
Synthesis, Identification, Computer-Aided Docking Studies, and ADMET Prediction of Novel Benzimidazo-1,2,3-triazole Based Molecules as Potential Antimicrobial Agents

 ,
,  , and
, and
Abstract
:1. Introduction
2. Results and Discussion
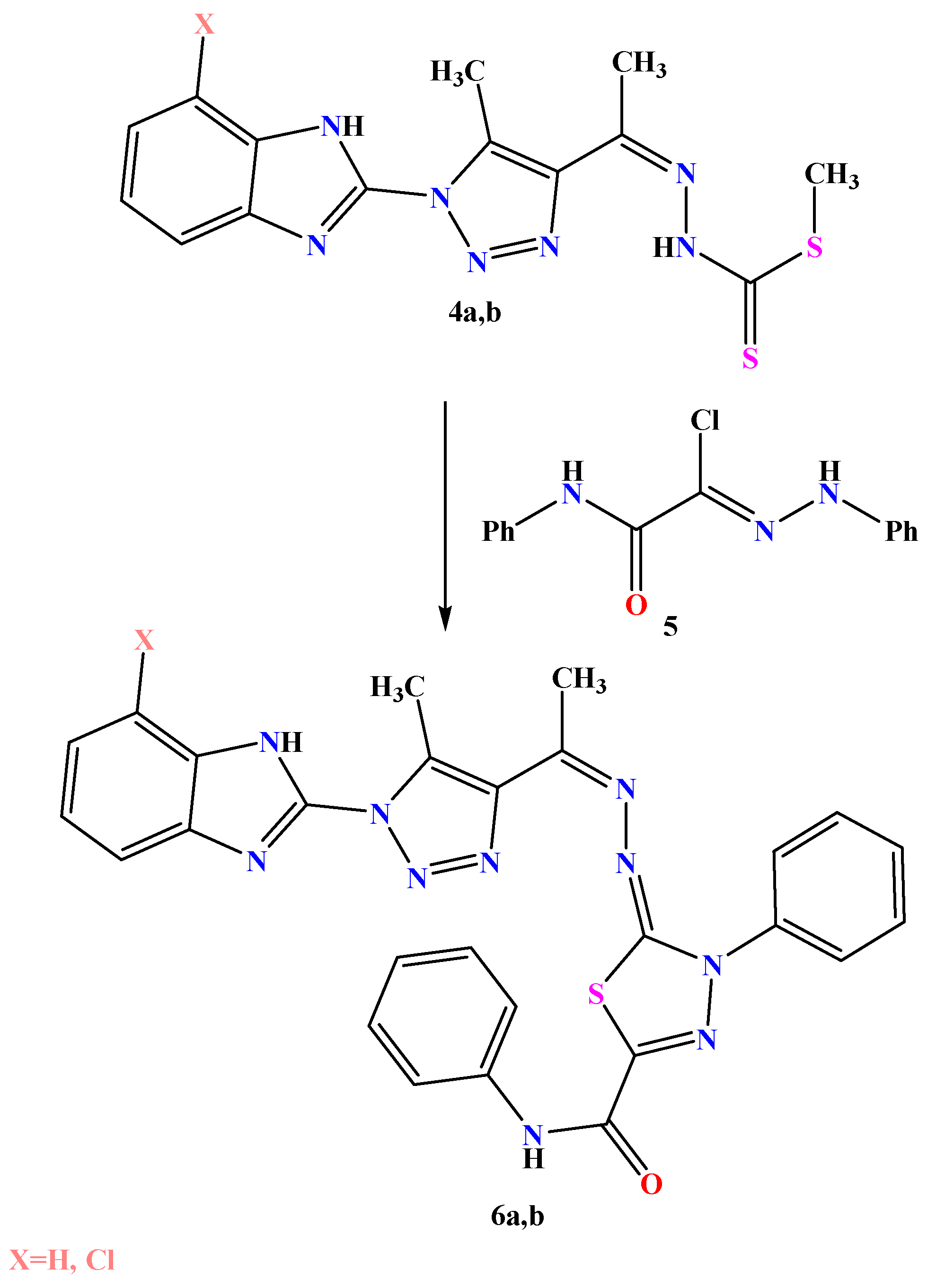
2.1. Chemistry
2.2. Computational Approach
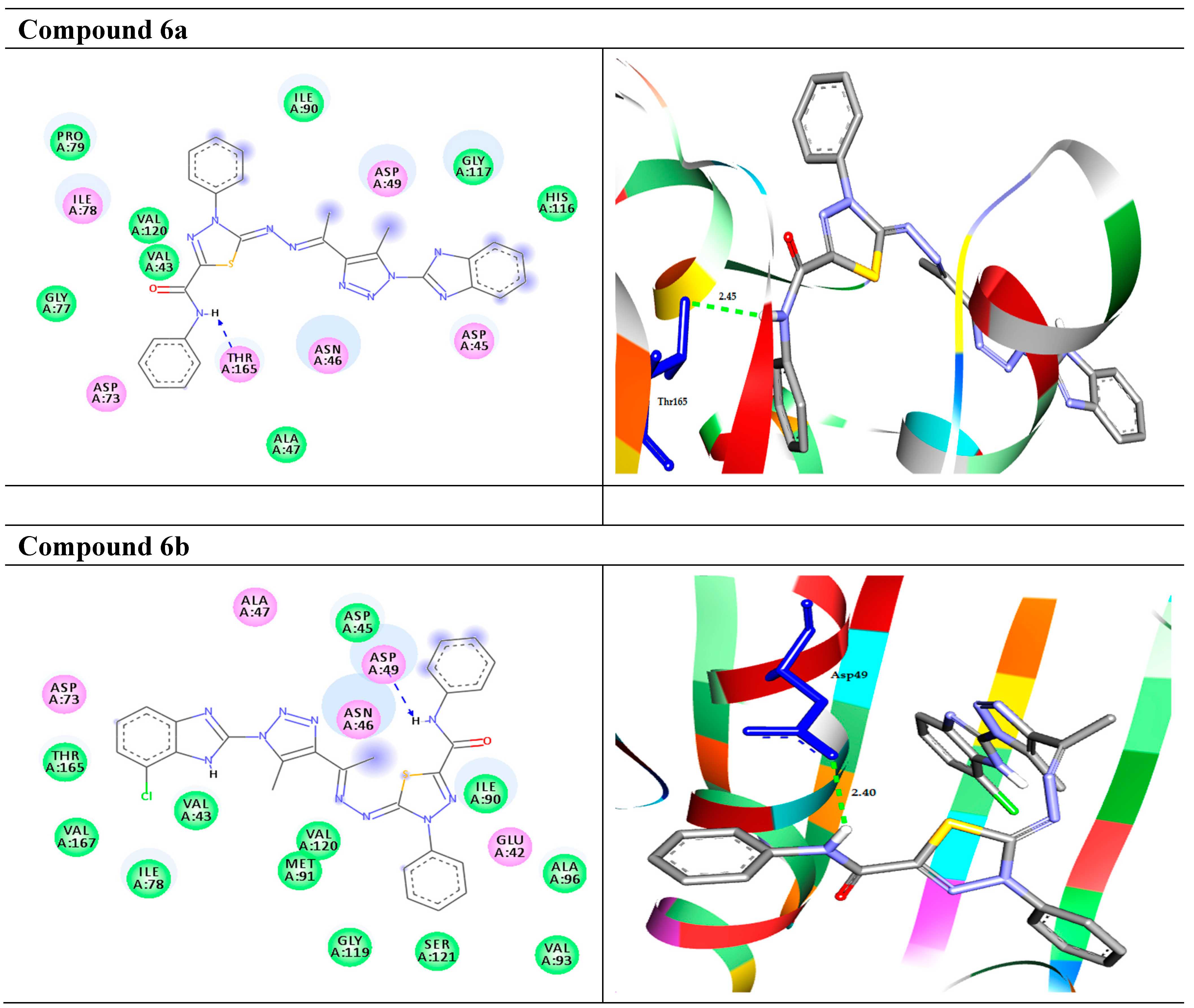
2.2.1. Molecular Docking Study
2.2.2. Predictive ADMAT and Drug-Likeness of the Compounds
2.3. Antimicrobial Activity
3. Experimental
3.1. Chemistry
3.1.1. Experimental Instrumentation
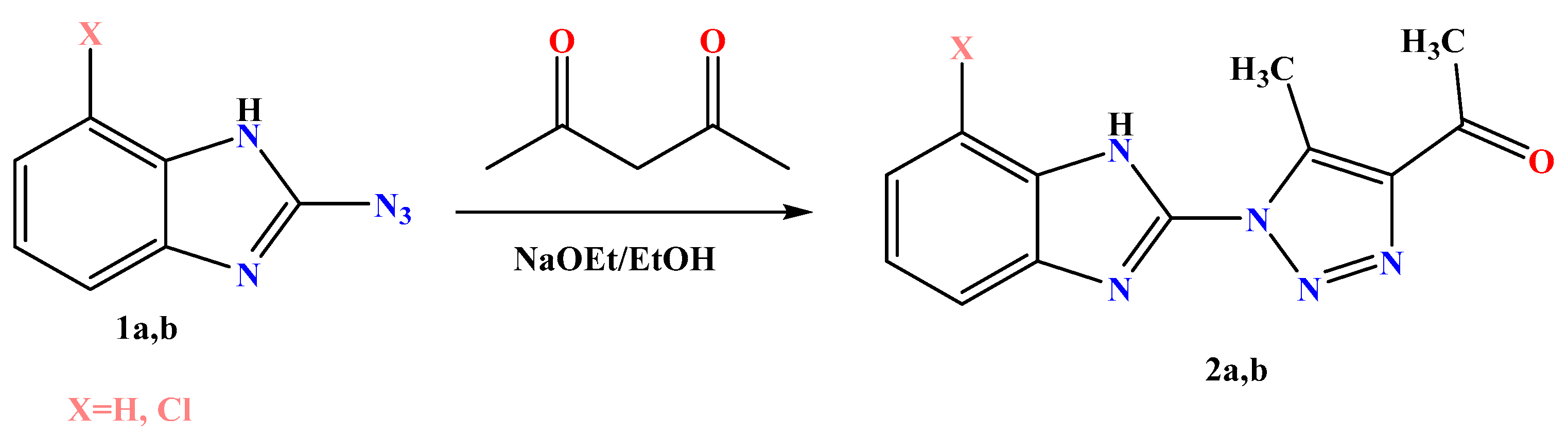
3.1.2. General Procedures for Synthesis of Derivatives 2a,b
- 1-(1-(1H-benzo[d]imidazol-2-yl)-5-methyl-1H-1,2,3-triazol-4-yl)ethan-1-one derivatives 2a
- 1-(1-(7-chloro-1H-benzo[d]imidazol-2-yl)-5-methyl-1H-1,2,3-triazol-4-yl) ethan-1-one 2b
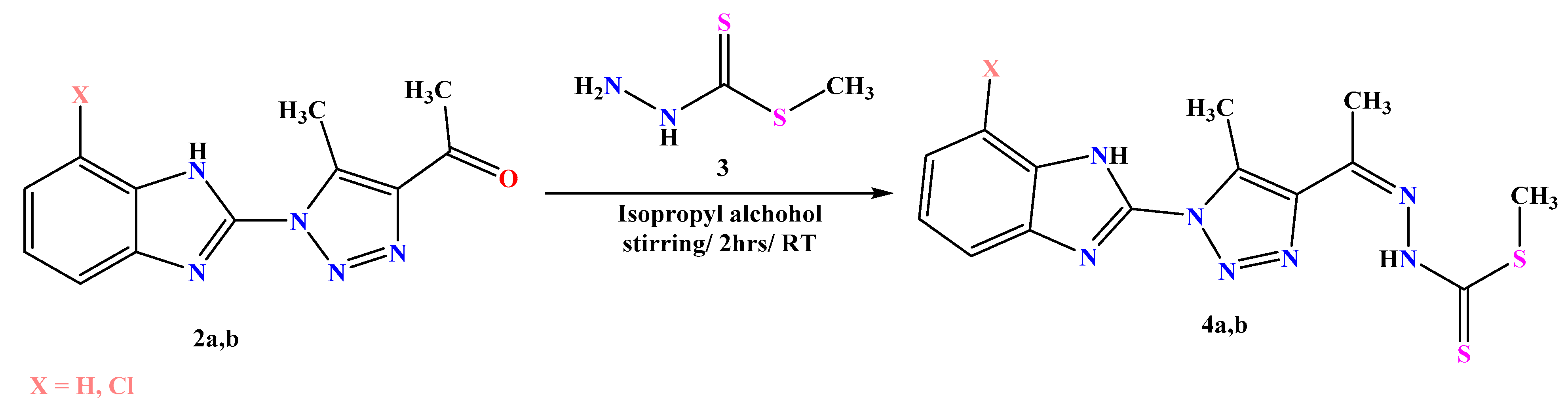
3.1.3. General Procedures for Synthesis of 4a,b
- Methyl-2-(1-(1-(1H-benzo[d]imidazol-2-yl)-5-methyl-1H-1,2,3-triazol-4-yl) ethylidene) hydrazine-1-carbodithioate 4a
- Methyl-2-(1-(1-(7-chloro-1H-benzo[d]imidazol-2-yl)-5-methyl-1H-1,2,3-triazol-4-yl) ethylidene) hydrazine-1-carbodithioate 4b
3.1.4. General Procedures for Synthesis of Compounds 6a,b
- 5-(1-(1-(1-H-benzo[d]imidazol-2-yl)-5-methyl-1H-1,2,3-triazol-4-yl)ethylidene) hydrazono)-N,4-diphenyl-4,5-dihydro-1,3,4-thiadiazole-2-carboxamide 6a
- 5-((1-(1-(7-chloro-1H-benzo[d]imidazol-2-yl)-5-methyl-1H-1,2,3-triazol-4-yl) ethylidene)hydrazono)-N,4-diphenyl-4,5-dihydro-1,3,4-thiadiazole-2-carboxamide 6b
3.2. Docking Study
3.3. Antimicrobial Activity
3.3.1. Antimicrobial Assay
3.3.2. Agar Diffusion Medium
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Hassan, M.A.; Seleem, M.A.; Younes, A.M.M.; Taha, M.M.; Abdel-Monsef, A.-B.H. Synthesis and spectral characterization of some heterocyclic nitrogen compounds. Eur. J. Chem. 2013, 4, 121–123. [Google Scholar] [CrossRef] [Green Version]
