
Quantitative Proteome and Transcriptome Dynamics Analysis Reveals Iron Deficiency Response Networks and Signature in Neuronal Cells


Abstract
:1. Introduction
2. Results
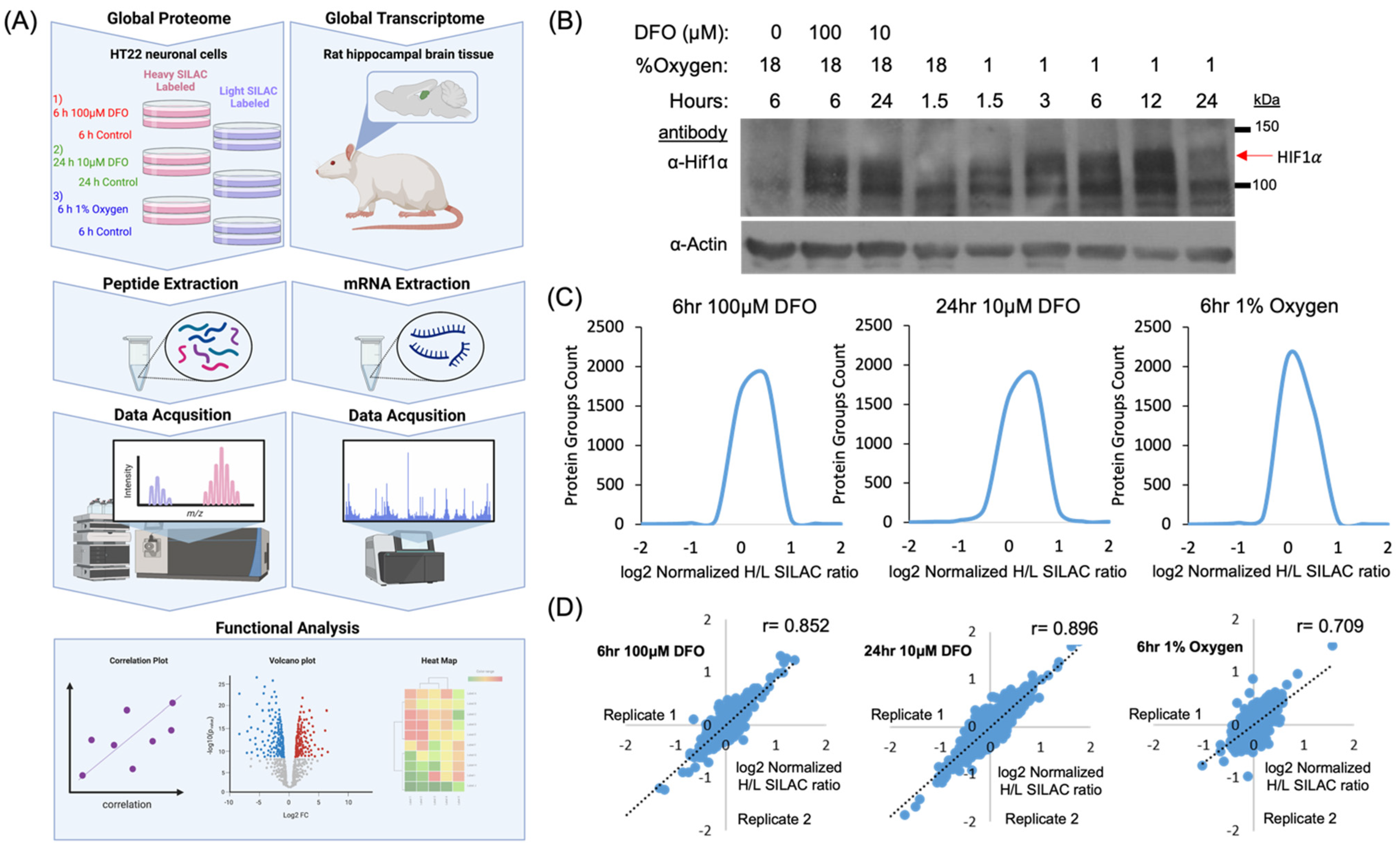
2.1. Experimental Strategy for the Quantification of the Iron and Oxygen Starvation-Dependent Proteome

2.2. Dynamics of Cellular Proteome in Response to Hypoxia and Iron Deficiency
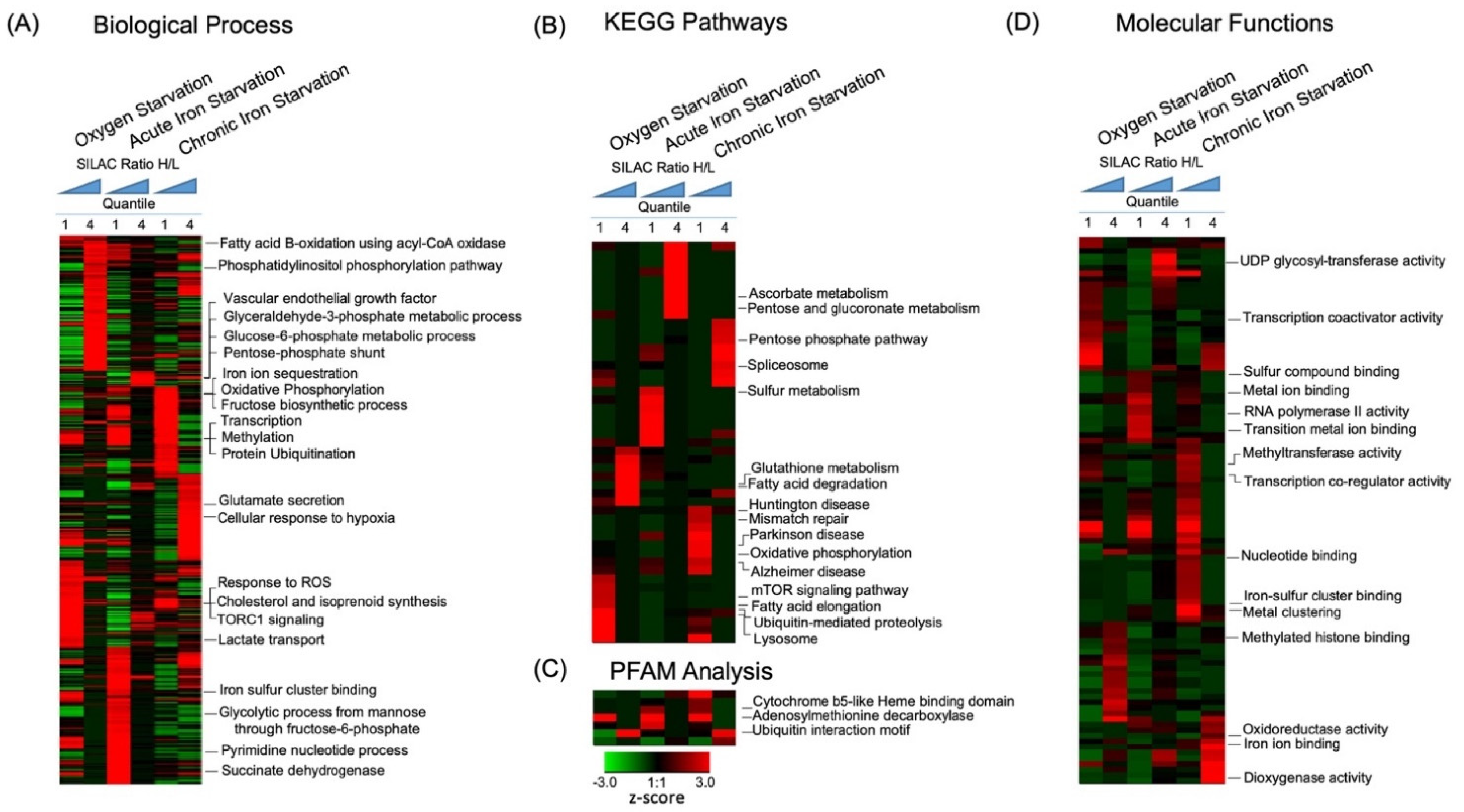
2.3. Dynamics of Biological Pathways Induced by Acute Hypoxia
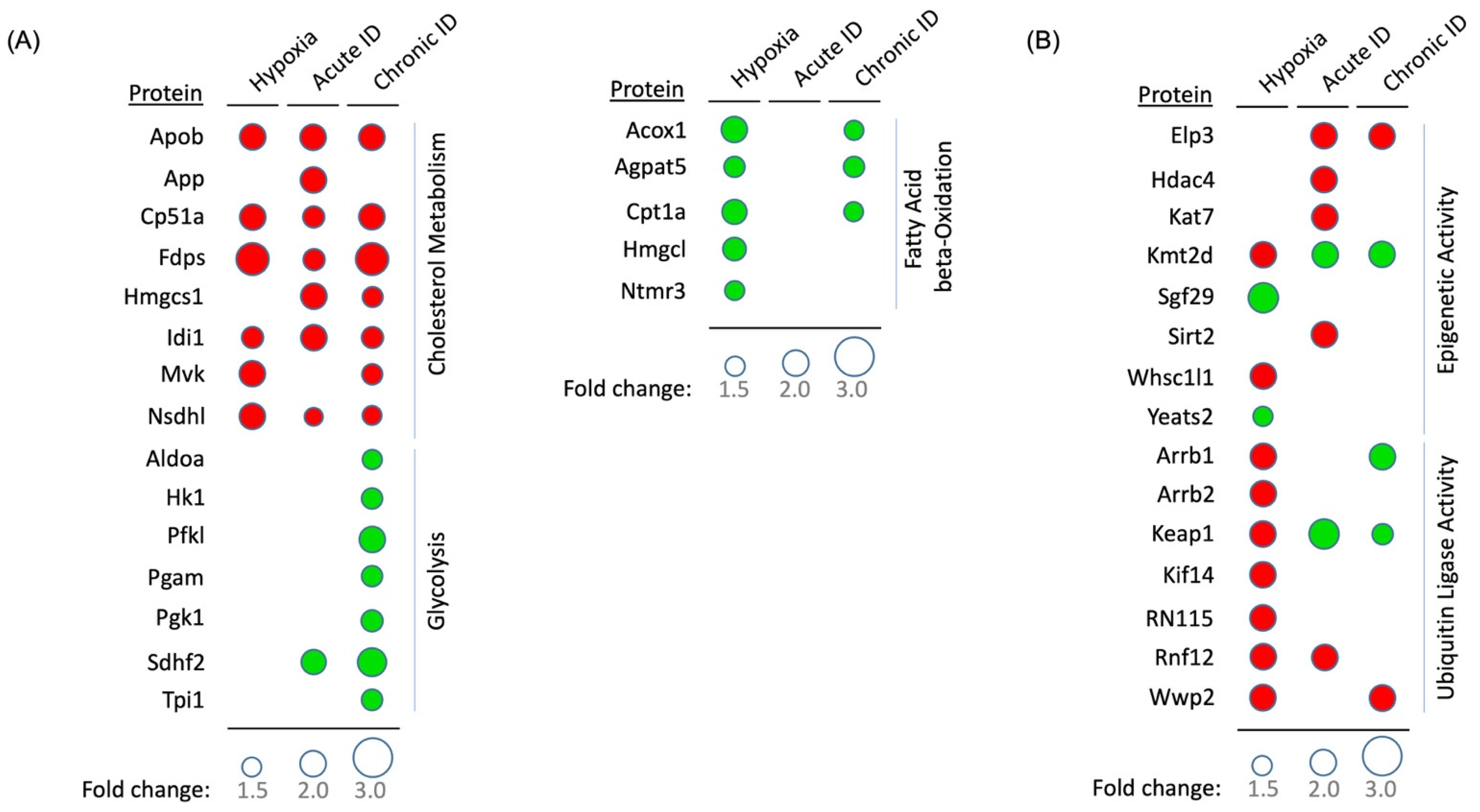
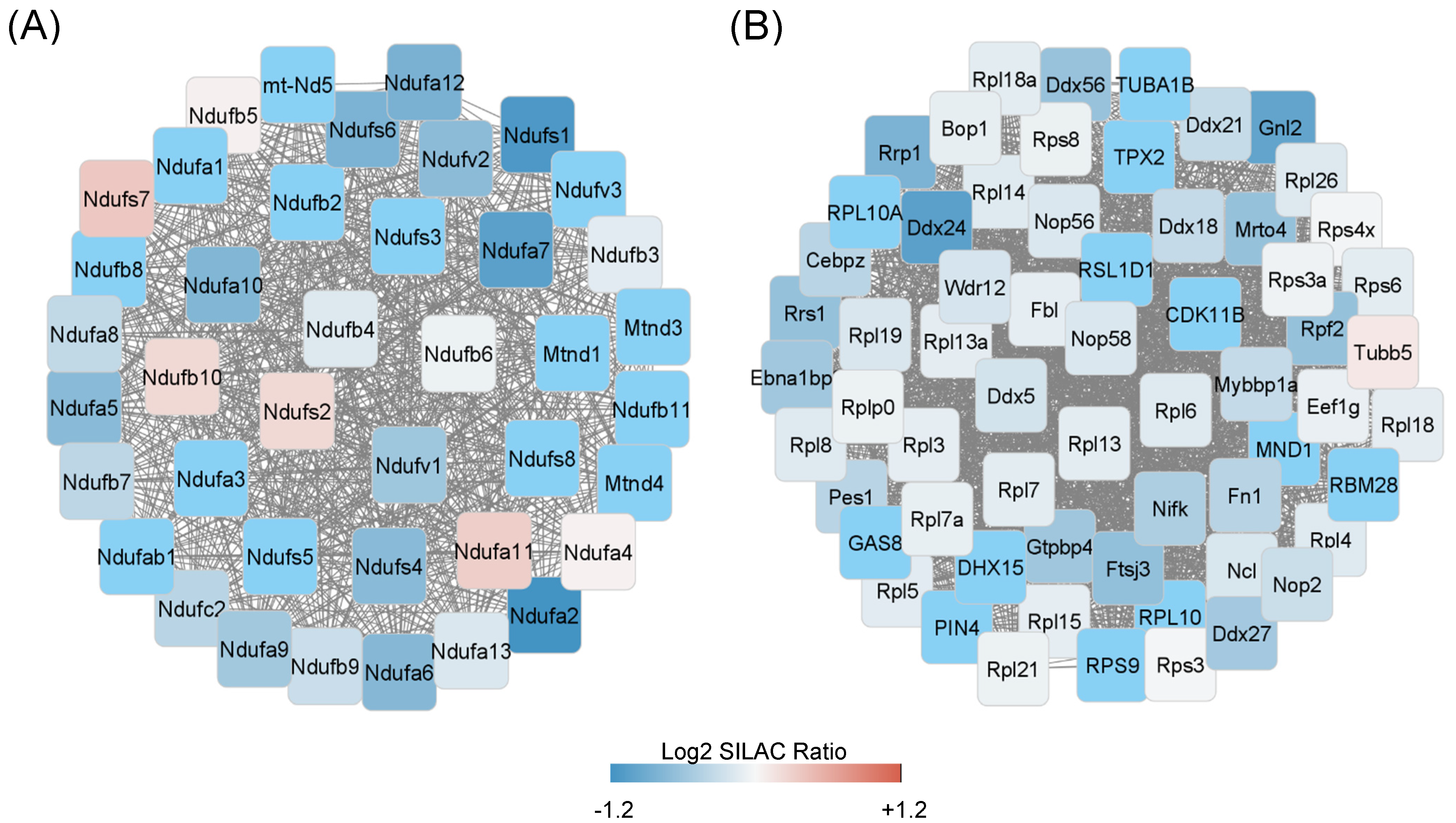
2.3.1. Metabolic Processes
2.3.2. Protein Synthesis, Folding, and Survival Pathways
2.3.3. Ubiquitination and Protein Degradation Pathways
2.3.4. Regulation of Epigenetic Pathways
2.3.5. Nutrient-Dependent Cellular Signaling
2.4. Identification of Specific Iron-Dependent Cellular Pathways in Neuronal Cells
2.4.1. Metal ion Binding Proteins and Processes
2.4.2. Effects of Iron Deficiency on Regulation of Cellular Metabolism
2.4.3. Iron-Dependent Regulation of Neuronal Signaling
2.5. Transcriptome Analysis of the Rat Hippocampus in Response to Iron Deficiency
2.6. Transcriptional Factor Activity Enrichment Analysis in Iron-Deficient Rat Brain Tissue and Neuronal Cell Line
3. Discussion
4. Materials and Methods
4.1. The P15 Rat Hippocampal Transcriptome
4.2. Quantitative Proteomics Analysis
4.2.1. Cell Culture
4.2.2. Cell Lysis
4.2.3. Peptide Preparation
4.2.4. Offline High pH Reverse-Phase HPLC Fractionation
4.2.5. LC-MS/MS Acquisition
4.2.6. Sequence Database Searching and Data Processing
4.3. Functional Annotation and Clustering
4.4. Transcription Factor Enrichment
4.5. Western Blotting
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Georgieff, M.K. The role of iron in neurodevelopment: Fetal iron deficiency and the developing hippocampus. Biochem. Soc. Trans. 