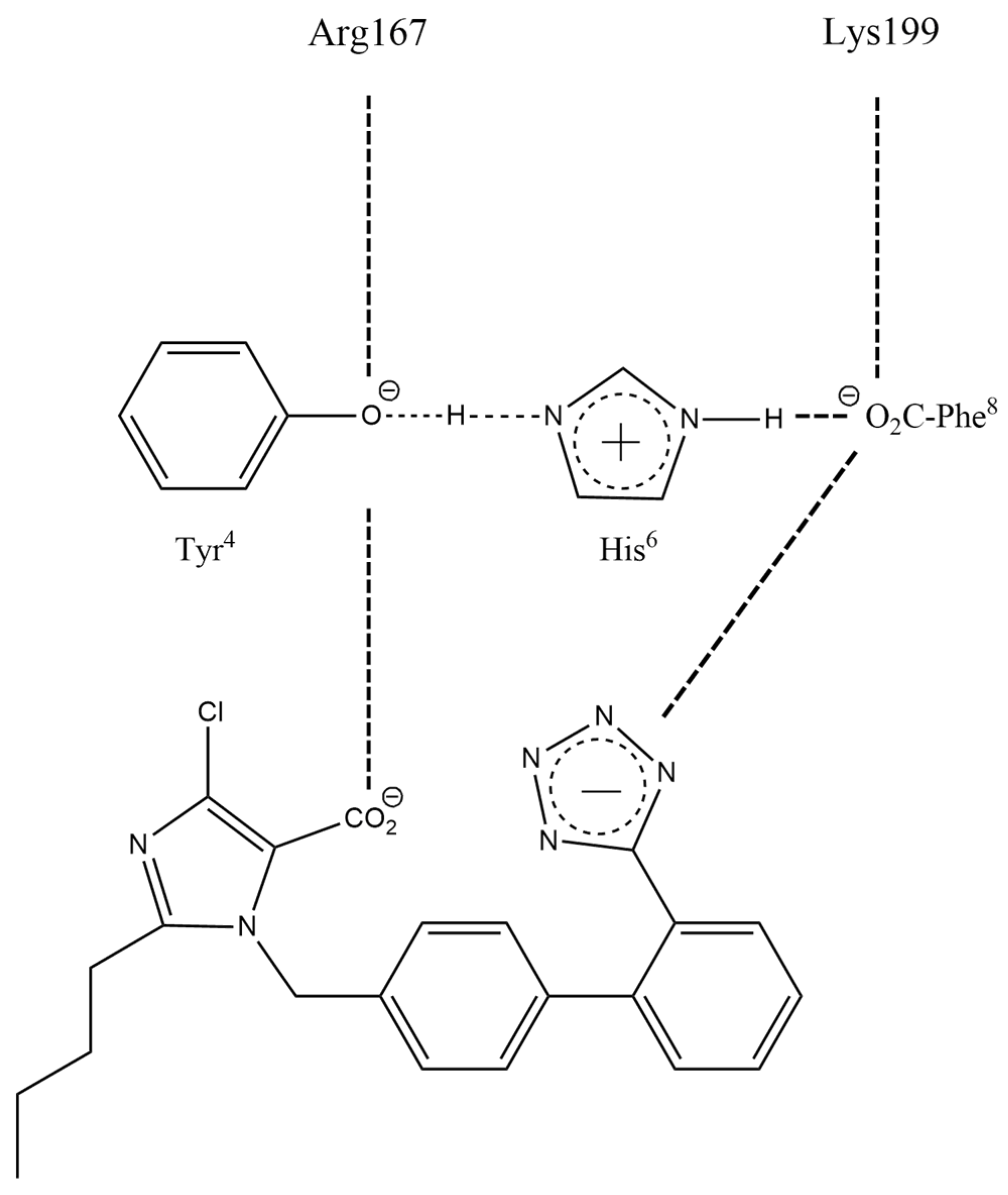
3.1.2. Charge-Relay Systems
3.1.3. Angiotensin Receptor Blockers (ARB Sartans)
3.1.4. SARS-CoV-2, ARBs, and Bisartans
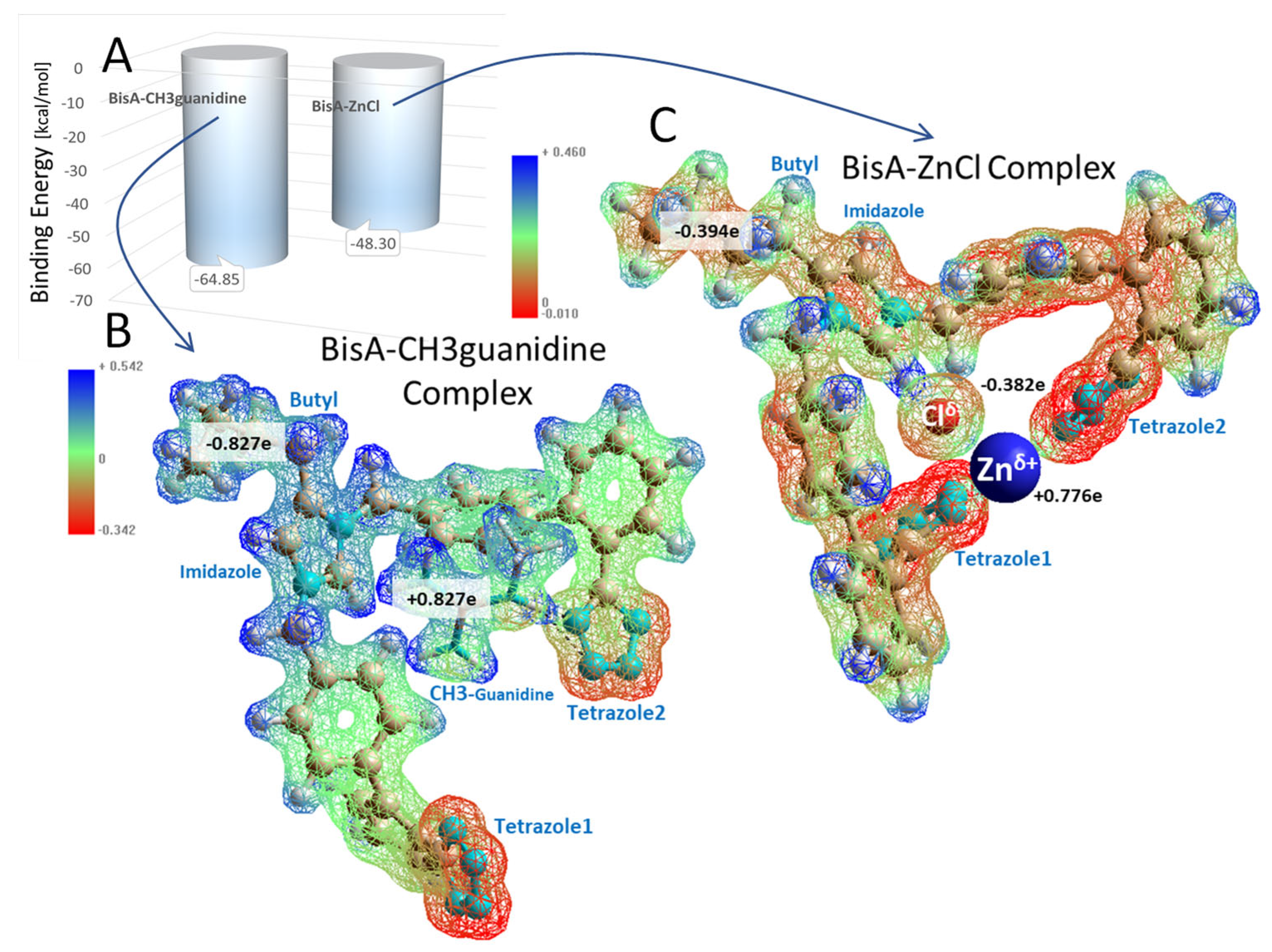
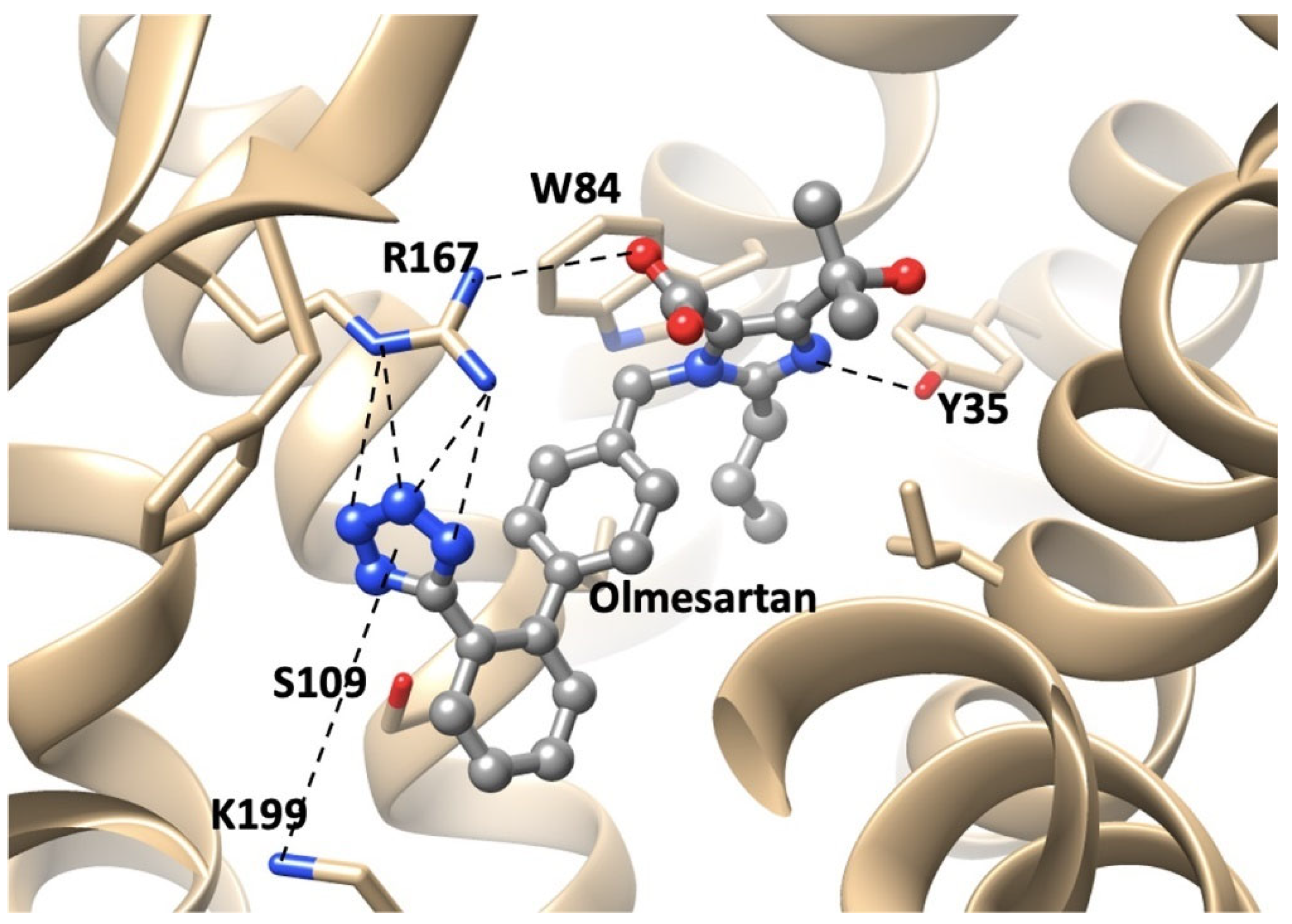
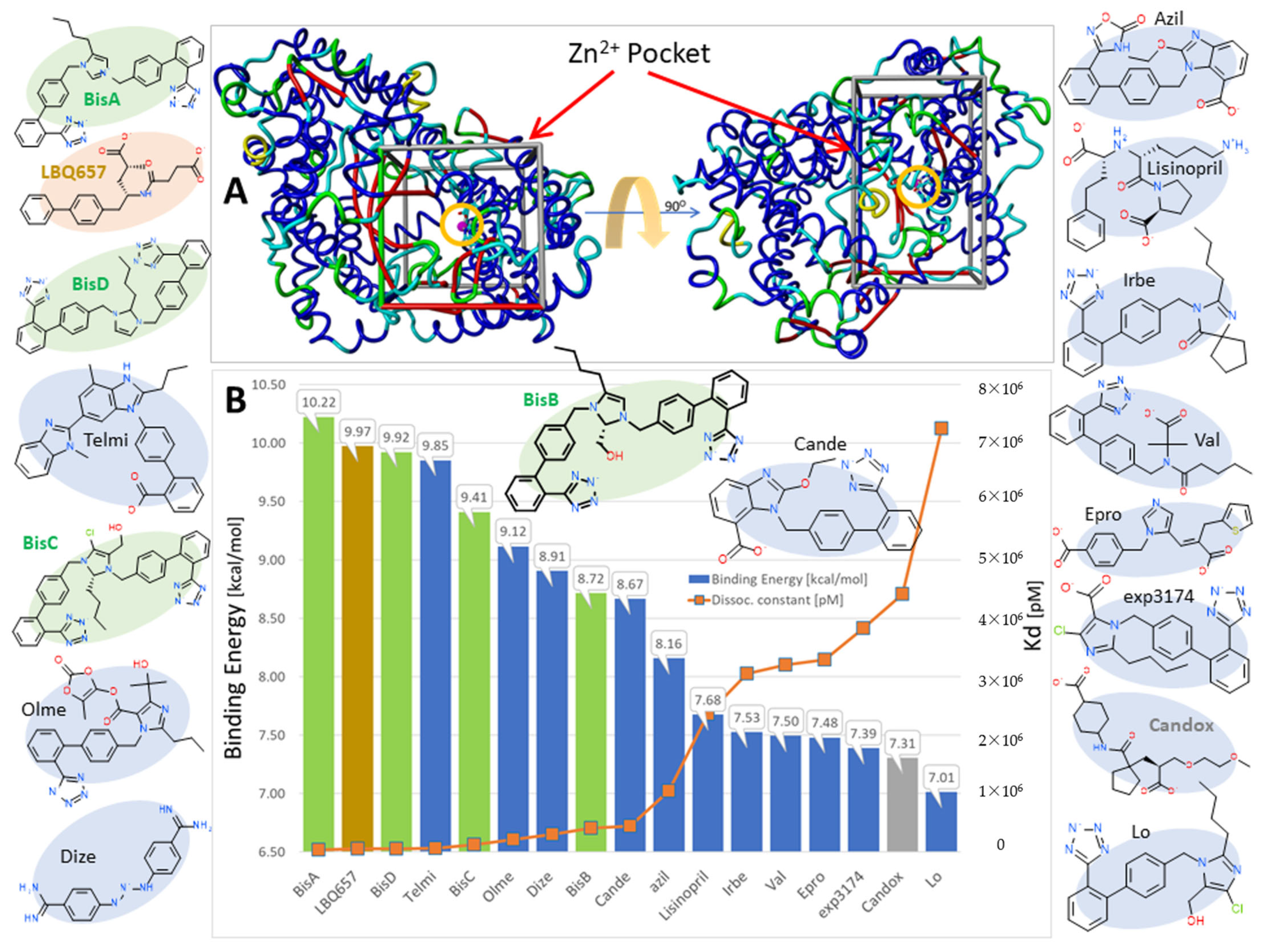
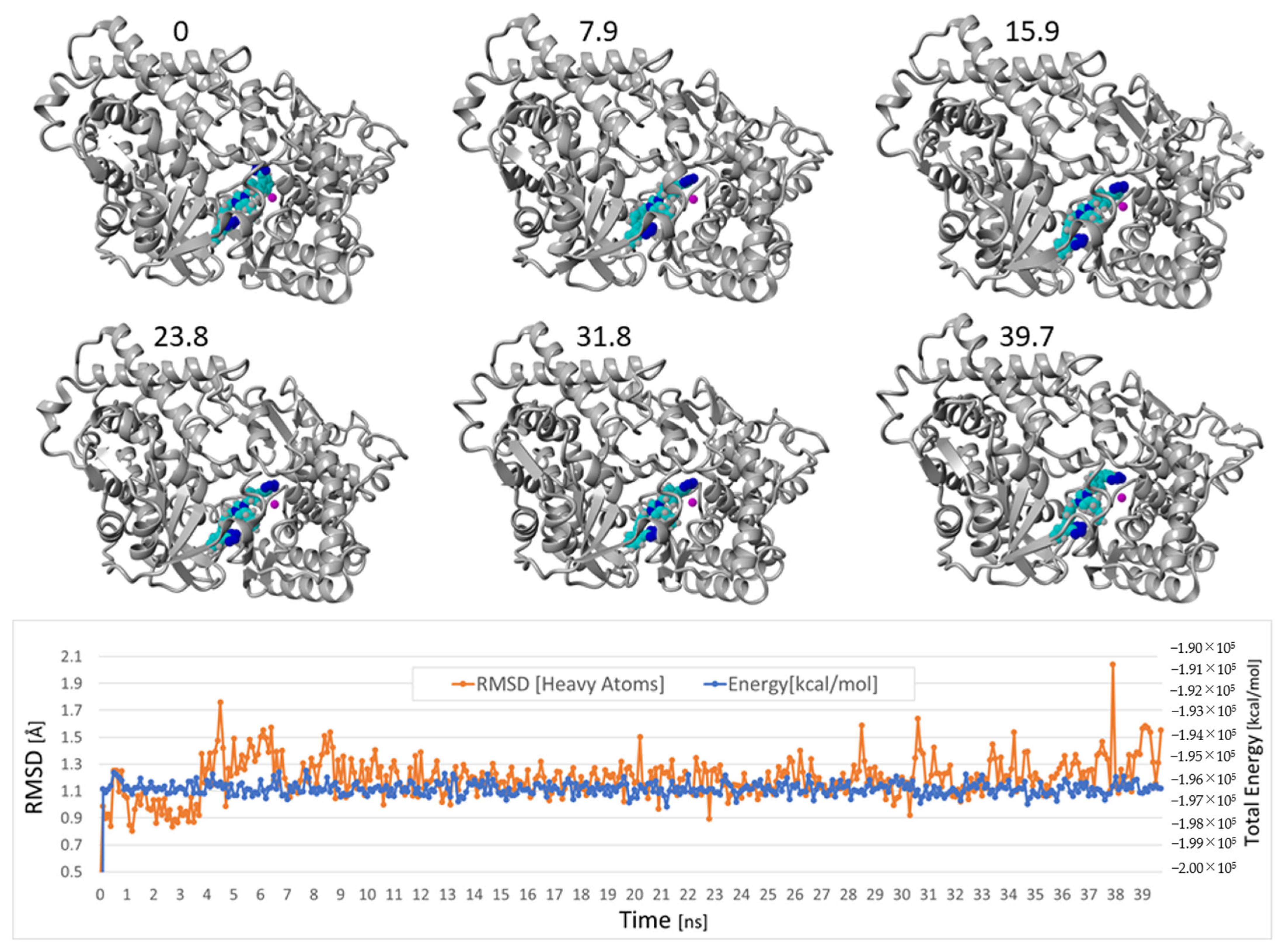
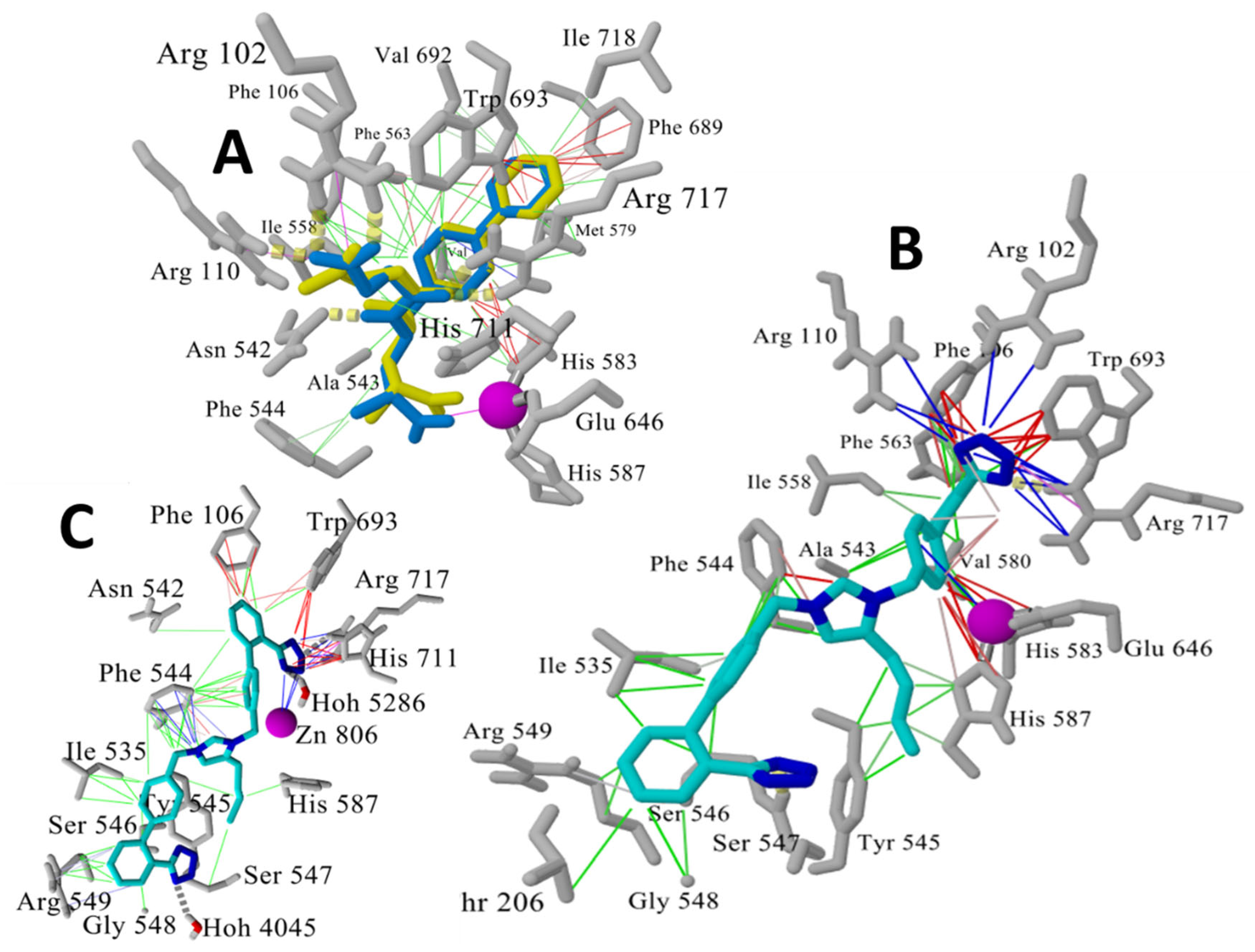
3.1.5. Bisartans Bound to AT1R
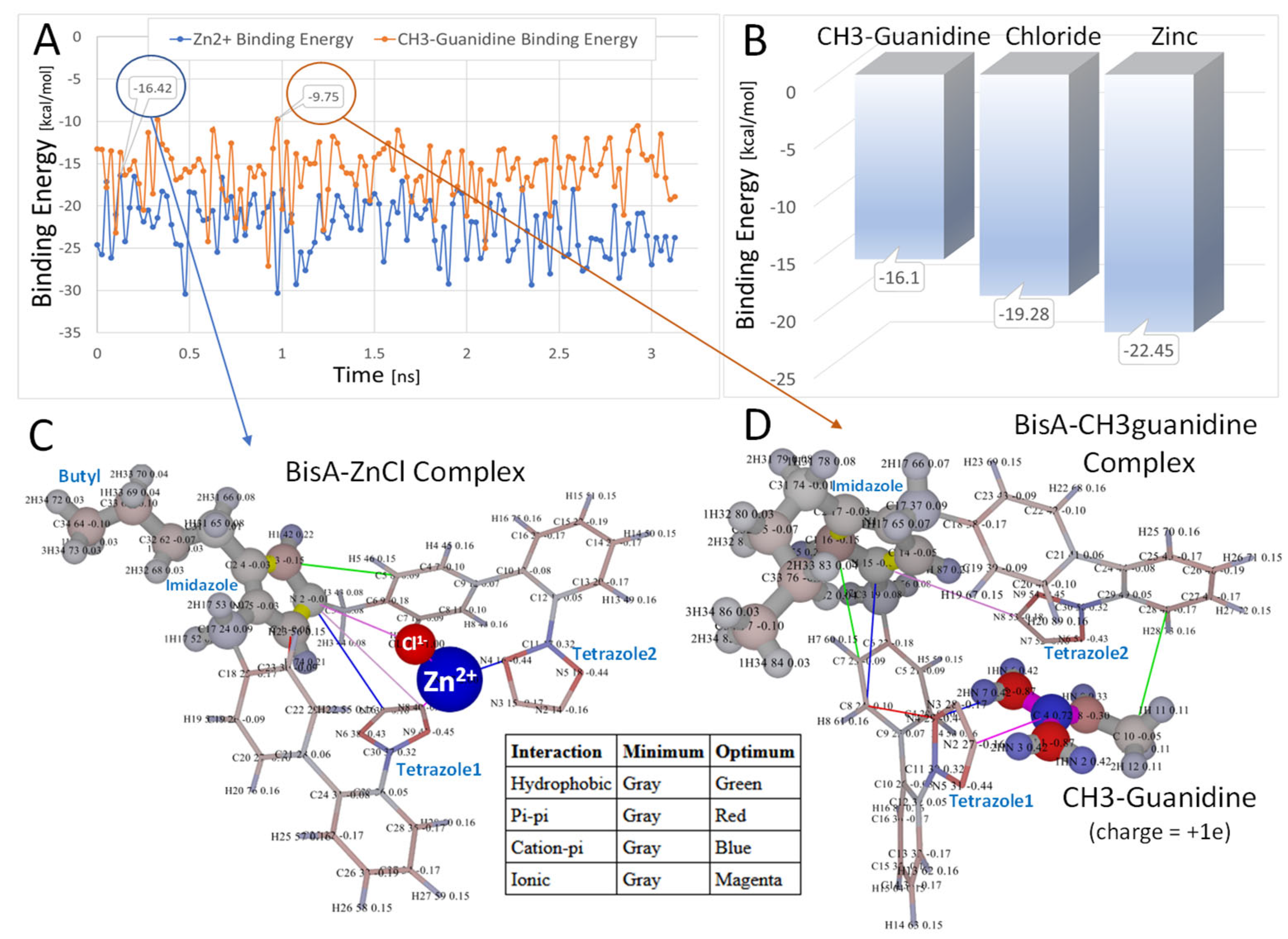
3.1.6. Bisartans Bound to NEP
3.1.7. ARBs: Potential Antiviral Drugs for the Treatment of COVID-19
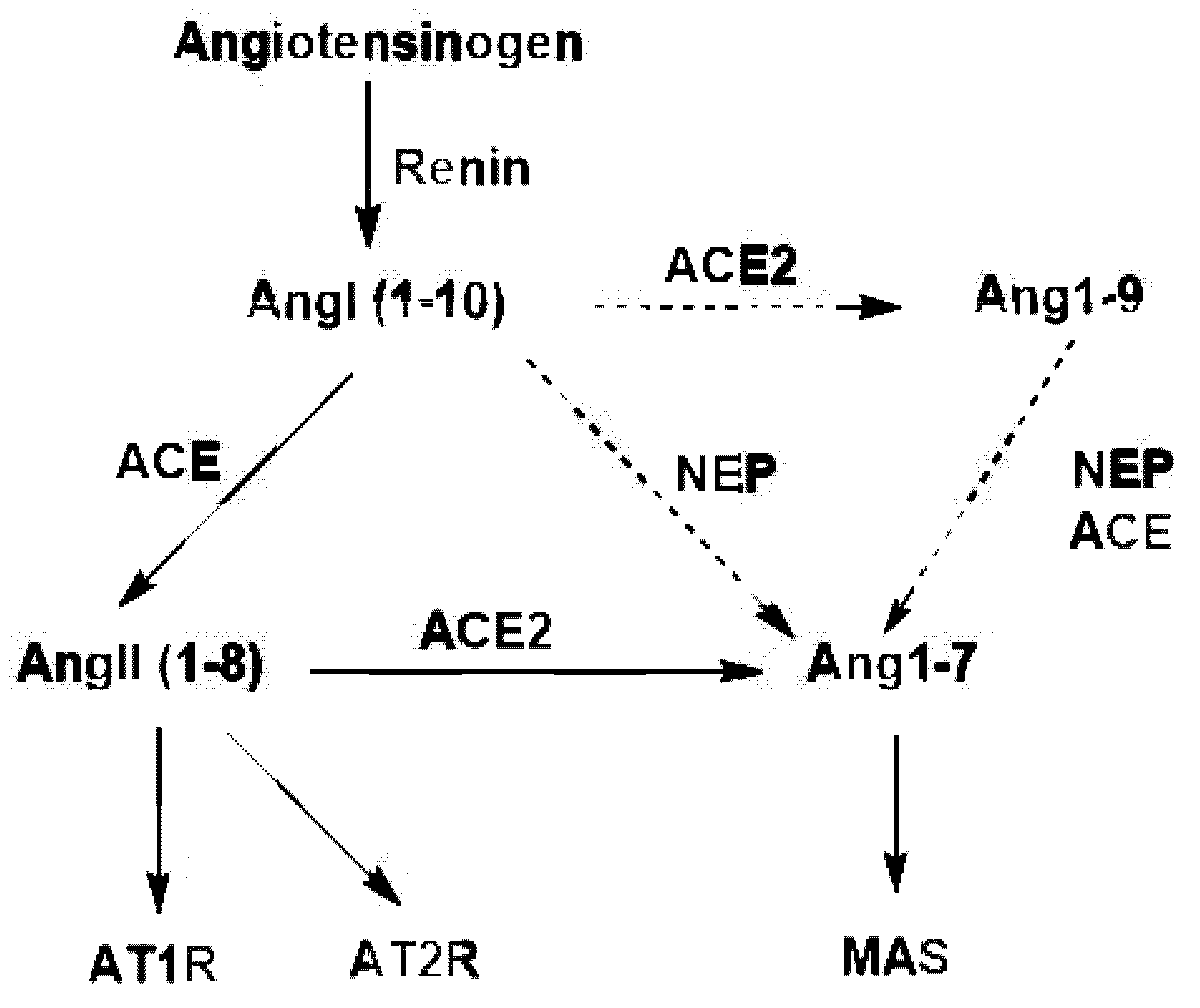
3.1.8. Effects of ACE, ACE2, and NEP in the Renin–Angiotensin System
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| ACE2 | Angiotensin Converting Enzyme 2 |
| Ala | Alanine |
| AMBER | Assisted model building with energy refinement |
| ANG II | Angiotensin II |
| AR1 | Androgen receptor 1 |
| ARBs | Angiotensin II receptor blockers |
| Arg | Arginine |
| AT1(2) | ANG II receptors type 1 (2) |
| Azil | Azilsartan |
| Bis | Bisartan |
| BPT | Biphenyl tetrazole |
| Cande | Candesartan |
| 3CLpro | 3C-like protease |
| COVID-19 | Coronavirus diseases |
| CRS | Charge-relay system |
| DIZE | Deminazene aceturate |
| DNA | Deoxyribonucleic acid |
| Epro | Eprosartan |
| EXP3174 | Losartan carboxylic acid |
| His | Histidine |
| ID | Identifier |
| Ile | Isoleucine |
| Irbe | Irbesartan |
| Leu | Leucine |
| Lo | Losartan |
| Lys | Lysine |
| MD | Molecular dynamics |
| mRNA | Messenger ribonucleic acid |
| NEP | Neprilysin |
| Olme | Olmesartan |
| PDB | Protein Data Bank |
| Phe | Phenylalanine |
| Pro | Proline |
| RAS | Renin–angiotensin system |
| RM1 | Recife model 1 |
| RMSD | Root-mean-square-deviation |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| Telmi | Telmisartan |
| TIP3P | Transferable intermolecular potential with 3 points |
| Trp | Tryptophan |
| Tyr | Tyrosine |
| UHF | Unrestricted Hartree–Fock |
| Val | Valsartan |
| YAMBER | Yet Another Model Building and Energy Refinement |
| YASARA | Yet Another Scientific Artificial Reality Application |
Nomenclature for Bisartans
References
- Bumpus, F.M.; Smeby, R.R.; Page, I.H. Angiotensin, the Renal Pressor Hormone. Circ. Res. 1961, 9, 762–767. [Google Scholar] [CrossRef] [Green Version]
- Reid, I.A.; Morris, B.J.; Ganong, W.F. The Renin-Angiotensin System. Annu. Rev. Physiol. 1978, 40, 377–410. [Google Scholar] [CrossRef] [PubMed]
- Izumi, Y.; Iwao, H. Chapter 186—Angiotensin II Peptides. In Handbook of Biologically Active Peptides, 2nd ed.; Academic Press: Cambridge, MA, USA, 2013; pp. 1369–1376. [Google Scholar] [CrossRef]