- El-Sheshtawy, H.S.; Abdelmonsef, A.H.; Abboudy, S.M.; Younes, A.M.M.; Taha, M.M.; Hassan, M.A. Synthesis, Structural, and Theoretical Studies of Quinazoline-2,4-dione Derivatives. Polycycl. Aromat. Compd. 2017, 39, 1–8. [Google Scholar] [CrossRef]
- Hassan, M.A.; Mohamed, A.; Younes, M.; Taha, M.M.; Haredy, A.-B.; Monsef, A. Synthesis and reactions of 3-aminotetrachloroquinazolin-2,4-dione. Eur. J. Chem. 2011, 2, 514–518. [Google Scholar] [CrossRef]
- Abdelmonsef, A.H.; Abdelhakeem, M.A.; Mosallam, A.M.; Temairk, H.; El-Naggar, M.; Okasha, H.; Rashdan, H.R.M. A Search for Anti-inflammatory Therapies: Synthesis, In silico Investigation of the Mode of Action and In vitro Analyses of New Quinazolin-2, 4-dione Derivatives Targeting Phosphodiesterase-4 Enzyme. J. Heterocycl. Chem. 2021. [Google Scholar] [CrossRef]
- Ahmad, N.; Azad, M.I.; Khan, A.R.; Azad, I. Benzimidazole as a Promising Antiviral Heterocyclic Scaffold: A Review. J. Sci. Arts 2021, 21, 273–284. [Google Scholar] [CrossRef]
- Ogurtsov, V.A.; Rakitin, O.A.; Rees, C.W.; Smolentsev, A.A. 4,5-Dichloro-1,2-dithiole-3-thione in the synthesis of benzimidazole, benzoxazole and benzothiazole derivatives of 1,3-dithioles. Mendeleev Commun. 2003, 13, 50–51. [Google Scholar] [CrossRef]
- Rashdan, H.R.M.; Abdelmonsef, A.H.; Shehadi, I.A.; Gomha, S.M.; Soliman, A.M.M.; Mahmoud, H.K. Synthesis, Molecular Docking Screening and Anti-Proliferative Potency Evaluation of Some New Imidazo[2,1-b]Thiazole Linked Thiadiazole Conjugates. Molecules 2020, 25, 4997. [Google Scholar] [CrossRef] [PubMed]
- Rashdan, H.R.M.; Shehadi, I.A.; Abdelmonsef, A.H. Synthesis, anticancer evaluation, computer-aided docking studies, and ADMET prediction of 1,2,3-triazolyl-pyridine hybrids as human aurora B kinase inhibitors. ACS Omega 2021, 6, 1445–1455. [Google Scholar] [CrossRef]
