
Anharmonicity and Spectra–Structure Correlations in MIR and NIR Spectra of Crystalline Menadione (Vitamin K3)

 ,
,  ,
,  ,
,  and
and
Abstract
:
1. Introduction
2. Results and Discussion
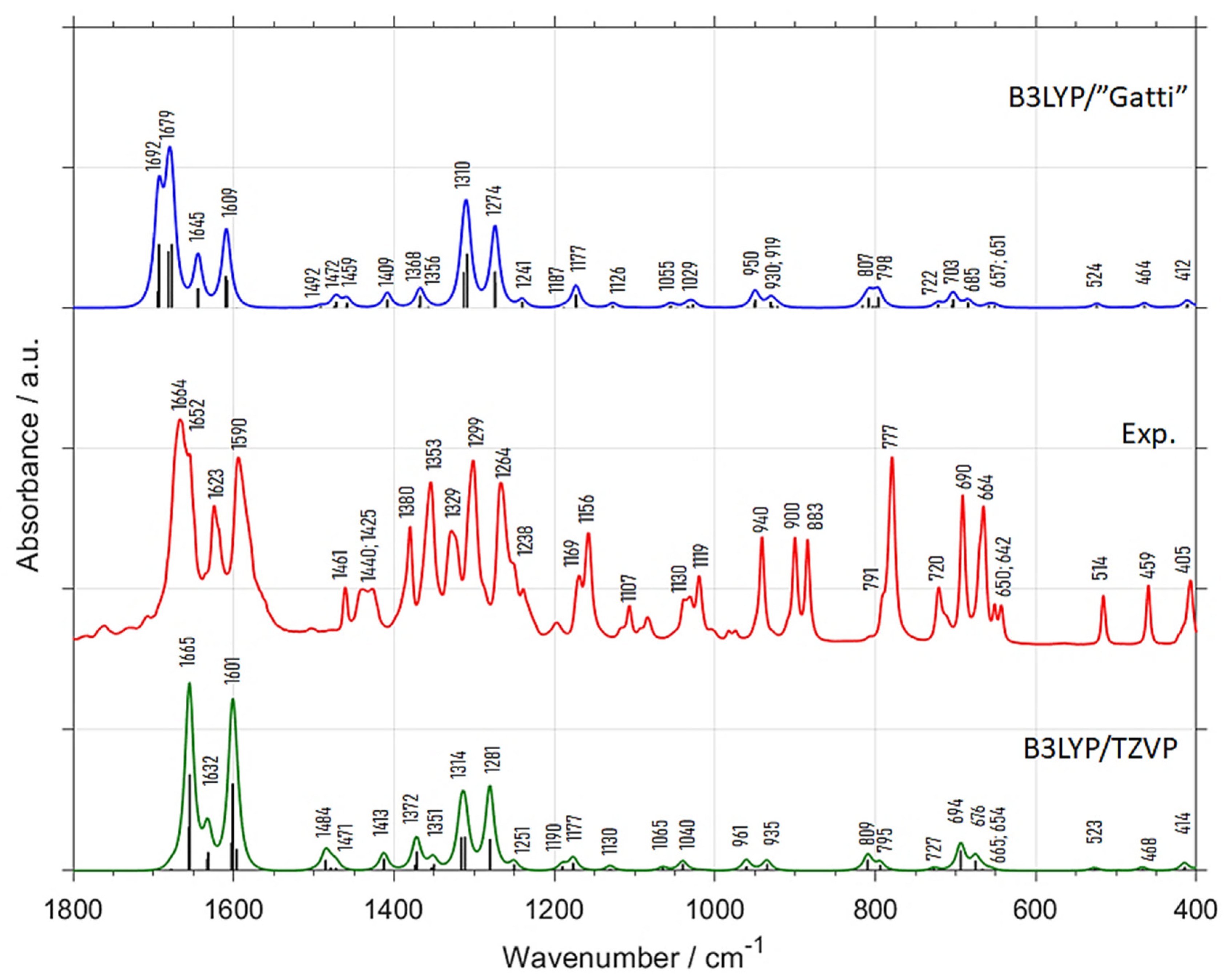
2.1. Comparison of Experimental and Theoretical (Harmonic Approximation within Periodic Model) MIR Spectra
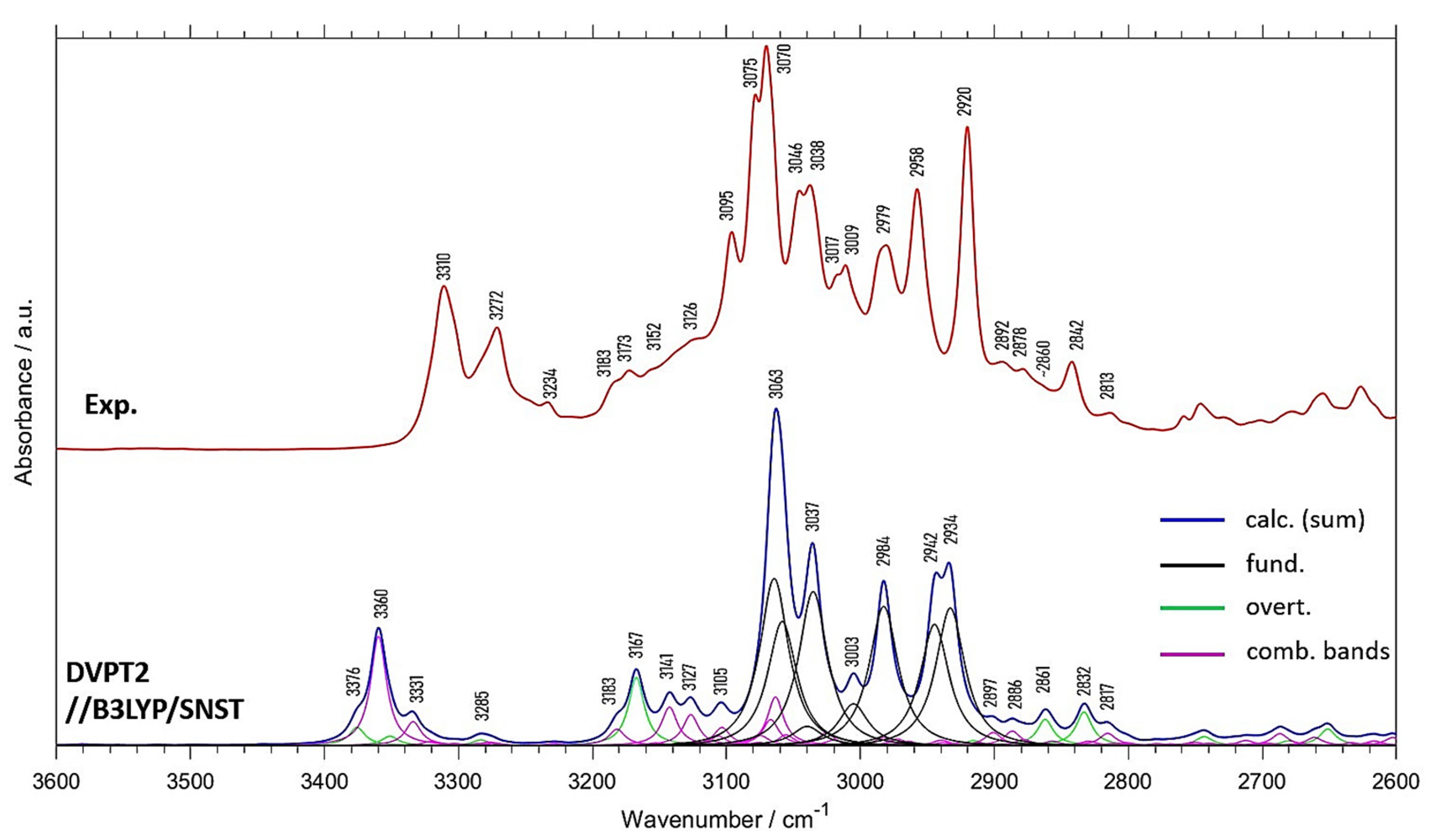
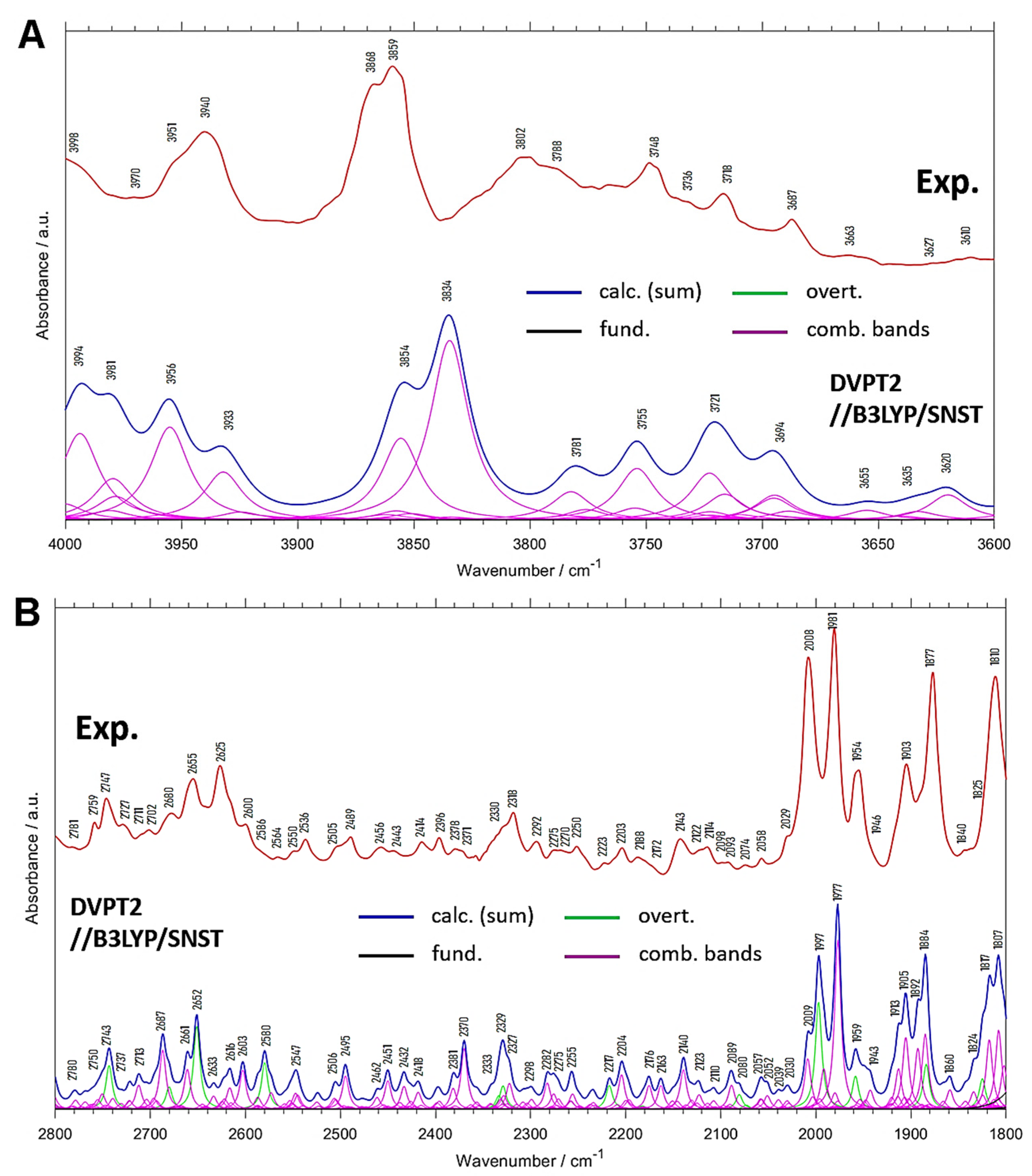
2.2. Overtones and Combination Bands in MIR Spectrum of Menadione
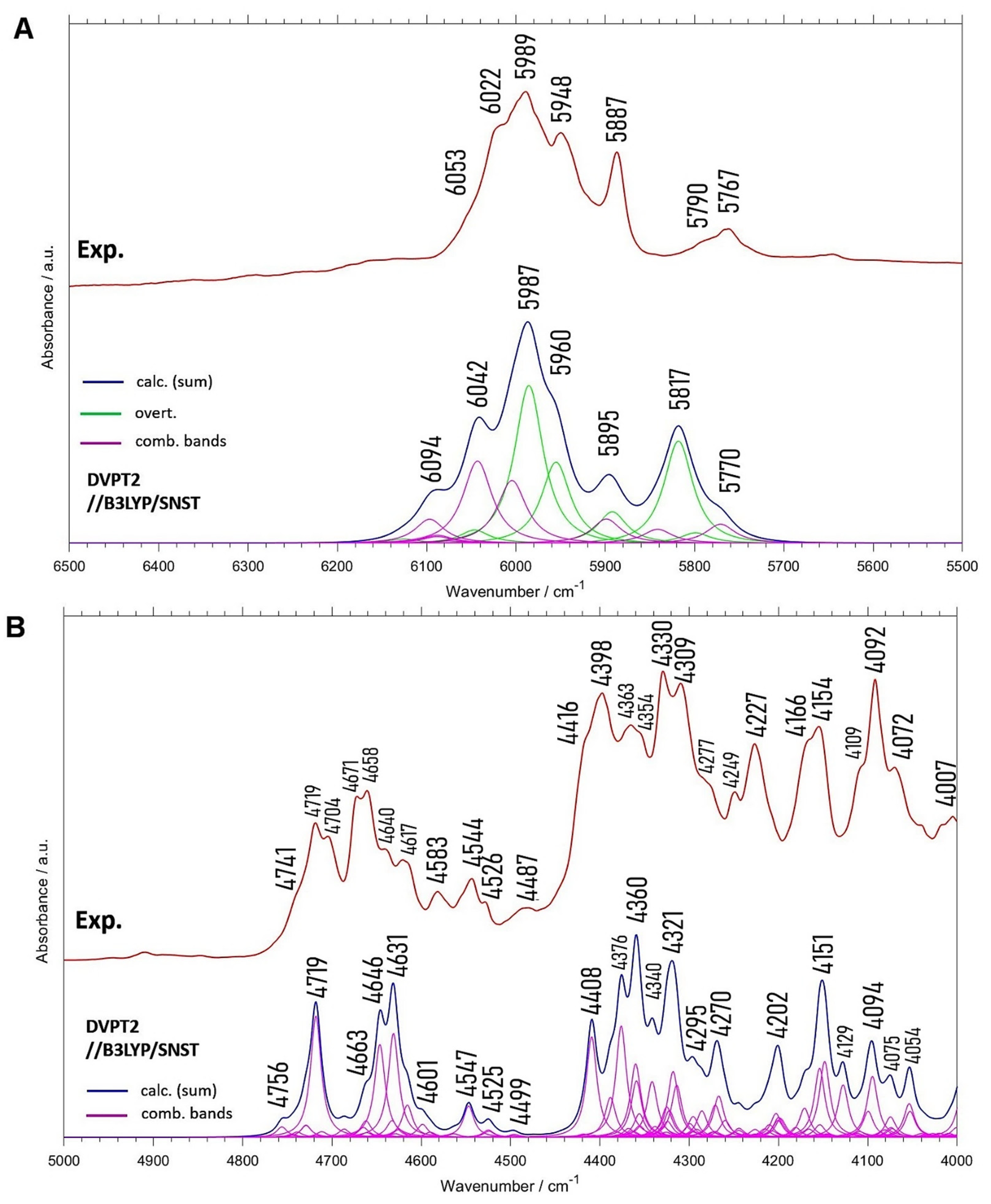
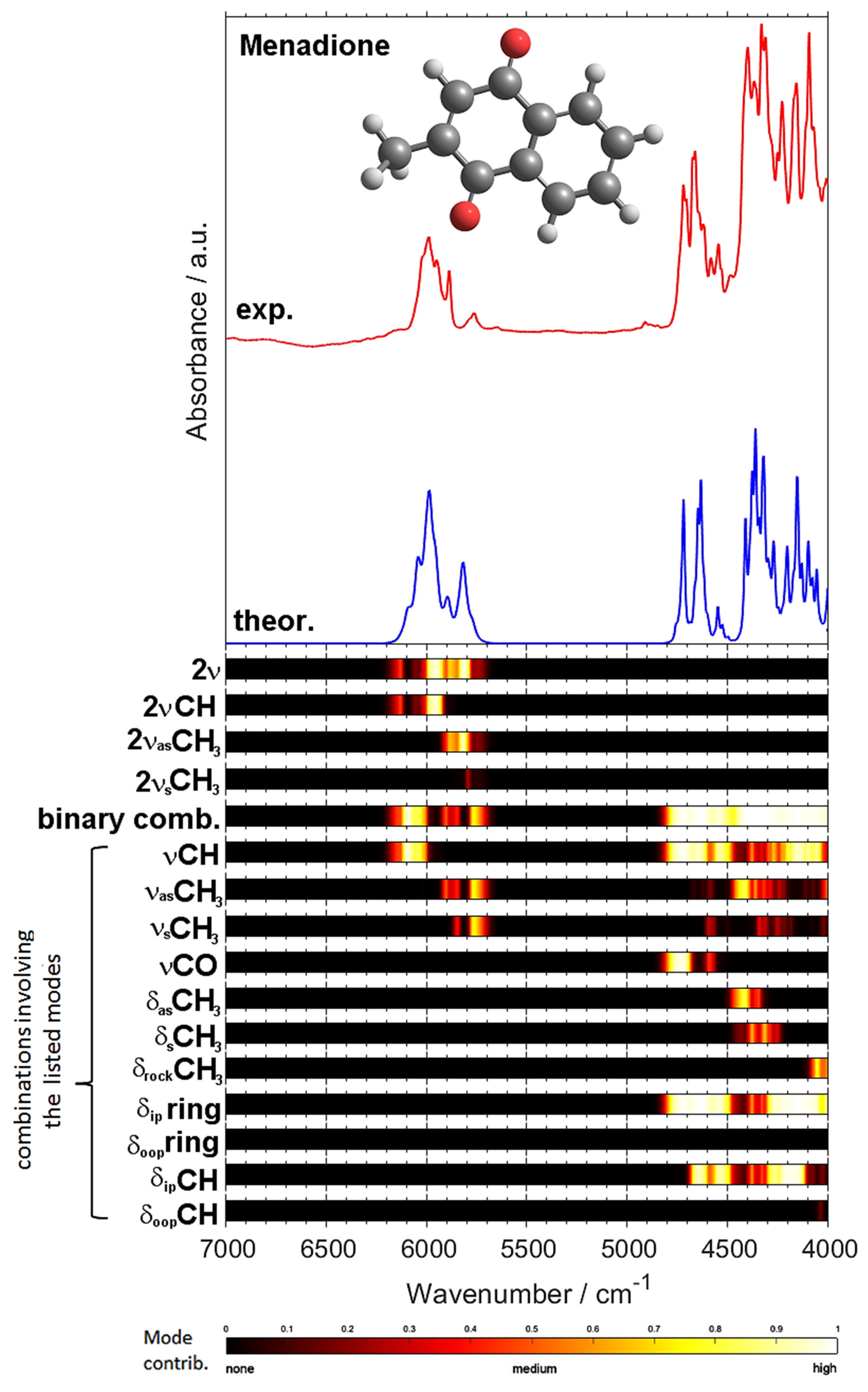
2.3. NIR Spectrum of Menadione
3. Materials and Methods
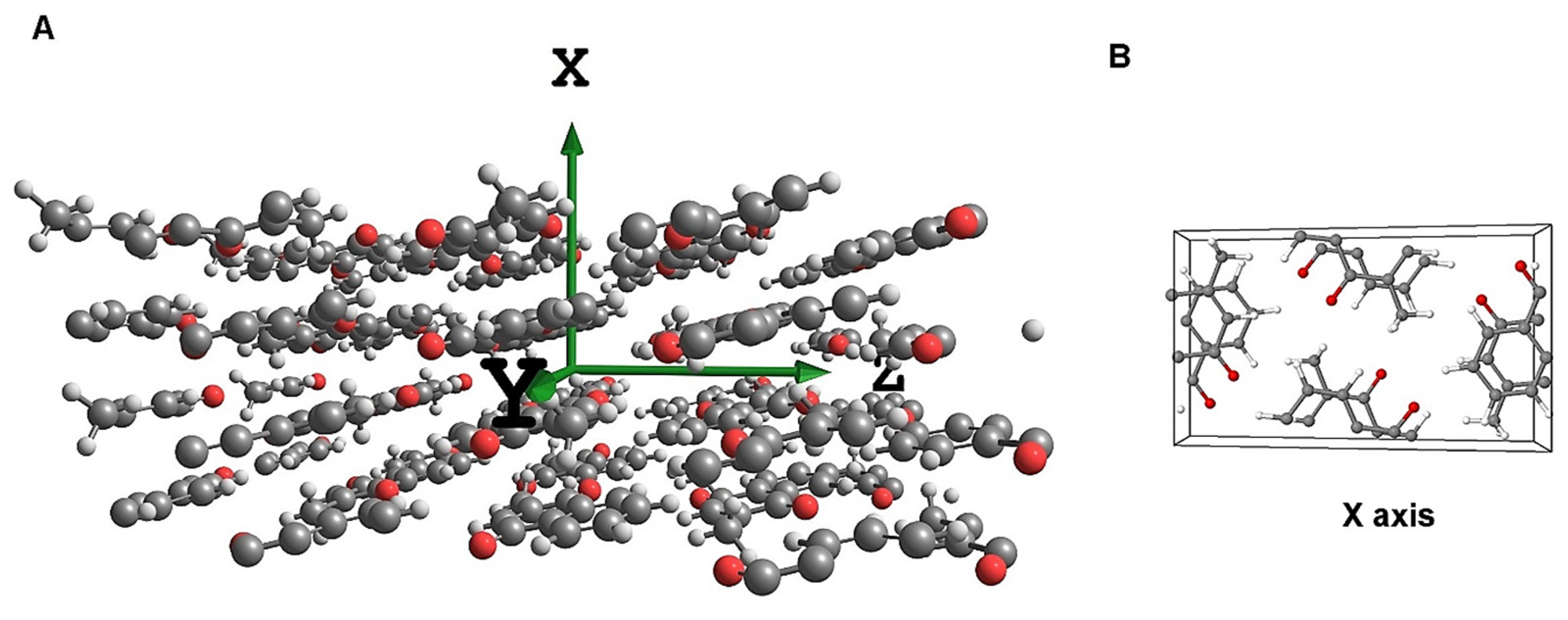
3.1. Experimental
3.2. Computational Procedures
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Walsh, C.T.; Tang, Y. The Chemical Biology of Human Vitamins; The Royal Society of Chemistry: London, UK, 2019. [Google Scholar]
- Liao, M.-L.; Seib, P.A. Chemistry of L-ascorbic acid related to foods. Food Chem. 1988, 30, 289–290. [Google Scholar] [CrossRef]
- Belzile, M.-N.; Godin, R.; Durantini, A.M.; Cosa, G. Monitoring chemical and biological electron transfer reactions with a fluorogenic vitamin K analogue probe. J. Am. Chem. Soc. 2016, 138, 16388–16397. [Google Scholar] [CrossRef]
- Bik, E.; Mateuszuk, L.; Stojak, M.; Chlopicki, S.; Baranska, M.; Majzner, K. Menadione-induced endothelial inflammation detected by Raman spectroscopy. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 1868, 2021, 118911. [Google Scholar] [CrossRef]
- Song, C.-Y.; Shen, H.-Z.; Wang, L.-C.; Zhao, J.-H.; Wang, F.-A. Solubilities of vitamin K3 in benzene, toluene, ethylbenzene, o-xylene, m-xylene, and p-xylene between (299.44 and 344.24) K. J. Chem. Eng. Data 2008, 53, 283–285. [Google Scholar] [CrossRef]
- Dubbs, M.D.; Gupta, R.B. Solubility of vitamin E (α-tocopherol) and vitamin K3 (menadione) in ethanol−water mixture. J. Chem. Eng. Data 1998, 43, 590–591. [Google Scholar] [CrossRef]
- Lu, X.; Nan, M.; Zhang, H.; Liu, X.; Yuan, H.; Yang, J. Investigation of the antioxidant property of ascorbic acid. J. Phys. Chem. C 2007, 111, 14998–15002. [Google Scholar] [CrossRef]
- Zhu, B.; Wang, J.-R.; Zhang, Q.; Mei, X. Improving dissolution and photostability of vitamin K3 via cocrystallization with naphthoic acids and sulfamerazine. Cryst. Growth Des. 2016, 16, 483–492. [Google Scholar] [CrossRef]
- Yang, Z.; Zhu, B.; Jiang, Y.; Zhang, Z.; Dai, W.; Mei, X. Different solid forms of vitamin K3 and their effect on the chemical stability. Cryst. Growth Des. 2021, 21, 528–535. [Google Scholar] [CrossRef]
- Yulia, M.; Suhandy, D.; Ogawa, Y.; Kondo, N. Investigation on the influence of temperature in l-ascorbic acid determination using FTIR-ATR terahertz spectroscopy: Calibration model with temperature compensation. Eng. Agric. Environ. Food 2014, 7, 148–154. [Google Scholar] [CrossRef]