- Ardaillou, R.; Chansel, D. Synthesis and effects of active fragments of angiotensin II. Kidney Int. 1997, 52, 1458–1468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, R.; Suzuki, J.-i.; Wakayama, K.; Maejima, Y.; Shimamura, M.; Koriyama, H.; Nakagami, H.; Kumagai, H.; Ikeda, Y.; Akazawa, H.; et al. A peptide vaccine targeting angiotensin II attenuates the cardiac dysfunction induced by myocardial infarction. Sci. Rep. 2017, 7, 43920. [Google Scholar] [CrossRef]
- Mogi, M. Clinical study on angiotensin II vaccination—The first big step. Hypertens. Res. 2021, 45, 162–163. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Wei, W. Angiotensin II in inflammation, immunity and rheumatoid arthritis. Clin. Exp. Immunol. 2015, 179, 137–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stegbauer, J.; Lee, D.-H.; Seubert, S.; Ellrichmann, G.; Manzel, A.; Kvakan, H.; Muller, D.N.; Gaupp, S.; Rump, L.C.; Gold, R.; et al. Role of the renin-angiotensin system in autoimmune inflammation of the central nervous system. Proc. Natl. Acad. Sci. USA 2009, 106, 14942–14947. [Google Scholar] [CrossRef] [Green Version]
- Platten, M.; Youssef, S.; Hur, E.M.; Ho, P.P.; Han, M.H.; Lanz, T.V.; Phillips, L.K.; Goldstein, M.J.; Bhat, R.; Raine, C.S.; et al. Blocking angiotensin-converting enzyme induces potent regulatory T cells and modulates TH1- and TH17-mediated autoimmunity. Proc. Natl. Acad. Sci. USA 2009, 106, 14948–14953. [Google Scholar] [CrossRef] [Green Version]
- Katsara, M.; Matsoukas, J.; Deraos, G.; Apostolopoulos, V. Towards immunotherapeutic drugs and vaccines against multiple sclerosis. Acta Biochim. Biophys. Sin. 2008, 40, 636–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apostolopoulos, V.; Rostami, A.; Matsoukas, J. The Long Road of Immunotherapeutics against Multiple Sclerosis. Brain Sci. 2020, 10, 288. [Google Scholar] [CrossRef]
- Hsueh, W.A.; Do, Y.S.; Anderson, P.W.; Law, R.E. Angiotensin II in Cell Growth and Matrix Production. Adv. Exp. Med. Biol. 1995, 377, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Ruiz-Ortega, M.; Lorenzo, O.; Ruperez, M.; Esteban, V.; Egido, J. Inflammation and angiotensin II. Int. J. Biochem. Cell Biol. 2003, 35, 881–900. [Google Scholar] [CrossRef]
- Ishikane, S.; Takahashi-Yanaga, F. The role of angiotensin II in cancer metastasis: Potential of renin-angiotensin system blockade as a treatment for cancer metastasis. Biochem. Pharmacol. 2018, 151, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Matsoukas, J.; Apostolopoulos, V.; Zulli, A.; Moore, G.; Kelaidonis, K.; Moschovou, K.; Mavromoustakos, T. From Angiotensin II to Cyclic Peptides and Angiotensin Receptor Blockers (ARBs): Perspectives of ARBs in COVID-19 Therapy. Molecules 2021, 26, 618. [Google Scholar] [CrossRef]
