
Prediction of Low-Dose Aspirin-Induced Gastric Toxicity Using Nuclear Magnetic Resonance Spectroscopy-Based Pharmacometabolomics in Rats

 ,
,  , , , and
, , , and
Abstract
:1. Introduction
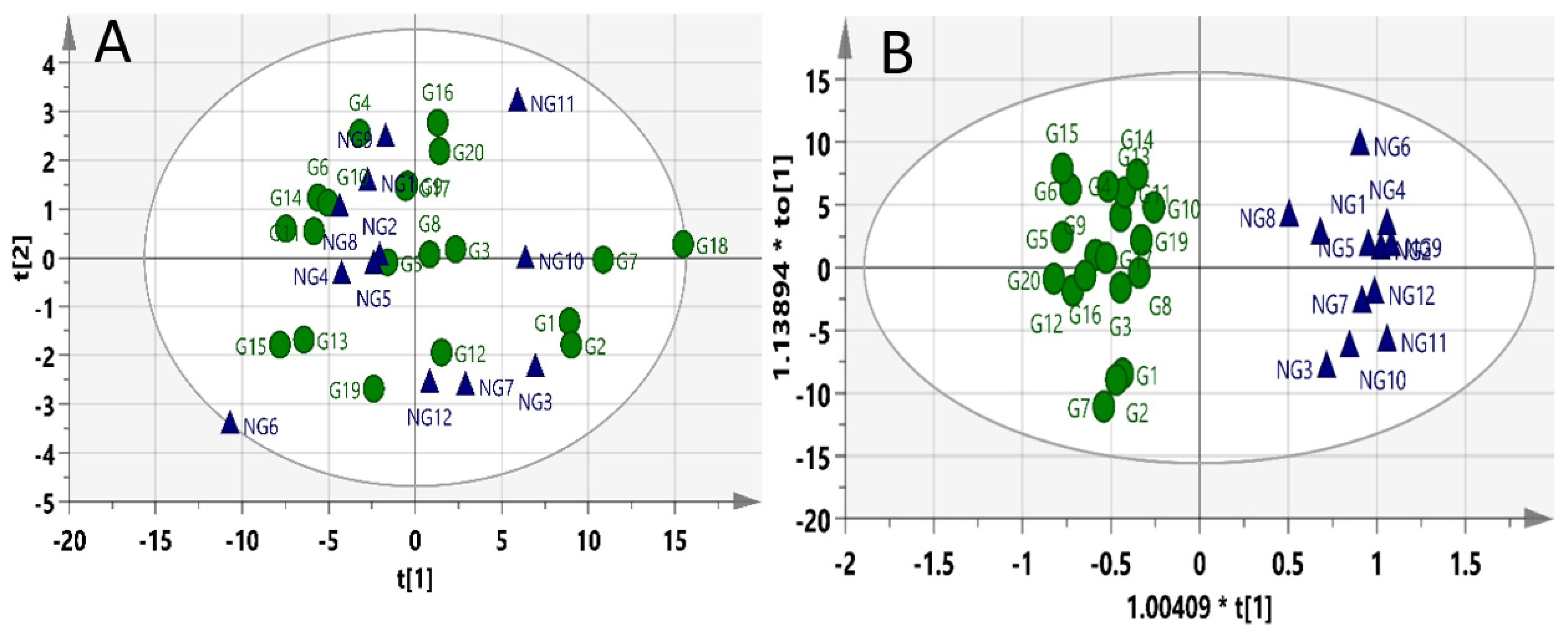
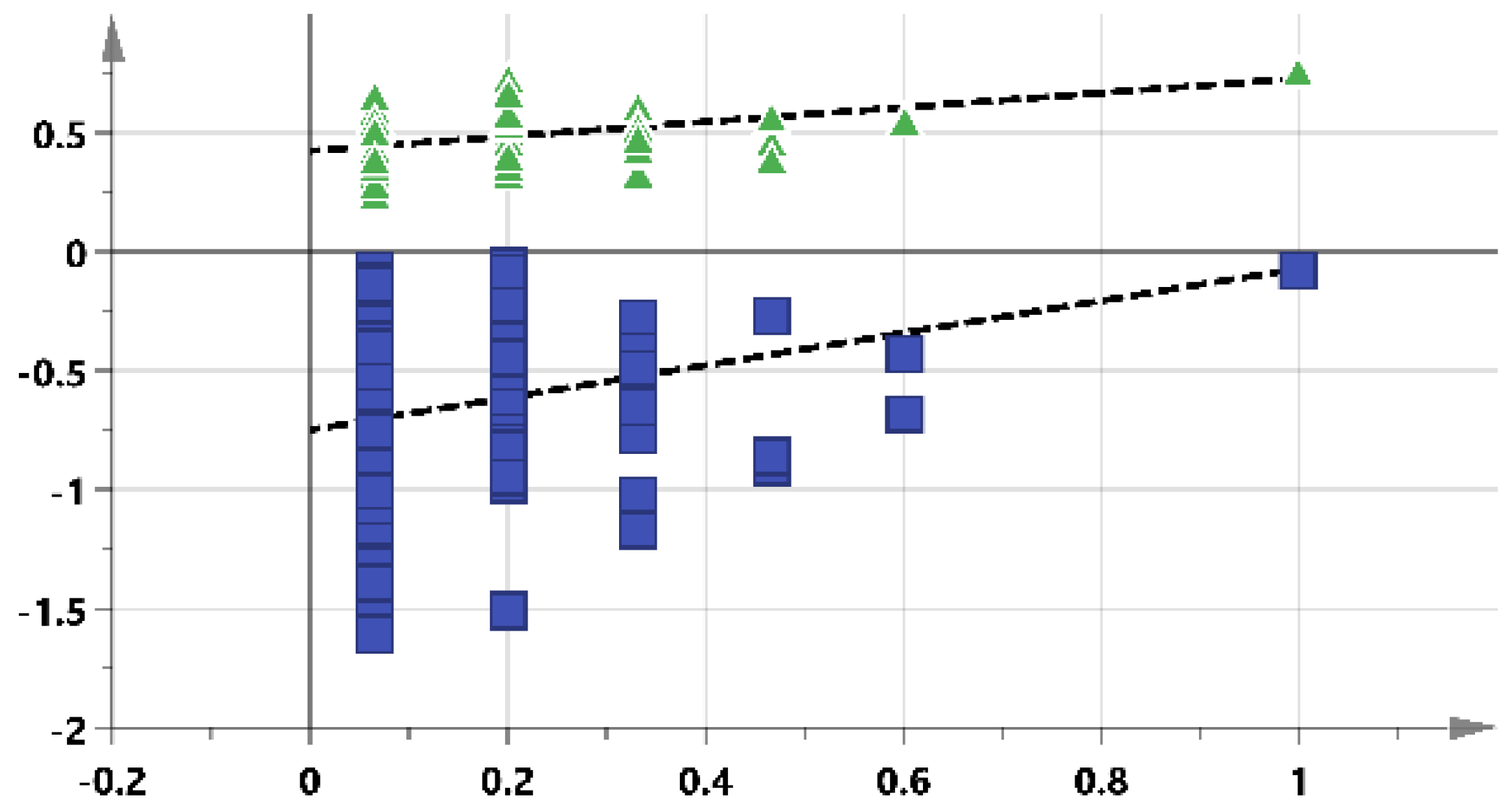
2. Results
2.1. Gastric Toxicity
2.2. Pre-Dose Profiling Models
2.3. Pre-Dosed Identification Models
2.4. Identification of Biomarkers to Predict LDA-Induced Gastric Toxicity
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Experimental Protocol
4.3. Sample Collection
4.4. Stomach Preparation
4.5. Pharmacometabolomics Analysis
4.6. Statistical Analysis
4.7. Metabolites Identification
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; et al. Heart Disease and Stroke Statistics—2018 Update: A Report From the American Heart Association. Circulation 2018, 137, e67–e492. [Google Scholar] [CrossRef] [PubMed]
- Levine, G.N.; Bates, E.R.; Bittl, J.A.; Brindis, R.G.; Fihn, S.D.; Fleisher, L.A.; Granger, C.B.; Lange, R.A.; Mack, M.J.; Mauri, L. 2016 ACC/AHA Guideline Focused Update on Duration of Dual Antiplatelet Therapy in Patients With Coronary Artery Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines: An Update of the 2011 ACCF/AHA/SCAI Guideline for Percutaneous Coronary Intervention, 2011 ACCF/AHA Guideline for Coronary Artery Bypass Graft Surgery, 2012 ACC/AHA/ACP/AATS/PCNA/SCAI/STS Guideline for the Diagnosis and Management of Patients With Stable Ischemic Heart Disease, 2013 ACCF/AHA Guideline for the Management of ST-Elevation Myocardial Infarction, 2014 AHA/ACC Guideline for the Management of Patients With Non–ST-Elevation Acute Coronary Syndromes, and 2014 ACC/AHA Guideline on Perioperative Cardiovascular Evaluation and Management of Patients Undergoing Noncardiac Surgery. Circulation 2016, 134, e123–e155. Available online: https://www.ahajournals.org/doi/10.1161/CIR.0000000000000404?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%20%200pubmed (accessed on 27 January 2022). [PubMed] [Green Version]