- Bichara, L.C.; Lanús, H.E.; Brandán, S.A. Stabilities of aqueous solutions of sucrose containing ascorbic and citric acids by using FTIR spectroscopy and physicochemical studies. J. Mol. Liq. 2014, 200, 448–459. [Google Scholar] [CrossRef]
- Bayari, S.; Atac, A.; Yurdakul, S. Coordination behaviour of nicotinamide: An infrared spectroscopic study. J. Mol. Struct. 2003, 655, 163–170. [Google Scholar] [CrossRef]
- Kokaislova, A.; Parchansky, V.; Matejka, P. Surface-enhanced infrared spectra of nicotinic acid and pyridoxine on copper substrates: What is the effect of temperature and deposition conditions? J. Phys. Chem. C 2015, 119, 26526–26539. [Google Scholar] [CrossRef]
- Machalska, E.; Zajac, G.; Gruca, A.; Zobi, F.; Baranska, M.; Kaczor, A. Resonance Raman optical activity shows unusual structural sensitivity for systems in resonance with multiple excited states: Vitamin B12 case. J. Phys. Chem. Lett. 2020, 11, 5037–5043. [Google Scholar] [CrossRef] [PubMed]
- Panicker, C.Y.; Varghese, H.T.; Philip, D. FT-IR, FT-Raman and SERS spectra of vitamin C. Spectrochim. Acta A 2006, 65, 802–804. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Kaur, S.; Saini, G.S.S. Experimental and theoretical vibrational analysis of vitamin C. Indian J. Pure Appl. Phys. 2007, 45, 714–720. [Google Scholar]
- Bichara, L.C.; Lanús, H.E.; Nieto, C.G.; Brandán, S.A. Density functional theory calculations of the molecular force field of l-ascorbic acid, vitamin C. J. Phys. Chem. A 2010, 114, 4997–5004. [Google Scholar] [CrossRef]
- Yadav, R.A.; Rani, P.; Kumar, M.; Singh, R.; Singh, P.; Singh, N.P. Experimental IR and Raman spectra and quantum chemical studies of molecular structures, conformers and vibrational characteristics of L-ascorbic acid and its anion and cation. Spectrochim. Acta A 2011, 84, 13–14. [Google Scholar] [CrossRef]
- Dabbagh, H.A.; Azami, F.; Farrokhpour, H.; Chermahini, A.N. UV-VIS, NMR and FT-IR spectra of tautomers of vitamin C. Experimental and DFT calculations. J. Chil. Chem. Soc. 2014, 59, 2588–2594. [Google Scholar] [CrossRef] [Green Version]
- Falk, M.; Wójcik, M.J. Effect of temperature and hydrogen-bond strength on the i.r. absorption of isolated OH oscillators: Crystalline L-ascorbic acid. Spectrochim. Acta A 1979, 35, 1117–1123. [Google Scholar] [CrossRef]
- Dimitrova, Y. Theoretical study of the changes in the vibrational characteristics arising from the hydrogen bonding between Vitamin C (l-ascorbic acid) and H2O. Spectrochim. Acta A 2006, 63, 427–437. [Google Scholar] [CrossRef]
- Brela, M.Z.; Wójcik, M.J.; Boczar, M.; Witek, L.; Yasuda, M.; Ozaki, Y. Car-Parrinello molecular Dynamics simulations of infrared spectra of crystalline vitamin C with analysis of double minimum proton potentials for medium-strong hydrogen bonds. J. Phys. Chem. B 2015, 119, 7922–7930. [Google Scholar] [CrossRef]