2008, 36, 1267–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozoff, B.; Beard, J.; Connor, J.; Felt, B.; Georgieff, M.; Schallert, T. Long-lasting neural and behavioral effects of iron deficiency in infancy. Nutr. Rev. 2006, 64, S34–S43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barks, A.; Hall, A.M.; Tran, P.V.; Georgieff, M.K. Iron as a model nutrient for understanding the nutritional origins of neuropsychiatric disease. Pediatr. Res. 2019, 85, 176–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peyssonnaux, C.; Nizet, V.; Johnson, R.S. Role of the hypoxia inducible factors HIF in iron metabolism. Cell Cycle 2008, 7, 28–32. [Google Scholar] [CrossRef] [Green Version]
- E Bickler, P.; Donohoe, P.H. Adaptive responses of vertebrate neurons to hypoxia. J. Exp. Biol. 2002, 205, 3579–3586. [Google Scholar] [CrossRef]
- Wang, C.-Y.; Babitt, J.L. Liver iron sensing and body iron homeostasis. Blood 2019, 133, 18–29. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2013, 10, 9–17. [Google Scholar] [CrossRef]
- Busl, K.M.; Greer, D.M. Hypoxic-ischemic brain injury: Pathophysiology, neuropathology and mechanisms. NeuroRehabilitation 2010, 26, 5–13. [Google Scholar] [CrossRef] [Green Version]
- Semenza, L.G.; Wang, G.L. A Nuclear Factor Induced by Hypoxia Via Denovo Protein-Synthesis Binds to the Human Erythropoietin Gene Enhancer at a Site Required for Transcriptional Activation. Mol. Cell. Biol. 1992, 12, 5447–5454. [Google Scholar]
- Wang, G.L.; Jiang, B.-H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Hypoxia-inducible factor 1: Oxygen homeostasis and disease pathophysiology. Trends Mol. Med. 2001, 7, 345–350. [Google Scholar] [CrossRef]
- Schofield, C.; Ratcliffe, P. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004, 5, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.S.; Thirunavukkarasu, M.; Rishi, M.T.; Sanchez, J.A.; Maulik, N.; Maulik, G. HIF–prolyl hydroxylases and cardiovascular diseases. Toxicol. Mech. Methods 2012, 22, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Ratcliffe, P. Oxygen sensing and hypoxia signalling pathways in animals: The implications of physiology for cancer. J. Physiol. 2013, 591, 2027–2042. [Google Scholar] [CrossRef] [PubMed]
- Shao, G.; Wang, Y.; Guan, S.; Burlingame, A.L.; Lu, F.; Knox, R.; Ferriero, D.M.; Jiang, X. Proteomic Analysis of Mouse Cortex Postsynaptic Density following Neonatal Brain Hypoxia-Ischemia. Dev. Neurosci. 2017, 39, 66–81. [Google Scholar] [CrossRef] [Green Version]