- Patrono, C. The multifaceted clinical readouts of platelet inhibition by low-dose aspirin. J. Am. Coll. Cardiol. 2015, 66, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Sostres, C.; Lanas, A. Gastrointestinal effects of aspirin. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Cryer, B.; Mahaffey, K.W. Gastrointestinal ulcers, role of aspirin, and clinical outcomes: Pathobiology, diagnosis, and treatment. J. Multidiscip. Healthc. 2014, 7, 137–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clayton, T.A.; Lindon, J.C.; Cloarec, O.; Antti, H.; Charuel, C.; Hanton, G.; Provost, J.-P.; Le Net, J.-L.; Baker, D.; Walley, R.J.; et al. Pharmaco-metabonomic phenotyping and personalized drug treatment. Nature 2006, 440, 1073–1077. [Google Scholar] [CrossRef] [PubMed]
- Szymańska, E.; Saccenti, E.; Smilde, A.K.; Westerhuis, J.A. Double-check: Validation of diagnostic statistics for PLS-DA models in metabolomics studies. Metabolomics 2012, 8, 3–16. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, L.; Kettaneh-Wold, N.; Trygg, J.; Wikström, C.; Wold, S. Multi-and Megavariate Data Analysis: Part I: Basic Principles and Applications; Umetrics Inc.: Goettingen, Germany, 2006. [Google Scholar]
- Hua-Lin, C.; Li, H.-D.; Yan, X.-Z.; Sun, B.; Zhang, Q.; Yan, M.; Zhang, W.-Y.; Jiang, P.; Zhu, R.-H.; Liu, Y.-P.; et al. Metabolomic Analysis of Biochemical Changes in the Plasma and Urine of First-Episode Neuroleptic-Naïve Schizophrenia Patients after Treatment with Risperidone. J. Proteome Res. 2012, 11, 4338–4350. [Google Scholar] [CrossRef]
- Triba, M.N.; Le Moyec, L.; Amathieu, R.; Goossens, C.; Bouchemal, N.; Nahon, P.; Rutledge, D.N.; Savarin, P. PLS/OPLS models in metabolomics: The impact of permutation of dataset rows on the K-fold cross-validation quality parameters. Mol. BioSyst. 2015, 11, 13–19. [Google Scholar] [CrossRef]
- Westerhuis, J.A.; Hoefsloot, H.C.; Smit, S.; Vis, D.J.; Smilde, A.K.; van Velzen, E.J.; van Duijnhoven, J.P.; van Dorsten, F.A. Assessment of PLSDA cross validation. Metabolomics 2008, 4, 81–89. [Google Scholar] [CrossRef] [Green Version]
- Baratloo, A.; Hosseini, M.; Negida, A.; El Ashal, G. Part 1: Simple definition and calculation of accuracy, sensitivity and specificity. Emergency 2015, 3, 48–49. [Google Scholar] [PubMed]