- Balakrishnan, G.; Mohandas, P.; Umapathy, S. Time-resolved resonance Raman spectroscopic studies on the radical anions of menaquinone and naphthoquinone. J. Phys. Chem. 1996, 100, 16475. [Google Scholar] [CrossRef]
- Balakrishnan, G.; Mohandas, P.; Umapathy, S. Ab initio studies on structure and vibrational spectra of ubiquinone and its radical anion. Spectrochim. Acta A 1997, 53, 1553–1561. [Google Scholar] [CrossRef]
- Grafton, A.K.; Boesch, S.E.; Wheeler, R.A. Structures and properties of vitamin K and its radical anion predicted by a hybrid Hartree-Fock/density functional method. J. Mol. Struct. THEOCHEM 1997, 392, 1–11. [Google Scholar] [CrossRef]
- Guo, Y.; Xue, Y.; Shu, X.; Yan, G.S. Density functional theory investigations on the conformational stability and vibrational spectra of the model compound of vitamin K. J. Mol. Struct. THEOCHEM 2007, 820, 40–47. [Google Scholar] [CrossRef]
- Kumar, M.; Yadav, R.A. Experimental IR and Raman spectra and quantum chemical studies of molecular structures, conformers and vibrational characteristics of nicotinic acid and its N-oxide. Spectrochim. Acta A 2011, 79, 1316–1325. [Google Scholar] [CrossRef]
- Koczoń, P.; Dobrowolski, J.C.; Lewandowski, W.; Mazurek, A.P. Experimental and theoretical IR and Raman spectra of picolinic, nicotinic and isonicotinic acids. J. Mol. Struct. 2003, 655, 89–95. [Google Scholar] [CrossRef]
- Beć, K.B.; Grabska, J.; Hofer, T.S. Introduction to quantum vibrational spectroscopy. In Near-Infrared Spectroscopy; Ozaki, Y., Huck, C.W., Tsuchikawa, S., Engelsen, S.B., Eds.; Springer: Singapore, 2021; pp. 83–110. [Google Scholar] [CrossRef]
- Ozaki, Y.; Beć, K.B.; Morisawa, Y.; Yamamoto, S.; Tanabe, I.; Huck, C.W.; Hofer, T.S. Advances, challenges and perspectives of quantum chemical approaches in molecular spectroscopy of the condensed phase. Chem. Soc. Rev. 2021. [Google Scholar] [CrossRef]
- Beć, K.B.; Huck, C.W. Breakthrough potential in near-infrared spectroscopy: Spectra simulation. A review of recent developments. Front. Chem. 2019, 7, 48. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Xiang, B.; Qu, L. Structure analysis of ascorbic acid using near-infrared spectroscopy and generalized two-dimensional correlation spectroscopy. J. Mol. Struct. 2006, 794, 12–17. [Google Scholar] [CrossRef]
- Grabska, J.; Beć, K.B.; Kirchler, C.G.; Ozaki, Y.; Huck, C.W. Distinct difference in sensitivity of NIR vs. IR bands of melamine to inter-molecular interactions with impact on analytical spectroscopy explained by anharmonic quantum mechanical study. Molecules 2019, 24, 1402. [Google Scholar] [CrossRef] [Green Version]