- Rashdan, H.R.M.; Farag, M.M.; El-Gendey, M.S.; Mounier, M.M. Toward rational design of novel anti-cancer drugs based on targeting, solubility, and bioavailability exemplified by 1,3,4-thiadiazole derivatives synthesized under solvent-free conditions. Molecules 2019, 24, 2371. [Google Scholar] [CrossRef] [Green Version]
- Rashdan, H.R.M.; Shehadi, I.A.; Abdelrahman, M.T.; Hemdan, B.A. Antibacterial Activities and Molecular Docking of Novel Sulfone Biscompound Containing Bioactive 1, 2, 3-Triazole Moiety. Molecules 2021, 26, 4817. [Google Scholar] [CrossRef]
- El-Naggar, M.; Abd El-All, A.S.; El-Naem, S.I.A.; Abdalla, M.M.; Rashdan, H.R.M. New potent 5α- Reductase and aromatase inhibitors derived from 1,2,3-triazole derivative. Molecules 2020, 25, 672. [Google Scholar] [CrossRef] [Green Version]
- Rashdan, H.R.M.; El-Naggar, M.; Abdelmonsef, A.H. Synthesis, Molecular Docking Studies and In Silico ADMET Screening of New Heterocycles Linked Thiazole Conjugates as Potent Anti-Hepatic Cancer Agents. Molecules 2021, 26, 1705. [Google Scholar] [CrossRef]
- Aouad, M.R.; Khan, D.J.O.; Said, M.A.; Al-Kaff, N.S.; Rezki, N.; Ali, A.A.; Bouqellah, N.; Hagar, M. Novel 1, 2, 3-Triazole Derivatives as Potential Inhibitors against Covid-19 Main Protease: Synthesis, Characterization, Molecular Docking and DFT Studies. Chem. Sel. 2021, 6, 3468. [Google Scholar] [CrossRef]
- Li, Y.; Geng, J.; Liu, Y.; Yu, S.; Zhao, G. Thiadiazole-a Promising Structure in Medicinal Chemistry. ChemMedChem 2013, 8, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.; Taya, P. Neetu. A review on the various biological activities of thiadiazole. Int. J. Pharm. Pharm. Sci. 2015, 7, 39–47. [Google Scholar]