- Takeuchi, K.; Ohishi, M.; Ota, S.; Suzumura, K.; Naraoka, H.; Ohata, T.; Seki, J.; Miyamae, Y.; Honma, M.; Soga, T. Metabolic Profiling to Identify Potential Serum Biomarkers for Gastric Ulceration Induced by Nonsteroid Anti-Inflammatory Drugs. J. Proteome Res. 2013, 12, 1399–1407. [Google Scholar] [CrossRef] [PubMed]
- Um, S.Y.; Park, J.H.; Chung, M.W.; Choi, K.H.; Lee, H.J. 1H-Nuclear magnetic resonance-based metabolic profiling of nonsteroidal anti-inflammatory drug-induced adverse effects in rats. J. Pharm. Biomed. Anal. 2016, 129, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Ohishi, M.; Endo, K.; Suzumura, K.; Naraoka, H.; Ohata, T.; Seki, J.; Miyamae, Y.; Honma, M.; Soga, T. Metabolomic analysis of the effects of omeprazole and famotidine on aspirin-induced gastric injury. Metabolomics 2014, 10, 995–1004. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vazquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.A.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Um, S.Y.; Park, J.H.; Chung, M.W.; Kim, K.-B.; Kim, S.H.; Choi, K.H.; Lee, H.J. Nuclear magnetic resonance-based metabolomics for prediction of gastric damage induced by indomethacin in rats. Anal. Chim. Acta 2012, 722, 87–94. [Google Scholar] [CrossRef]
- Wu, N.; Yang, M.; Gaur, U.; Xu, H.; Yao, Y.; Li, D. Alpha-Ketoglutarate: Physiological Functions and Applications. Biomol. Ther. 2016, 24, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Silagy, C.A.; McNeil, J.J.; Donnan, G.A.; Tonkin, A.M.; Worsam, B.; Campion, K. Adverse effects of low-dose aspirin in a healthy elderly population. Clin. Pharmacol. Ther. 1993, 54, 84–89. [Google Scholar] [CrossRef]
- Li, J.-P.; Guo, J.-M.; Hua, Y.-Q.; Zhu, K.Y.; Tang, Y.-P.; Zhao, B.-C.; Jia, L.-F.; Zhao, J.; Tang, Z.-S.; Duan, J.-A. The mixture of Salvia miltiorrhiza–Carthamus tinctorius (Danhong injection) alleviates low-dose aspirin induced gastric mucosal damage in rats. Phytomedicine 2016, 23, 662–671. [Google Scholar] [CrossRef]
- Coen, M.; Goldfain-Blanc, F.; Rolland-Valognes, G.; Walther, B.; Robertson, D.G.; Holmes, E.; Lindon, J.C.; Nicholson, J.K. Pharmacometabonomic Investigation of Dynamic Metabolic Phenotypes Associated with Variability in Response to Galactosamine Hepatotoxicity. J. Proteome Res. 2012, 11, 2427–2440. [Google Scholar] [CrossRef] [PubMed]