- Xia, J.-F.; Li, X.-J.; Li, P.-W.; Ma, Q.; Ding, X.-X. Application of wavelet transform in the prediction of navel orange vitamin C content by near-infrared spectroscopy. Agric. Sci. China 2007, 6, 1067–1073. [Google Scholar] [CrossRef]
- Dos Santos Garcia, V.A.; da Silva, M.R.; Seixas, F.A.V. Rapid analisys of vitamin-C content in acerola extract by FT-NIR spectroscopy. Rev. Tecnol. 2013, 22, 13–21. [Google Scholar]
- Blanco, M.; Coello, J.; Iturriaga, H.; Maspoch, S.; de la Pezuela, C. Determination of ascorbic acid in pharmaceutical preparations by near infrared reflectance spectroscopy. Talanta 1993, 40, 1671–1676. [Google Scholar] [CrossRef]
- Yang, H.; Irudayaraj, J. Rapid determination of vitamin C by NIR, MIR and FT-Raman techniques. J. Pharm. Pharmacol. 2002, 54, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Maqbool, M.A.; Aslam, M.; Akbar, W.; Iqbal, Z. Biological importance of vitamins for human health: A review. J. Agric. Basic Sci. 2017, 2, 50–58. [Google Scholar]
- Chen, Y.; Morisawa, Y.; Futami, Y.; Czarnecki, M.A.; Wang, H.-S.; Ozaki, Y. Combined IR/NIR and density functional theory calculations analysis of the solvent effects on frequencies and intensities of the fundamental and overtones of the C=O stretching vibrations of acetone and 2-hexanone. J. Phys. Chem. A 2014, 118, 2576–2583. [Google Scholar] [CrossRef]
- Beć, K.B.; Grabska, J.; Ozaki, Y.; Hawranek, J.P.; Huck, C.W. Influence of non-fundamental modes on mid-infrared spectra. Anharmonic DFT study of aliphatic ethers. J. Phys. Chem. A 2017, 121, 1412–1424. [Google Scholar] [CrossRef]
- Beć, K.B.; Grabska, J.; Czarnecki, M.A.; Huck, C.W.; Wójcik, M.J.; Nakajima, T.; Ozaki, Y. IR spectra of crystalline nucleobases: Combination of periodic harmonic calculations with anharmonic corrections based on finite models. J. Phys. Chem. B 2019, 123, 10001–10013. [Google Scholar] [CrossRef]
- Singh, S.; Czarnecki, M.A. How much anharmonicity is in vibrational spectra of CH3I and CD3I? Spectrochim. Acta A 2021, 248, 119176. [Google Scholar] [CrossRef]
- Singh, S.; Szostak, R.; Czarnecki, M.A. Vibrational intensities and anharmonicity in MIR, NIR and Raman spectra of liquid CHCl3, CDCl3, CHBr3 and CDBr3: Spectroscopic and theoretical study. J. Mol. Liq. 2021, 336, 116277. [Google Scholar] [CrossRef]
- Beć, K.B.; Grabska, J.; Ozaki, Y.; Czarnecki, M.A.; Huck, C.W. Simulated NIR spectra as sensitive markers of the structure and interactions in nucleobases. Sci. Rep. 2019, 9, 17398. [Google Scholar] [CrossRef] [PubMed]
- Corinti, D.; Chiavarino, B.; Scuderi, D.; Fraschetti, C.; Filippi, A.; Fornarini, S.; Crestoni, M.E. Molecular properties of bare and microhydrated vitamin B5–calcium complexes. Int. J. Mol. Sci. 2021, 22, 692. [Google Scholar] [CrossRef]
- Ebrahimi, S.; Dabbagh, H.A.; Eskandari, K. Nature of intramolecular interactions of vitamin C in view of interacting quantum atoms: The role of hydrogen bond cooperativity on geometry. Phys. Chem. Chem. Phys. 2016, 18, 18278–18288. [Google Scholar] [CrossRef]