- Hernández, R.; Blanco, S.; Peragón, J.; Pedrosa, J.; Peinado, M.A. Hypobaric Hypoxia and Reoxygenation Induce Proteomic Profile Changes in the Rat Brain Cortex. NeuroMolecular Med. 2013, 15, 82–94. [Google Scholar] [CrossRef]
- Zhou, Y.; Bhatia, I.; Cai, Z.; He, Q.-Y.; Cheung, P.-T.; Chiu, J.-F. Proteomic Analysis of Neonatal Mouse Brain: Evidence for Hypoxia- and Ischemia-Induced Dephosphorylation of Collapsin Response Mediator Proteins. J. Proteome Res. 2008, 7, 2507–2515. [Google Scholar] [CrossRef]
- Bastian, T.W.; Von Hohenberg, W.C.; Mickelson, D.J.; Lanier, L.M.; Georgieff, M.K. Iron Deficiency Impairs Developing Hippocampal Neuron Gene Expression, Energy Metabolism, and Dendrite Complexity. Dev. Neurosci. 2016, 38, 264–276. [Google Scholar] [CrossRef] [Green Version]
- Bastian, T.W.; Rao, R.; Tran, P.V.; Georgieff, M.K. The Effects of Early-Life Iron Deficiency on Brain Energy Metabolism. Neurosci. Insights 2020, 15. [Google Scholar] [CrossRef]
- Guengerich, F.P. Introduction: Metals in Biology: α-Ketoglutarate/Iron-Dependent Dioxygenases. J. Biol. Chem. 2015, 290, 20700–20701. [Google Scholar] [CrossRef] [Green Version]
- Erber, L.N.; Luo, A.; Gong, Y.; Beeson, M.; Tu, M.; Tran, P.; Chen, Y. Iron Deficiency Reprograms Phosphorylation Signaling and Reduces O-GlcNAc Pathways in Neuronal Cells. Nutrients 2021, 13, 179. [Google Scholar] [CrossRef] [PubMed]
- Kezele, T.G.; Ćurko-Cofek, B. Age-Related Changes and Sex-Related Differences in Brain Iron Metabolism. Nutrients 2020, 12, 2601. [Google Scholar] [CrossRef]
- Kautz, L.; Meynard, D.; Monnier, A.; Darnaud, V.; Bouvet, R.; Wang, R.H.; Roth, M.P. Iron regulates phosphorylation of Smad1/5/8 and gene expression of Bmp6, Smad7, Id1, and Atoh8 in the mouse liver. Blood 2008, 112, 1503–1509. [Google Scholar] [CrossRef]