- Ott, K.-H.; Aranibar, N. Nuclear magnetic resonance metabonomics: Methods for drug discovery and development. Metab. Methods Protoc. 2007, 358, 247–271. [Google Scholar]
- Szabo, S.; Trier, J.S.; Brown, A.; Schnoor, J.; Homan, H.D.; Bradford, J.C. A quantitative method for assessing the extent of experimental gastric erosions and ulcers. J. Pharmacol. Methods 1985, 13, 59–66. [Google Scholar] [CrossRef]
- Emwas, A.-H.M.; Salek, R.; Griffin, J.; Merzaban, J. NMR-based metabolomics in human disease diagnosis: Applications, limitations, and recommendations. Metabolomics 2013, 9, 1048–1072. [Google Scholar] [CrossRef]
- Beckonert, O.; Keun, H.C.; Ebbels, T.M.; Bundy, J.; Holmes, E.; Lindon, J.C.; Nicholson, J.K. Metabolic profiling, metabolomic and metabonomic procedures for NMR spectroscopy of urine, plasma, serum and tissue extracts. Nat. Protoc. 2007, 2, 2692–2703. [Google Scholar] [CrossRef]
- Tran, T.N.; Afanador, N.L.; Buydens, L.M.C.; Blanchet, L. Interpretation of variable importance in Partial Least Squares with Significance Multivariate Correlation (sMC). Chemom. Intell. Lab. Syst. 2014, 138, 153–160. [Google Scholar] [CrossRef]
- Musharraf, S.G.; Siddiqui, A.J.; Shamsi, T.; Choudhary, M.I.; Rahman, A.U. Serum Metabonomics of Acute Leukemia using Nuclear Magnetic Resonance Spectroscopy. Sci. Rep. 2016, 6, 30693. [Google Scholar] [CrossRef] [Green Version]
- Blasco, H.; Błaszczyński, J.; Billaut, J.C.; Nadal-Desbarats, L.; Pradat, P.F.; Devos, D.; Moreau, C.; Andres, C.R.; Emond, P.; Corcia, P.; et al. Comparative analysis of targeted metabolomics: Dominance-based rough set approach versus orthogonal partial least square-discriminant analysis. J. Biomed. Inform. 2015, 53, 291–299. [Google Scholar] [CrossRef]
- Caboni, P.; Liori, B.; Kumar, A.; Santoru, M.L.; Asthana, S.; Pieroni, E.; Fais, A.; Era, B.; Cacace, E.; Ruggiero, V.; et al. Metabolomics Analysis and Modeling Suggest a Lysophosphocholines-PAF Receptor Interaction in Fibromyalgia. PLoS ONE 2014, 9, e107626. [Google Scholar] [CrossRef] [Green Version]