- Oliviera, L.B.A.; Colherinhas, G.; Castro, M.A. Spectroscopic properties of vitamin E models in solution. Chem. Phys. Lett. 2015, 628, 49–53. [Google Scholar] [CrossRef]
- Sato, H.; Murakami, R.; Padermshoke, A.; Hirose, F.; Senda, K.; Noda, I.; Ozaki, Y. Infrared spectroscopy studies of CH···O hydrogen bondings and thermal behavior of biodegradable poly(hydroxyalkanoate). Macromolecules 2004, 37, 7203–7213. [Google Scholar] [CrossRef]
- Guo, L.; Sato, H.; Hashimoto, T.; Ozaki, Y. FTIR study on hydrogen-bonding interactions in biodegradable polymer blends of poly(3-hydroxybutyrate) and poly(4-vinylphenol). Macromolecules 2010, 43, 3897–3902. [Google Scholar] [CrossRef]
- Beć, K.B.; Futami, Y.; Wójcik, M.J.; Nakajima, T.; Ozaki, Y. Spectroscopic and computational study of acetic acid and its cyclic dimer in the near-infrared region. J. Phys. Chem. A 2016, 120, 6170–6183. [Google Scholar] [CrossRef]
- Grabska, J.; Ishigaki, M.; Beć, K.B.; Wójcik, M.J.; Ozaki, Y. Correlations between structure and near-infrared spectra of saturated and unsaturated carboxylic acids. Insight from anharmonic density functional theory calculations. J. Phys. Chem. A 2017, 121, 3437–3451. [Google Scholar] [CrossRef]
- Grabska, J.; Beć, K.B.; Ishigaki, M.; Wójcik, M.J.; Ozaki, Y. Spectra-structure correlations of saturated and unsaturated medium-chain fatty acids. Near-infrared and anharmonic DFT study of hexanoic acid and sorbic acid. Spectrochim. Acta A 2017, 185, 35–44. [Google Scholar] [CrossRef]
- Grabska, J.; Beć, K.B.; Ishigaki, M.; Huck, C.W.; Ozaki, Y. NIR spectra simulations by anharmonic DFT-saturated and unsaturated long-chain fatty acids. J. Phys. Chem. B 2018, 122, 6931–6944. [Google Scholar] [CrossRef] [PubMed]
- Czarnecki, M.A.; Morisawa, Y.; Katsumoto, Y.; Takaya, T.; Singh, S.; Sato, H.; Ozaki, Y. Solvent effect on competition between weak and strong interactions in phenol solutions studied by near-infrared spectroscopy and DFT calculations. Phys. Chem. Chem. Phys. 2021, 23, 19188–19194. [Google Scholar] [CrossRef] [PubMed]
- Bernard-Houplain, M.C.; Sandorfy, C. Low-temperature infrared study of sterically hindered associated alcohols. Chem. Phys. Lett. 1974, 27, 154–156. [Google Scholar] [CrossRef]
- Sandorfy, C. Chapter 13. In The Hydrogen Bond. Recent Developments in Theory and Experiments; Schuster, P., Zundel, G., Sandorfy, C., Eds.; North-Holland Publ. Co: Amsterdam, The Netherlands, 1976; pp. 615–654. [Google Scholar]
- Czarnecki, M.A.; Morisawa, Y.; Futami, Y.; Ozaki, Y. Advances in Molecular Structure and Interaction Studies Using Near-Infrared Spectroscopy. Chem. Rev. 2015, 115, 9707–9744. [Google Scholar] [CrossRef] [PubMed]