- Ridgway, H.; Moore, G.J.; Mavromoustakos, T.; Tsiodras, S.; Ligielli, I.; Kelaidonis, K.; Chasapis, C.T.; Gadanec, L.K.; Zulli, A.; Apostolopoulos, V.; et al. Discovery of a new generation of angiotensin receptor blocking drugs: Receptor mechanisms and in silico binding to enzymes relevant to SARS-CoV-2. Comput. Struct. Biotechnol. J. 2022, 20, 2091–2111. [Google Scholar] [CrossRef] [PubMed]
- Apostolopoulos, V.; Bojarska, J.; Chai, T.-T.; Elnagdy, S.; Kaczmarek, K.; Matsoukas, J.; New, R.; Parang, K.; Lopez, O.P.; Parhiz, H.; et al. A Global Review on Short Peptides: Frontiers and Perspectives. Molecules 2021, 26, 430. [Google Scholar] [CrossRef]
- Matsoukas, J.; Cordopatis, P.; Belte, U.; Goghari, M.H.; Ganter, R.C.; Franklin, K.J.; Moore, G.J. Importance of the N-terminal domain of the type II angiotensin antagonist sarmesin for receptor blockade. J. Med. Chem. 1988, 31, 1418–1421. [Google Scholar] [CrossRef]
- Matsoukas, J.M.; Agelis, G.; Hondrelis, J.; Yamdagni, R.; Wu, Q.; Ganter, R.; Moore, D.; Moore, G.J.; Smith, J.R. Synthesis and biological activities of angiotensin II, sarilesin, and sarmesin analogs containing Aze or Pip at position 7. J. Med. Chem. 1993, 36, 904–911. [Google Scholar] [CrossRef]
- Hondrelis, J.; Lonergan, G.; Voliotis, S.; Matsoukas, J. One pot synthesis and conformation of N-t-butyloxycarbonyl, O-Phenacyl derivatives of proline and other secondary amino acids. Tetrahedron 1990, 46, 565–576. [Google Scholar] [CrossRef]
- Polevaya, L.; Mavromoustakos, T.; Zoumboulakis, P.; Grdadolnik, S.G.; Roumelioti, P.; Giatas, N.; Mutule, I.; Keivish, T.; Vlahakos, D.V.; Iliodromitis, E.K.; et al. Synthesis and study of a cyclic angiotensin II antagonist analogue reveals the role of π*–π* interactions in the C-terminal aromatic residue for agonist activity and its structure resemblance with AT1 non-peptide antagonists. Bioorg. Med. Chem. 2001, 9, 1639–1647. [Google Scholar] [CrossRef]
- Kaltenecker, C.C.; Domenig, O.; Kopecky, C.; Antlanger, M.; Poglitsch, M.; Berlakovich, G.; Kain, R.; Stegbauer, J.; Rahman, M.; Hellinger, R.; et al. Critical Role of Neprilysin in Kidney Angiotensin Metabolism. Circ. Res. 2020, 127, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Domenig, O.; Manzel, A.; Grobe, N.; Kaltenecker, C.; Kovarik, J.; Stegbauer, J.; Gurley, S.B.; Antlanger, M.; Elased, K.; Saemann, M.; et al. The role of neprilysin in angiotensin 1-7 formation in the kidney. J. Hypertens. 2015, 33, e114–e115. [Google Scholar] [CrossRef]