- Lee, S.M.; Lee, S.B.; Prywes, R.; Vulpe, C.D. Iron deficiency upregulates Egr1 expression. Genes Nutr. 2015, 10, 468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schachtschneider, K.M.; Liu, Y.; Rund, L.A.; Madsen, O.; Johnson, R.W.; Groenen, M.A.M.; Schook, L.B. Impact of neonatal iron deficiency on hippocampal DNA methylation and gene transcription in a porcine biomedical model of cognitive development. BMC Genom. 2016, 17, 856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saletta, F.; Rahmanto, Y.S.; Noulsri, E.; Richardson, D. Iron Chelator-Mediated Alterations in Gene Expression: Identification of Novel Iron-Regulated Molecules That Are Molecular Targets of Hypoxia-Inducible Factor-1α and p53. Mol. Pharmacol. 2010, 77, 443–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, S.-E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable Isotope Labeling by Amino Acids in Cell Culture, SILAC, as a Simple and Accurate Approach to Expression Proteomics. Mol. Cell. Proteom. 2002, 1, 376–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagara, Y.; Schubert, D. The Activation of Metabotropic Glutamate Receptors Protects Nerve Cells from Oxidative Stress. J. Neurosci. 1998, 18, 6662–6671. [Google Scholar] [CrossRef]
- Porter, J.; Rafique, R.; Srichairatanakool, S.; Davis, B.A.; Shah, F.T.; Hair, T.; Evans, P. Recent Insights into Interactions of Deferoxamine with Cellular and Plasma Iron Pools: Implications for Clinical Use. Ann. N. Y. Acad. Sci. 2005, 1054, 155–168. [Google Scholar] [CrossRef]
- Zaman, K.; Ryu, H.; Hall, D.; O’Donovan, K.; Lin, K.I.; Miller, M.P.; Marquis, J.C.; Baraban, J.M.; Semenza, G.L.; Ratan, R.R. Protection from oxidative stress-induced apoptosis in cortical neuronal cultures by iron chelators is associated with enhanced DNA binding of hypoxia-inducible factor-1 and ATF-1/CREB and increased expression of glycolytic enzymes, p21(waf1/cip1), and erythropoietin. J. Neurosci. 1999, 19, 9821–9830. [Google Scholar]
- Bastian, T.W.; Von Hohenberg, W.C.; Georgieff, M.K.; Lanier, L.M. Chronic Energy Depletion due to Iron Deficiency Impairs Dendritic Mitochondrial Motility during Hippocampal Neuron Development. J. Neurosci. 2019, 39, 802–813. [Google Scholar] [CrossRef] [Green Version]