- Huck, C.W.; Beć, K.B.; Grabska, J.; Ozaki, Y. Quantum chemical calculation of NIR spectra of practical materials. NIR News 2017, 28, 13–20. [Google Scholar] [CrossRef]
- Grabska, J.; Beć, K.B.; Ozaki, Y.; Huck, C.W. Temperature drift of conformational equilibria of butyl alcohols studied by near-infrared spectroscopy and fully anharmonic DFT. J. Phys. Chem. A 2017, 121, 1950–1961. [Google Scholar] [CrossRef]
- Beć, K.B.; Grabska, J.; Huck, C.W.; Czarnecki, M.A. Spectra–structure correlations in isotopomers of ethanol (CX3CX2OX.; X = H, D): Combined near-infrared and anharmonic computational study. Molecules 2019, 24, 2189. [Google Scholar] [CrossRef] [Green Version]
- Beć, K.B.; Grabska, J.; Huck, C.W.; Czarnecki, M.A. Effect of conformational isomerism on NIR spectra of ethanol isotopologues. Spectroscopic and anharmonic DFT study. J. Mol. Liq. 2020, 310, 113271. [Google Scholar] [CrossRef]
- Pulay, P.; Fogarasi, G.; Pang, F.; Boggs, J.E. Systematic ab initio gradient calculation of molecular geometries, force constants, and dipole moment derivatives. J. Am. Chem. Soc. 1979, 101, 2550–2560. [Google Scholar] [CrossRef]
- Beć, K.B.; Grabska, J.; Badzoka, J.; Huck, C.W. Spectra-structure correlations in NIR region of polymers from quantum chemical calculations. The cases of aromatic ring, C=O, C≡N and C-Cl functionalities. Spectrochim. Acta A 2021, 262, 120085. [Google Scholar] [CrossRef]
- Weyer, L.G.; Lo, S.C. Spectra–structure correlations in the near-infrared. In Handbook of Vibrational Spectroscopy; Chalmers, J.M., Griffiths, P.R., Eds.; Wiley: Chichester, UK, 2002; Volume 3, pp. 1817–1837. [Google Scholar]
- Dovesi, R.; Saunders, V.R.; Roetti, C.; Orlando, R.; Zicovich-Wilson, C.M.; Pascale, F.; Civalleri, B.; Doll, K.; Harrison, N.M.; Bush, I.J.; et al. CRYSTAL09 User’s Manual; University of Torino: Torino, Italy, 2009. [Google Scholar]
- Cambridge Structural Database. Available online: https://www.ccdc.cam.ac.uk/ (accessed on 20 January 2021).
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Gatti, C.; Saunders, V.R.; Roetti, C. Crystal-field effects on the topological properties of the electron-density in molecular-crystals. The case of urea. J. Chem. Phys. 1994, 101, 10686–10696. [Google Scholar] [CrossRef]
- Grabska, J.; Czarnecki, M.A.; Beć, K.B.; Ozaki, Y. Spectroscopic and quantum mechanical calculation study of the effect of isotopic substitution on NIR spectra of methanol. J. Phys. Chem. A 2017, 121, 7925–7936. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.02; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Barone, V.; Biczysko, M.; Bloino, J. Fully anharmonic IR and Raman spectra of medium-size molecular systems: Accuracy and interpretation. J. Phys. Chem. Chem. Phys. 2014, 16, 1759–1787. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parameterization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S. Density functional theory with London dispersion corrections. WIREs 2011, 1, 211–228. [Google Scholar] [CrossRef]