- Moore, G.J.; Franklin, K.J.; Nystrom, D.M.; Goghari, M.H. Structure–desensitization relationships of angiotensin analogues in the rat isolated uterus. Can. J. Physiol. Pharmacol. 1985, 63, 966–971. [Google Scholar] [CrossRef]
- Moore, G.J.; Scanlon, M.N. Methods for analyzing and interpreting cooperativity in dose-response curves—I. Antagonist effects on angiotensin receptors in smooth muscle. Gen. Pharmacol. Vasc. Syst. 1989, 20, 193–198. [Google Scholar] [CrossRef]
- Scanlon, M.N.; Koziarz, P.; Moore, G.J. The relationship between homotropic and heterotropic cooperativity for angiotensin receptors in smooth muscle. Gen. Pharmacol. Vasc. Syst. 1990, 21, 59–65. [Google Scholar] [CrossRef]
- Moore, G.J. Methods for analyzing and interpreting cooperativity in dose-response curves—II. Partial agonists acting on muscarinic receptors in smooth muscle. Gen. Pharmacol. Vasc. Syst. 1989, 20, 199–203. [Google Scholar] [CrossRef]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Christie, C.H.; Dalenberg, K.; Di Costanzo, L.; Duarte, J.M.; et al. RCSB Protein Data Bank: Powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 2021, 49, D437–D451. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Modeling 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Nielsen, J.E.; Spronk, C.A.E.M.; Vriend, G. Fast empirical pKa prediction by Ewald summation. J. Mol. Graph. Model. 2006, 25, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Vriend, G. YASARA View—Molecular graphics for all devices—From smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef] [PubMed]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins Struct. Funct. Bioinform. 2006, 65, 712–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Moore, G.J.; Ganter, R.C.; Franklin, K.J. Angiotensin ‘antipeptides’: (−)messenger RNA complementary to human angiotensin II (+)messenger RNA encodes an angiotensin receptor antagonist. Biochem. Biophys. Res. Commun. 1989, 160, 1387–1391. [Google Scholar] [CrossRef]
- Blow, D.M.; Birktoft, J.J.; Hartley, B.S. Role of a Buried Acid Group in the Mechanism of Action of Chymotrypsin. Nature 1969, 221, 337–340. [Google Scholar] [CrossRef]
- Moore, G.J. Designing peptide mimetics. Trends Pharmacol. Sci. 1994, 15, 124–129. [Google Scholar] [CrossRef]