- Samadpour, A.N.; Merrikh, H. DNA gyrase activity regulates DnaA-dependent replication initiation in Bacillus subtilis. Mol. Microbiol. 2018, 108, 115–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reece, R.J.; Maxwell, A.; Wang, J.C. DNA gyrase: Structure and function. Crit. Rev. Biochem. Mol. Biol. 1991, 26, 335–375. [Google Scholar] [CrossRef] [PubMed]
- El-Saghier, A.M.; El-Naggar, M.; Hussein, A.H.M.; El-Adasy, A.-B.A.; Olish, M.; Abdelmonsef, A.H. Eco-Friendly Synthesis, Biological Evaluation, and In Silico Molecular Docking Approach of Some New Quinoline Derivatives as Potential Antioxidant and Antibacterial Agents. Front. Chem. 2021, 9, 1–14. [Google Scholar] [CrossRef]
- Chopra, B.; Dhingra, A.K.; Prasad, D.N.; Bhardwaj, S.; Dubey, S. Synthesis and in silico molecular docking studies on substituted piperic acid derivatives as inhibitors of bacterial DNA gyrase. Curr. Comput. Aided Drug Des. 2020, 16, 281–294. [Google Scholar] [CrossRef]
- Halim, S.A.; Sikandari, A.G.; Khan, A.; Wadood, A.; Fatmi, M.Q.; Csuk, R.; Al-Harrasi, A. Structure-Based Virtual Screening of Tumor Necrosis Factor-α Inhibitors by Cheminformatics Approaches and Bio-Molecular Simulation. Biomolecules 2021, 11, 329. [Google Scholar] [CrossRef]
- Halim, S.A.; Waqas, M.; Khan, A.; Al-Harrasi, A. In Silico Prediction of Novel Inhibitors of SARS-CoV-2 Main Protease through Structure-Based Virtual Screening and Molecular Dynamic Simulation. Pharmaceuticals 2021, 14, 896. [Google Scholar] [CrossRef]
- Abo-Bakr, A.M.; Alsoghier, H.M.; Abdelmonsef, A.H. Molecular docking, modeling, semiempirical calculations studies and in vitro evaluation of new synthesized pyrimidin-imide derivatives. J. Mol. Struct. 2022, 1249, 131548. [Google Scholar] [CrossRef]
- Abdelmonsef, A.H.; Mosallam, A.M. Synthesis, in vitro biological evaluation and in silico docking studies of new quinazolin-2,4-dione analogues as possible anticarcinoma agents. J. Heterocycl. Chem. 2020, 57, 1637–1654. [Google Scholar] [CrossRef]