- Siriwardana, G.; Seligman, P.A. Iron depletion results in Src kinase inhibition with associated cell cycle arrest in neuroblastoma cells. Physiol. Rep. 2015, 3, e12341. [Google Scholar] [CrossRef] [PubMed]
- Mu, S.; Guo, S.; Wang, X.; Zhan, Y.; Li, Y.; Jiang, Y.; Zhang, R.; Zhang, B. Effects of deferoxamine on the osteogenic differentiation of human periodontal ligament cells. Mol. Med. Rep. 2017, 16, 9579–9586. [Google Scholar] [CrossRef] [PubMed]
- Yarosz, E.L.; Ye, C.; Kumar, A.; Black, C.; Choi, E.-K.; Seo, Y.-A.; Chang, C.-H. Cutting Edge: Activation-Induced Iron Flux Controls CD4 T Cell Proliferation by Promoting Proper IL-2R Signaling and Mitochondrial Function. J. Immunol. 2020, 204, 1708–1713. [Google Scholar] [CrossRef]
- Yang, F.; Shen, Y.; Camp, D.G.; Smith, R.D. High-pH reversed-phase chromatography with fraction concatenation for 2D proteomic analysis. Expert Rev. Proteom. 2012, 9, 129–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlson, E.S.; Stead, J.; Neal, C.R.; Petryk, A.; Georgieff, M.K. Perinatal iron deficiency results in altered developmental expression of genes mediating energy metabolism and neuronal morphogenesis in hippocampus. Hippocampus 2007, 17, 679–691. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 27, 29–34. [Google Scholar] [CrossRef] [Green Version]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, K.; Qi, J.; Ronai, Z. The Ubiquitin Ligase Siah2 and the Hypoxia Response. Mol. Cancer Res. 2009, 7, 443–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koumenis, C. ER Stress, Hypoxia Tolerance and Tumor Progression. Curr. Mol. Med. 2006, 6, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Sabatini, D.M. Twenty-five years of mTOR: Uncovering the link from nutrients to growth. Proc. Natl. Acad. Sci. USA 2017, 114, 11818–11825. [Google Scholar] [CrossRef] [Green Version]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004, 18, 2893–2904. [Google Scholar] [CrossRef] [Green Version]