- Matsoukas, J.M.; Hondrelis, J.; Keramida, M.; Mavromoustakos, T.; Makriyannis, A.; Yamdagni, R.; Wu, Q.; Moore, G.J. Role of the NH2-terminal domain of angiotensin II (ANG II) and [Sar1]angiotensin II on conformation and activity. NMR evidence for aromatic ring clustering and peptide backbone folding compared with [des-1,2,3]angiotensin II. J. Biol. Chem. 1994, 269, 5303–5312. [Google Scholar]
- Matsoukas, J.M.; Bigam, G.; Zhou, N.; Moore, G.J.I. 1H-NMR studies of [Sar1]angiotensin II conformation by nuclear Overhauser effect spectroscopy in the rotating frame (ROESY): Clustering of the aromatic rings in dimethylsulfoxide. Peptides 1990, 11, 359–366. [Google Scholar] [CrossRef]
- Zhang, H.; Unal, H.; Desnoyer, R.; Han, G.W.; Patel, N.; Katritch, V.; Karnik, S.S.; Cherezov, V.; Stevens, R.C. Structural Basis for Ligand Recognition and Functional Selectivity at Angiotensin Receptor. J. Biol. Chem. 2015, 290, 29127–29139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Unal, H.; Gati, C.; Han, G.W.; Liu, W.; Zatsepin, N.A.; James, D.; Wang, D.; Nelson, G.; Weierstall, U.; et al. Structure of the Angiotensin Receptor Revealed by Serial Femtosecond Crystallography. Cell 2015, 161, 833–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asada, H.; Inoue, A.; Ngako Kadji, F.M.; Hirata, K.; Shiimura, Y.; Im, D.; Shimamura, T.; Nomura, N.; Iwanari, H.; Hamakubo, T.; et al. The Crystal Structure of Angiotensin II Type 2 Receptor with Endogenous Peptide Hormone. Structure 2020, 28, 418–425.e4. [Google Scholar] [CrossRef] [PubMed]
- Moore, G.J.; Matsoukas, J.M. Angiotensin as a model for hormone—Receptor interactions. Biosci. Rep. 1985, 5, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Takezako, T.; Unal, H.; Karnik, S.S.; Node, K. Current topics in angiotensin II type 1 receptor research: Focus on inverse agonism, receptor dimerization and biased agonism. Pharmacol. Res. 2017, 123, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Qi, F.; Bao, L.; Du, L.; Liu, S.; et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020, 30, 343–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agelis, G.; Resvani, A.; Koukoulitsa, C.; Tůmová, T.; Slaninová, J.; Kalavrizioti, D.; Spyridaki, K.; Afantitis, A.; Melagraki, G.; Siafaka, A.; et al. Rational design, efficient syntheses and biological evaluation of N, N ′-symmetrically bis-substituted butylimidazole analogs as a new class of potent Angiotensin II receptor blockers. Eur. J. Med. Chem. 2013, 62, 352–370. [Google Scholar] [CrossRef] [Green Version]
- Fatouros, P.R.; Roy, U.; Sur, S. Modeling Substrate Coordination to Zn-Bound Angiotensin Converting Enzyme 2. bioRxiv 2021, preprint. [Google Scholar] [CrossRef]