- Noser, A.A.; El-Naggar, M.; Donia, T.; Abdelmonsef, A.H. Synthesis, In Silico and In Vitro Assessment of New Quinazolinones as Anticancer Agents via Potential AKT Inhibition. Molecules 2020, 25, 4780. [Google Scholar] [CrossRef] [PubMed]
- Noser, A.A.; Abdelmonsef, A.H.; El-naggar, M.; Salem, M.M. New Amino Acid Schiff Bases as Anticancer Agents via Potential Mitochondrial Complex I-Associated Hexokinase Inhibition and Targeting AMP-Protein Kinases/mTOR Signaling Pathway. Molecules 2021, 26, 5332. [Google Scholar] [CrossRef]
- Haredi Abdelmonsef, A.; Eldeeb Mohamed, M.; El-Naggar, M.; Temairk, H.; Mohamed Mosallam, A. Novel Quinazolin-2,4-Dione Hybrid Molecules as Possible Inhibitors Against Malaria: Synthesis and in silico Molecular Docking Studies. Front. Mol. Biosci. 2020, 7, 105. [Google Scholar] [CrossRef]
- Sobhi, M.G.; Abdelhady, H.A.; Doaa, Z.H.; Abdelmonsef, A.H.; El-Naggar, M.; Elaasser, M.M.; Mahmoud, H.K. Thiazole-Based Thiosemicarbazones: Synthesis, Cytotoxicity Evaluation and Molecular Docking Study. Drug Des. Devel. Ther. 2021, 2021, 659–677. [Google Scholar] [CrossRef]
- Shehadi, I.A.; Rashdan, H.R.M.; Abdelmonsef, A.H. Homology Modeling and Virtual Screening Studies of Antigen MLAA-42 Protein: Identification of Novel Drug Candidates against Leukemia—An In Silico Approach. Comput. Math. Methods Med. 2020, 2020, 8196147. [Google Scholar] [CrossRef] [Green Version]
- Haredi Abdelmonsef, A. Computer-aided identification of lung cancer inhibitors through homology modeling and virtual screening. Egypt. J. Med. Hum. Genet. 2019, 20, 1–14. [Google Scholar] [CrossRef] [Green Version]
- El-Maghraby, A.M.; Abdelmonsef, A.H. Synthesis, characterization and in silico molecular docking studies of novel chromene derivatives as Rab23 inhibitors. Egypt. J. Chem. 2020, 63, 1341–1358. [Google Scholar] [CrossRef]