- Risthaus, T.; Steinmetz, M.; Grimme, S. Implementation of nuclear gradients of range-separated hybrid density functionals and benchmarking on rotational constants for organic molecules. J. Comput. Chem. 2014, 35, 1509. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Steinmetz, M. Effects of London dispersion correction in density functional theory on the structures of organic molecules in the gas phas. Phys. Chem. Chem. Phys. 2013, 15, 16031–16042. [Google Scholar] [CrossRef]
- Folmsbee, D.; Hutchison, G. Assessing conformer energies using electronic structure and machine learning methods. Quantum Chem. 2021, 121, e26381. [Google Scholar] [CrossRef]
- Beć, K.B.; Futami, Y.; Wójcik, M.J.; Ozaki, Y. A spectroscopic and theoretical study in the near-infrared region of low concentration aliphatic alcohols. Phys. Chem. Chem. Phys. 2016, 18, 13666–13682. [Google Scholar] [CrossRef]
- Bradley, M.S. Lineshapes in IR and Raman spectroscopy: A primer. Spectroscopy 2015, 30, 42–46. [Google Scholar]
- Yoshida, H.; Ehara, A.; Matsuura, H. Density functional vibrational analysis using wavenumber-linear scale factors. Chem. Phys. Lett. 2000, 325, 477–483. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental Position (int.) (a) | B3LYP/”Gatti” | B3LYP/TZVP | Position Difference (Exp—Calc) | Assignment (c) | |||
|---|---|---|---|---|---|---|---|
| Position | Int. (b) | Position | Int. (b) | B3LYP/”Gatti” | B3LYP/TZVP | ||
| 1664 (vs) | 1692 | 82.5 | 1665 | 100.0 | −28 | −1 | δring, νC=O |
| 1652 (vs) | 1679 | 100.0 | - | - | −27 | - | δring, νC=O |
| 1623 (m) | 1645 | 33.3 | 1632 | 26.5 | −22 | −9 | δring (νCC), νC=O |
| 1590 (s) | 1609 | 49.1 | 1601 | 89.7 | −19 | −11 | δring, δCH |
| 1461 (w) | 1492 | 1.8 | 1484 | 11.8 | −31 | −23 | δring, δCH, δasCH3 |
| 1440 (w) | 1472 | 7.0 | 1471 | 5.9 | −32 | −31 | δasCH3 |
| 1425 (w) | 1459 | 5.3 | 1413 | 8.8 | −34 | 12 | δasCH3 |
| 1380 (m) | 1409 | 8.8 | 1372 | 17.6 | −29 | 8 | δsCH3 |
| 1353 (m) | 1368 | 12.3 | 1351 | 8.8 | −15 | 2 | δring, δCH |
| 1329 (m) | 1356 | 3.5 | - | - | −27 | - | δring, δCH |
| 1299 (s) | 1310 | 66.7 | 1314 | 41.2 | −11 | −15 | δring, δCH |
| 1264 (m) | 1274 | 50.9 | 1281 | 44.1 | −10 | −17 | δring, δCH |
| 1238 (w) | 1241 | 5.3 | 1251 | 4.4 | −3 | −13 | δring, δCH |
| 1169 (w) | 1187 | 1.8 | 1190 | 4.4 | −18 | −21 | δring, δCH |
| 1156 (m) | 1177 | 1.8 | 1177 | 7.4 | −21 | −21 | δring, δCH |
| 1107 (vw) | 1126 | 14.0 | 1130 | 2.9 | −19 | −23 | δring, δCH |
| 1030 (vw) | 1055 | 3.5 | 1065 | 1.5 | −25 | −35 | γCH, ρCH3 |
| 1019 (w) | 1029 | 3.5 | 1040 | 7.4 | −10 | −21 | γCH, ρCH3 |
| 940 (w) | 950 | 5.3 | 961 | 5.9 | −10 | −21 | δring |