- Wingler, L.M.; McMahon, C.; Staus, D.P.; Lefkowitz, R.J.; Kruse, A.C. Distinctive Activation Mechanism for Angiotensin Receptor Revealed by a Synthetic Nanobody. Cell 2019, 176, 479–490.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, G.J.; Pires, J.M.; Kelaidonis, K.; Gadanec, L.K.; Zulli, A.; Apostolopoulos, V.; Matsoukas, J.M. Receptor Interactions of Angiotensin II and Angiotensin Receptor Blockers—Relevance to COVID-19. Biomolecules 2021, 11, 979. [Google Scholar] [CrossRef] [PubMed]
- Jhund, P.S.; McMurray, J.J.V. The neprilysin pathway in heart failure: A review and guide on the use of sacubitril/valsartan. Heart 2016, 102, 1342–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiering, N.; D’Arcy, A.; Villard, F.; Ramage, P.; Logel, C.; Cumin, F.; Ksander, G.M.; Wiesmann, C.; Karki, R.G.; Mogi, M. Structure of neprilysin in complex with the active metabolite of sacubitril. Sci. Rep. 2016, 6, 27909. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Huang, Z.; Lin, L.; Lv, J. Coronavirus Disease 2019 (COVID-19) and Cardiovascular Disease: A Viewpoint on the Potential Influence of Angiotensin-Converting Enzyme Inhibitors/Angiotensin Receptor Blockers on Onset and Severity of Severe Acute Respiratory Syndrome Coronavirus 2 Infection. J. Am. Heart Assoc. 2020, 9. [Google Scholar] [CrossRef]
- Moreno, M.; Bataller, R. Cytokines and Renin-Angiotensin System Signaling in Hepatic Fibrosis. Clin. Liver Dis. 2008, 12, 825–852. [Google Scholar] [CrossRef]
- Ruiz-Ortega, M.; Ruperez, M.; Lorenzo, O.; Esteban, V.; Blanco, J.; Mezzano, S.; Egido, J. Angiotensin II regulates the synthesis of proinflammatory cytokines and chemokines in the kidney. Kidney Int. 2002, 62, S12–S22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Zhu, L.; Cai, J.; Lei, F.; Qin, J.-J.; Xie, J.; Liu, Y.-M.; Zhao, Y.-C.; Huang, X.; Lin, L.; et al. Association of Inpatient Use of Angiotensin-Converting Enzyme Inhibitors and Angiotensin II Receptor Blockers With Mortality Among Patients With Hypertension Hospitalized With COVID-19. Circ. Res. 2020, 126, 1671–1681. [Google Scholar] [CrossRef] [PubMed]
- Abassi, Z.A.; Skorecki, K.; Heyman, S.N.; Kinaneh, S.; Armaly, Z. COVID-19 infection and mortality: A physiologist’s perspective enlightening clinical features and plausible interventional strategies. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2020, 318, L1020–L1022. [Google Scholar] [CrossRef] [Green Version]
- Abassi, Z.A.; Skorecki, K.; Heyman, S.N.; Kinaneh, S.; Armaly, Z. Reply to Letter to the Editor: “Don’t judge too RAShly: The multifaceted role of the renin-angiotensin system and its therapeutic potential in COVID-19”. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2020, 318, L1029–L1030. [Google Scholar] [CrossRef]