- HA, A.; SP, L. Human Rab8b Protein as a Cancer Target—An In Silico Study. J. Comput. Sci. Syst. Biol. 2016, 9, 132–149. [Google Scholar] [CrossRef] [Green Version]
- Rondla, R.; PadmaRao, L.S.; Ramatenki, V.; Haredi-Abdel-Monsef, A.; Potlapally, S.R.; Vuruputuri, U. Selective ATP competitive leads of CDK4: Discovery by 3D-QSAR pharmacophore mapping and molecular docking approach. Comput. Biol. Chem. 2017, 71, 224–229. [Google Scholar] [CrossRef]
- Abdelmonsef, A.H.; Dulapalli, R.; Dasari, T.; Padmarao, L.S.; Mukkera, T.; Vuruputuri, U. Identification of Novel Antagonists for Rab38 Protein by Homology Modeling and Virtual Screening. Comb. Chem. High Throughput Screen. 2016, 19, 875–892. [Google Scholar] [CrossRef]
- Dasari, T.; Kondagari, B.; Dulapalli, R.; Abdelmonsef, A.H.; Mukkera, T.; Padmarao, L.S.; Malkhed, V.; Vuruputuri, U. Design of novel lead molecules against RhoG protein as cancer target–a computational study. J. Biomol. Struct. Dyn. 2017, 35, 3119–3139. [Google Scholar] [CrossRef]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The protein data bank. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002, 58, 899–907. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dallakyan, S.; Olson, A.J. Small-Molecule Library Screening by Docking with PyRx. In Chemical Biology; Springer: Berlin/Heidelberg, Germany, 2015; Volume 1263, pp. 243–250. ISBN 9780123944474. [Google Scholar]
- Metzger, B.E.; Contreras, M.; Sacks, D.A.; Watson, W.; Dooley, S.L.; Foderaro, M.; Niznik, C.; Bjaloncik, J.; Catalano, P.M.; Dierker, L. Hyperglycemia and adverse pregnancy outcomes. N. Engl. J. Med. 2008, 358, 1991–2002. [Google Scholar] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2D Structure | Binding Energy kcal/mol | Docked Complex (Amino Acid–Ligand) Interactions | Distance (Å) | |
|---|---|---|---|---|
| Ciprofloxacin |  | −7.4 | H-bond Arg76:NH1—ciprofloxacin Thr165:OG1—ciprofloxacin Val43:O—ciprofloxacin | 3.00 2.74 2.04 |