- Regazzetti, C.; Peraldi, P.; Grémeaux, T.; Najem-Lendom, R.; Ben-Sahra, I.; Cormont, M.; Bost, F.; Le Marchand-Brustel, Y.; Tanti, J.-F.; Giorgetti-Peraldi, S. Hypoxia Decreases Insulin Signaling Pathways in Adipocytes. Diabetes 2009, 58, 95–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.L.; Semenza, G.L. Desferrioxamine induces erythropoietin gene expression and hypoxia-inducible factor 1 DNA-binding activity: Implications for models of hypoxia signal transduction. Blood 1993, 82, 3610–3615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wingert, R.A.; Galloway, J.L.; Barut, B.; Foott, H.; Fraenkel, P.; Axe, J.L.; Weber, G.J.; Dooley, K.; Davidson, A.J.; Schmidt, B.; et al. Deficiency of glutaredoxin 5 reveals Fe–S clusters are required for vertebrate haem synthesis. Nature 2005, 436, 1035–1039. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Vasaikar, S.; Shi, Z.; Greer, M.; Zhang, B. WebGestalt 2017: A more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 2017, 45, W130–W137. [Google Scholar] [CrossRef] [PubMed]
- Torti, F.M.; Torti, S.V. Regulation of ferritin genes and protein. Blood 2002, 99, 3505–3516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chepelev, N.L.; Willmore, W.G. Regulation of iron pathways in response to hypoxia. Free Radic. Biol. Med. 2011, 50, 645–666. [Google Scholar] [CrossRef]
- Mimata, H.; Nomura, T.; Sato, F.; Yamasaki, M. Metallothionein is up-regulated under hypoxia and promotes the survival of human prostate cancer cells. Oncol. Rep. 2007, 18, 1145–1153. [Google Scholar] [CrossRef] [Green Version]
- Au, H.C.; Scheffler, I.E. Promoter analysis of the human succinate dehydrogenase iron-protein gene--both nuclear respiratory factors NRF-1 and NRF-2 are required. JBIC J. Biol. Inorg. Chem. 1998, 251, 164–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pon, J.R.; Marra, M.A. MEF2 transcription factors: Developmental regulators and emerging cancer genes. Oncotarget 2016, 7, 2297–2312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iolascon, A.; De Falco, L. Mutations in the Gene Encoding DMT1: Clinical Presentation and Treatment. Semin. Hematol. 2009, 46, 358–370. [Google Scholar] [CrossRef]