- Dambha-Miller, H.; Albasri, A.; Hodgson, S.; Wilcox, C.R.; Khan, S.; Islam, N.; Little, P.; Griffin, S.J. Currently prescribed drugs in the UK that could upregulate or downregulate ACE2 in COVID-19 disease: A systematic review. BMJ Open 2020, 10, e040644. [Google Scholar] [CrossRef]
- Meng, J.; Xiao, G.; Zhang, J.; He, X.; Ou, M.; Bi, J.; Yang, R.; Di, W.; Wang, Z.; Li, Z.; et al. Renin-angiotensin system inhibitors improve the clinical outcomes of COVID-19 patients with hypertension. Emerg. Microbes Infect. 2020, 9, 757–760. [Google Scholar] [CrossRef] [PubMed]
- Ni, W.; Yang, X.; Yang, D.; Bao, J.; Li, R.; Xiao, Y.; Hou, C.; Wang, H.; Liu, J.; Yang, D.; et al. Role of angiotensin-converting enzyme 2 (ACE2) in COVID-19. Crit. Care 2020, 24, 422. [Google Scholar] [CrossRef]
- Rico-Mesa, J.S.; White, A.; Anderson, A.S. Outcomes in Patients with COVID-19 Infection Taking ACEI/ARB. Curr. Cardiol. Rep. 2020, 22, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sriram, K.; Loomba, R.; Insel, P.A. Targeting the renin−angiotensin signaling pathway in COVID-19: Unanswered questions, opportunities, and challenges. Proc. Natl. Acad. Sci. USA 2020, 117, 29274–29282. [Google Scholar] [CrossRef] [PubMed]
- Vaduganathan, M.; Vardeny, O.; Michel, T.; McMurray, J.J.V.; Pfeffer, M.A.; Solomon, S.D. Renin–Angiotensin–Aldosterone System Inhibitors in Patients with COVID-19. N. Engl. J. Med. 2020, 382, 1653–1659. [Google Scholar] [CrossRef]
- Warner, F.J.; Rajapaksha, H.; Shackel, N.; Herath, C.B. ACE2: From protection of liver disease to propagation of COVID-19. Clin. Sci. 2020, 134, 3137–3158. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, B.; Batool, M.; Ain, Q.u.; Kim, M.S.; Choi, S. Exploring the Binding Mechanism of PF-07321332 SARS-CoV-2 Protease Inhibitor through Molecular Dynamics and Binding Free Energy Simulations. Int. J. Mol. Sci. 2021, 22, 9124. [Google Scholar] [CrossRef] [PubMed]
- Berteotti, A.; Vacondio, F.; Lodola, A.; Bassi, M.; Silva, C.; Mor, M.; Cavalli, A. Predicting the Reactivity of Nitrile-Carrying Compounds with Cysteine: A Combined Computational and Experimental Study. ACS Med. Chem. Lett. 2014, 5, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.C.; Fadl, S.; Villanueva, A.J.; Rabeh, W.M. Catalytic Dyad Residues His41 and Cys145 Impact the Catalytic Activity and Overall Conformational Fold of the Main SARS-CoV-2 Protease 3-Chymotrypsin-Like Protease. Front. Chem. 2021, 9. [Google Scholar] [CrossRef]