| 1a |  | −6.4 | H-bond Val71:O—compound 1a arene-σ Asn46:CB—compound 1a | 1.87 3.91 |
| 1b |  | −6.5 | arene-σ Asn46:CB—compound 1b | 3.93 |
| 2a |  | −7.9 | H-bond Ala47:N—compound 2a Thr165:OG1—compound 2a Thr165:OG1—compound 2a arene-cation Arg76:NH1—compound 2a | 2.99 2.98 2.95 4.06 |
| 2b |  | −8.1 | H-bond Ala47:N—compound 2b Thr165:OG1—compound 2b Thr165:OG1—compound 2b arene-cation Arg76:NH1—compound 2b | 2.99 2.95 2.94 4.02 |
| 4a |  | −7.0 | H-bond Thr34:OG1—compound 4a Thr34:OG1—compound 4a | 2.98 3.00 |
| 4b |  | −7.1 | H-bond Asp49:OD2—compound 4b | 2.20 |
| 6a |  | −9.7 | H-bond Thr165:OG1—compound 6a | 2.45 |
| 6b |  | −9.8 | H-bond Asp49:OD1—compound 6b | 2.40 |
| Ciprofloxacin | Compound 1a | Compound 1b | Compound 2a | Compound 2b | Compound 4a | Compound 4b | Compound 6a | Compound 6b | |
|---|---|---|---|---|---|---|---|---|---|
| Molecular Weight (g/mol) | 331.13 | 159.15 | 193.60 | 241.25 | 275.70 | 345.46 | 379.90 | 534.61 | 569.05 |
| BBB permeant | No | No | Yes | No | Yes | No | No | No | No |
| %Human Intestinal Absorption (HIA+) | 97.95 | 99.51 | 99.57 | 100.00 | 100.00 | 99.20 | 99.32 | 100.00 | 100.00 |
| logp | −0.7 | 2.80 | 3.43 | 1.72 | 2.35 | 2.80 | 3.43 | 3.91 | 4.38 |
| TPSA A2 | 74.57 | 78.44 | 78.44 | 76.47 | 76.47 | 83.79 | 83.79 | 131.05 | 131.05 |
| HBA | 6 | 5 | 5 | 6 | 6 | 7 | 7 | 11 | 2 |
| HBD | 2 | 1 | 1 | 1 | 1 | 2 | 2 | 2 | 6 |
| N rotatable | 3 | 1 | 1 | 2 | 2 | 5 | 5 | 6 | 1 |
| N violations | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 |
| Volume A3 | 285.46 | 133.76 | 147.29 | 209.43 | 222.97 | 288.66 | 302.20 | 454.50 | 468.04 |
| Carcinogenicity | Noncarcinogenic | Noncarcinogenic | Noncarcinogenic | Noncarcinogenic | Noncarcinogenic | Noncarcinogenic | Noncarcinogenic | Noncarcinogenic | Noncarcinogenic |
| GI absorption | High | High | High | High | High | Low | Low | Low | Low |