- Izquierdo-García, J.L.; Villa, P.; Kyriazis, A.; del Puerto-Nevado, L.; Pérez-Rial, S.; Rodriguez, I.; Hernandez, N.; Ruiz-Cabello, J. Descriptive review of current NMR-based metabolomic data analysis packages. Prog. Nucl. Magn. Reson. Spectrosc. 2011, 59, 263–270. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Significant Metabolites (VIP > 1) | OPLSDA Score Plot Model | Multivariate Analysis | ||||
|---|---|---|---|---|---|---|
| Goodness-of-Fit (R2Y) | Goodness-of-Prediction (Q2Y) | Sensitivity (%) | Specificity (%) | Accuracy (%) | ||
| Profiling model | 72 | 0.947 | −0.185 | 100 | 100 | 100 |
| Identification model | 10 | 0.726 | 0.142 | 95 | 100 | 96.88 |
| Compound | Molecular Formula | Multiplicity | Chemical Shift |
|---|---|---|---|
| Citrate | C6H5O7 | Doublet | 2.677–2.718 |
| Doublet | 2.531–2.571 | ||
| Methylamine | CH3NH2 | Singlet | 2.680–2.700 |
| Trimethylamine N-Oxide | C3H9NO | Singlet | 3.277–3.284 |
| Hippurate | C9H9NO3 | Doublet | 3.965–3.981 |
| Triplet | 7.539–7.571 | ||
| Doublet | 7.825–7.850 | ||
| Triplet | 7.626–7.656 | ||
| Alpha-keto-glutarate | C5H6O5 | Triplet | 2.431–2.459 |
| Triplet | 2.996–3.024 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sha’aban, A.; Zainal, H.; Khalil, N.A.; Abd Aziz, F.; Ch’ng, E.S.; Teh, C.-H.; Mohammed, M.; Ibrahim, B. Prediction of Low-Dose Aspirin-Induced Gastric Toxicity Using Nuclear Magnetic Resonance Spectroscopy-Based Pharmacometabolomics in Rats. Molecules 2022, 27, 2126. https://doi.org/10.3390/molecules27072126
Sha’aban A, Zainal H, Khalil NA, Abd Aziz F, Ch’ng ES, Teh C-H, Mohammed M, Ibrahim B. Prediction of Low-Dose Aspirin-Induced Gastric Toxicity Using Nuclear Magnetic Resonance Spectroscopy-Based Pharmacometabolomics in Rats. Molecules. 2022; 27(7):2126. https://doi.org/10.3390/molecules27072126
Chicago/Turabian StyleSha’aban, Abubakar, Hadzliana Zainal, Nor Azlina Khalil, Fatimatuzzahra’ Abd Aziz, Ewe Seng Ch’ng, Chin-Hoe Teh, Mustapha Mohammed, and Baharudin Ibrahim. 2022. "Prediction of Low-Dose Aspirin-Induced Gastric Toxicity Using Nuclear Magnetic Resonance Spectroscopy-Based Pharmacometabolomics in Rats" Molecules 27, no. 7: 2126. https://doi.org/10.3390/molecules27072126