- Owen, D.R.; Allerton, C.M.N.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An oral SARS-CoV-2 M pro inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. [Google Scholar] [CrossRef]
- Ridgway, H.; Chasapis, C.T.; Kelaidonis, K.; Ligielli, I.; Moore, G.J.; Gadanec, L.K.; Zulli, A.; Apostolopoulos, V.; Mavromoustakos, T.; Matsoukas, J.M. Understanding the Driving Forces That Trigger Mutations in SARS-CoV-2: Mutational Energetics and the Role of Arginine Blockers in COVID-19 Therapy. Viruses 2022, 14, 1029. [Google Scholar] [CrossRef]
- Matsoukas, J.M.; Gadanec, L.K.; Zulli, A.; Apostolopoulos, V.; Kelaidonis, K.; Ligielli, I.; Moschovou, K.; Georgiou, N.; Plotas, P.; Chasapis, C.T.; et al. Diminazene Aceturate Reduces Angiotensin II Constriction and Interacts with the Spike Protein of Severe Acute Respiratory Syndrome Coronavirus 2. Biomedicines 2022, 10, 1731. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Uterus | 1.4 | 0.8 * |
| Portal vein | 1.7 | 0.8 * |
| Aorta | 1.9 | 0.9 * |
| SUPERAGONIST | Sar-Arg-Val-Tyr-Ile-His-Pro-Phe |
| ANTAGONIST (SARMESIN) | Sar-Arg-Val-Tyr(Me)-Ile-His-Pro-Phe |
| (Surmountable) | |
| INVERSE AGONIST (SARILESIN) | Sar-Arg-Val-Tyr-Ile-His-Pro-Ile |
| (Insurmountable) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moore, G.J.; Ridgway, H.; Kelaidonis, K.; Chasapis, C.T.; Ligielli, I.; Mavromoustakos, T.; Bojarska, J.; Matsoukas, J.M. Actions of Novel Angiotensin Receptor Blocking Drugs, Bisartans, Relevant for COVID-19 Therapy: Biased Agonism at Angiotensin Receptors and the Beneficial Effects of Neprilysin in the Renin Angiotensin System. Molecules 2022, 27, 4854. https://doi.org/10.3390/molecules27154854
Moore GJ, Ridgway H, Kelaidonis K, Chasapis CT, Ligielli I, Mavromoustakos T, Bojarska J, Matsoukas JM. Actions of Novel Angiotensin Receptor Blocking Drugs, Bisartans, Relevant for COVID-19 Therapy: Biased Agonism at Angiotensin Receptors and the Beneficial Effects of Neprilysin in the Renin Angiotensin System. Molecules. 2022; 27(15):4854. https://doi.org/10.3390/molecules27154854
Chicago/Turabian StyleMoore, Graham J., Harry Ridgway, Konstantinos Kelaidonis, Christos T. Chasapis, Irene Ligielli, Thomas Mavromoustakos, Joanna Bojarska, and John M. Matsoukas. 2022. "Actions of Novel Angiotensin Receptor Blocking Drugs, Bisartans, Relevant for COVID-19 Therapy: Biased Agonism at Angiotensin Receptors and the Beneficial Effects of Neprilysin in the Renin Angiotensin System" Molecules 27, no. 15: 4854. https://doi.org/10.3390/molecules27154854