| Bioavailability score | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.17 | 0.17 |
| Compounds | Microorganism Inhibition Zone Diameters Using the Agar Diffusion Method (mm) | ||||
|---|---|---|---|---|---|
| Gram (+ve) Bacteria | Gram (−ve) Bacteria | Fungi | |||
| Staphylococcus aureus | Escherichia coli | Pseudomonas aeruginosa | Aspergillus niger | Candida albicans | |
| 2a | 15 ± 0.14 | 12 ± 1.08 | 22 ± 1.01 | −ve | −ve |
| 2b | −ve | 5 ± 0.2 | −ve | 30 ± 1.16 | 27 ± 1.1 |
| 4a | 23 ± 0.8 | −ve | 13 ± 0.65 | −ve | −ve |
| 4b | −ve | −ve | 12 ± 08 | 14 ± 0.15 | 19 ± 1.04 |
| 6a | 24 ± 0.6 | 25 ± 0.9 | 17 ± 0.75 | 20 ± 0.9 | 16 ± 0.89 |
| 6b | 29 ± 1.2 | 21 ± 1.14 | 19 ± 0.79 | 18 ± 0.12 | 14 ± 0.58 |
| Ciprofloxacin | 20 ± 0.9 | 23 ± 1.02 | 21 ± 0.9 | −ve | −ve |
| Nystatin | −ve | −ve | −ve | 22 ± 0.18 | 23 ± 1.15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rashdan, H.R.M.; Abdelmonsef, A.H.; Abou-Krisha, M.M.; Yousef, T.A. Synthesis, Identification, Computer-Aided Docking Studies, and ADMET Prediction of Novel Benzimidazo-1,2,3-triazole Based Molecules as Potential Antimicrobial Agents. Molecules 2021, 26, 7119. https://doi.org/10.3390/molecules26237119
Rashdan HRM, Abdelmonsef AH, Abou-Krisha MM, Yousef TA. Synthesis, Identification, Computer-Aided Docking Studies, and ADMET Prediction of Novel Benzimidazo-1,2,3-triazole Based Molecules as Potential Antimicrobial Agents. Molecules. 2021; 26(23):7119. https://doi.org/10.3390/molecules26237119
Chicago/Turabian StyleRashdan, Huda R. M., Aboubakr H. Abdelmonsef, Mortaga M. Abou-Krisha, and Tarek A. Yousef. 2021. "Synthesis, Identification, Computer-Aided Docking Studies, and ADMET Prediction of Novel Benzimidazo-1,2,3-triazole Based Molecules as Potential Antimicrobial Agents" Molecules 26, no. 23: 7119. https://doi.org/10.3390/molecules26237119