- Tran, P.V.; Kennedy, B.C.; Pisansky, M.T.; Won, K.-J.; Gewirtz, J.C.; Simmons, R.; Georgieff, M.K. Prenatal Choline Supplementation Diminishes Early-Life Iron Deficiency–Induced Reprogramming of Molecular Networks Associated with Behavioral Abnormalities in the Adult Rat Hippocampus123. J. Nutr. 2016, 146, 484–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lien, Y.-C.; E Condon, D.; Georgieff, M.K.; A Simmons, R.; Tran, P.V. Dysregulation of Neuronal Genes by Fetal-Neonatal Iron Deficiency Anemia Is Associated with Altered DNA Methylation in the Rat Hippocampus. Nutrients 2019, 11, 1191. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Fan, Y.; Wu, P.; Huang, Z.; Li, X.; Zhao, L.; Ji, Y.; Zhu, M. Parkinson’s Disease Dementia: Synergistic Effects of Alpha-Synuclein, Tau, Beta-Amyloid, and Iron. Front. Aging Neurosci 2021, 13, 743754. [Google Scholar] [CrossRef]
- Choi, W.-S.; Palmiter, R.D.; Xia, Z. Loss of mitochondrial complex I activity potentiates dopamine neuron death induced by microtubule dysfunction in a Parkinson’s disease model. J. Cell Biol. 2011, 192, 873–882. [Google Scholar] [CrossRef]
- Cabello-Rivera, D.; Sarmiento-Soto, H.; López-Barneo, J.; Muñoz-Cabello, A.M. Mitochondrial Complex I Function Is Essential for Neural Stem/Progenitor Cells Proliferation and Differentiation. Front. Neurosci. 2019, 13, 664. [Google Scholar] [CrossRef] [Green Version]
- Estrella, N.L.; Desjardins, C.A.; Nocco, S.E.; Clark, A.L.; Maksimenko, Y.; Naya, F.J. MEF2 Transcription Factors Regulate Distinct Gene Programs in Mammalian Skeletal Muscle Differentiation. J. Biol. Chem. 2015, 290, 1256–1268. [Google Scholar] [CrossRef] [Green Version]
- Flavell, S.W.; Cowan, C.W.; Kim, T.-K.; Greer, P.L.; Lin, Y.; Paradis, S.; Griffith, E.C.; Hu, L.S.; Chen, C.; Greenberg, M.E. Activity-Dependent Regulation of MEF2 Transcription Factors Suppresses Excitatory Synapse Number. Science 2006, 311, 1008–1012. [Google Scholar] [CrossRef] [Green Version]
- Du, M.; Perry, R.L.S.; Nowacki, N.B.; Gordon, J.W.; Salma, J.; Zhao, J.; Aziz, A.; Chan, J.; Siu, K.W.M.; McDermott, J.C. Protein Kinase A Represses Skeletal Myogenesis by Targeting Myocyte Enhancer Factor 2D. Mol. Cell. Biol. 2008, 28, 2952–2970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A Peptide Search Engine Integrated into the MaxQuant Environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef]
- Falcon, S.; Gentleman, R. Using GOstats to test gene lists for GO term association. Bioinformatics 2007, 23, 257–258. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CELLULAR FUNCTIONS | HYPOXIA | ACUTE ID | CHRONIC ID |
|---|---|---|---|
| TRANSCRIPTION | Decrease | Decrease | Decrease |
| PROTEIN SYNTHESIS | Decrease | Decrease | Decrease |
| MTORC1 SIGNALING | Decrease | Decrease | |
| PROTEIN DEGRADATION | |||
| UBIQUITIN MEDIATED | Decrease | ||
| ENDOPLASMIC RETICULUM-ASSOCIATED | Increase | ||
| AUTOPHAGY-MEDIATED | Decrease | Decrease | |
| EPIGENETIC PROCESSES | |||
| HISTONE DEACETYLASE | Decrease | ||
| HISTONE ACETYLTRANSFERASE | Increase | Decrease | |
| HISTONE METHYLATION | Decrease | Increase | Increase |
| IRON ION REGULATION | |||
| IRON ION HOMEOSTASIS | Decrease | Decrease | |
| CELLULAR METABOLISM | |||
| CHOLESTEROL BIOSYNTHESIS | Decrease | Decrease | Decrease |
| FATTY ACID METABOLISM | Decrease | ||
| BETA OXIDATION | Increase | ||
| GLYCOLYSIS | Increase | Increase | |
| ELECTRON-TRANSPORT CHAIN | Decrease | ||
| PROTEIN KINASE B SIGNALING | Decrease | ||
| VEGF SIGNALING | Increase | Increase | |
| GLUTAMATE SECRETION | Increase |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Erber, L.; Liu, S.; Gong, Y.; Tran, P.; Chen, Y. Quantitative Proteome and Transcriptome Dynamics Analysis Reveals Iron Deficiency Response Networks and Signature in Neuronal Cells. Molecules 2022, 27, 484. https://doi.org/10.3390/molecules27020484
Erber L, Liu S, Gong Y, Tran P, Chen Y. Quantitative Proteome and Transcriptome Dynamics Analysis Reveals Iron Deficiency Response Networks and Signature in Neuronal Cells. Molecules. 2022; 27(2):484. https://doi.org/10.3390/molecules27020484
Chicago/Turabian StyleErber, Luke, Shirelle Liu, Yao Gong, Phu Tran, and Yue Chen. 2022. "Quantitative Proteome and Transcriptome Dynamics Analysis Reveals Iron Deficiency Response Networks and Signature in Neuronal Cells" Molecules 27, no. 2: 484. https://doi.org/10.3390/molecules27020484