| 900 (w) | 930 | 10.5 | 935 | 5.9 | −30 | −35 | γring, γCH, ρCH3 |
| 883 (w) | 919 | 7.0 | - | - | −36 | - | γring, γCH, ρCH3 |
| 791 (w) | 807 | 12.3 | 809 | 8.8 | −16 | −18 | γring, γCH |
| 777 (s) | 798 | 12.3 | 795 | 5.9 | −21 | −18 | γring, γCH |
| 720 (w) | 722 | 3.5 | 727 | 1.5 | −2 | −7 | δring |
| 690 (m) | 703 | 10.5 | 694 | 14.7 | −13 | −4 | γring |
| 664 (m) | 685 | 5.3 | 676 | 8.8 | −21 | −12 | γring |
| 650 (vw) | 657 | 1.8 | 665 | 2.9 | −7 | −15 | δring |
| 642 (vw) | 651 | 1.8 | 654 | 1.5 | −9 | −12 | δring |
| 514 (w) | 524 | 3.5 | 523 | 1.5 | −10 | −9 | δring |
| 459 (w) | 464 | 3.5 | 468 | 1.5 | −5 | −9 | δring |
| 405 (w) | 412 | 5.3 | 414 | 4.4 | −7 | −9 | δring |
| Experimental Position | Exp. Intensity (Normalized) | Calc. Position | Calc. Intensity (Normalized) | Position Difference (Exp—Calc) | Assignment (a) |
|---|---|---|---|---|---|
| - | 3376 | 11.0 | - | 2(δring, νC=O) | |
| 3310 | 40.7 | 3360 | 35.1 | −50 | (δring, νC=O) + (δring, νC=O); 2(δring, νC=O) |
| 3272 | 30.4 | 3331 | 10.2 | −59 | (δring, νC=O) + (δring, νC=O) |
| 3234 | 11.9 | 3285 | 3.5 | −51 | 2(δring, νC=O) |
| 3183 | 16.7 | 3183 | 8.8 | 0 | (δring, δCH) + (δring, δCH) |
| 3173 | 19.8 | 3167 | 22.9 | 6 | 2(δring, δCH) |
| 3152 | 19.9 | 3141 | 15.9 | 11 | δCH + (δring, νC=O) |
| 3126 | 27.7 | 3127 | 14.3 | −1 | δCH + (δring, νC=O) |
| 3095 | 54.1 | 3105 | 12.9 | −10 | δCH + (δring, δCH) |
| 3075; 3070 | 88.0; 100.0 | 3063 | 100.0 | 12; 7 | νCH; δsymCH3 + (δring, νC=O) |
| 3046; 3038 | 64.1; 65.6 | 3037 | 60.2 | 9; 1 | νCH |
| 3017; 3009 | 43.5; 45.8 | 3003 | 21.6 | 14; 6 | νCH |
| 2979 | 50.6 | 2984 | 49.1 | −5 | νasCH3 |
| 2958 | 64.5 | 2942 | 51.6 | 16 | νasCH3 |
| 2920 | 79.5 | 2934 | 54.9 | −14 | νsCH3 |
| 2892 | 21.9 | 2897 | 8.7 | −5 | δring + (δring, δCH) |
| 2878 | 20.2 | 2886 | 7.9 | −8 | (δring, δCH) + (δring, δCH) |
| ~2860 | 16.1 | 2861 | 10.9 | −1 | 2δasymCH3 |
| 2842 | 21.8 | 2832 | 12.4 | 10 | 2δasymCH3 |
| 2813 | 9.3 | 2817 | 6.7 | −4 | δsymCH3 + δasymCH3 |
| Experimental Position | Calc. Position | Calc. Intensity (a) | Position Difference (Exp—Calc) | Assignment (b) |
|---|---|---|---|---|
| 3998 | 3994 | 1.6 | 4 | (δring, ρCH3) + νasCH3 |
| 3970 | 3981 | 1.5 | −11 | δring + νCH |
| 3951 | 3956 | 1.5 | −5 | ρCH3 + νsCH3 |
| 3940 | 3933 | 0.9 | 7 | (δring, ρCH3) + νsCH3 |
| 3868 | 3854 | 1.6 | 14 | (δring, ρCH3) + νCH |
| 3859 | 3834 | 2.5 | 25 | (δring, ρCH3) + νCH |
| 3802; 3788 | 3781 | 0.6 | 21; 7 | δring + νCH |
| 3748 | 3755 | 0.9 | −7 | δring + νCH |
| 3718 | 3721 | 1.2 | −3 | δring + νCH |
| 3687 | 3694 | 0.8 | −7 | δring + νCH |
| 3663 | 3655 | 0.2 | 8 | δring + νasCH3 |
| 3627 | 3635 | 0.2 | −8 | δring + νasCH3 |
| 3610 | 3620 | 0.4 | −10 | (γring, ρCH3) + νasCH3 |
| Experimental Position | Calc. Position | Calc. Intensity (a) | Position Difference (Exp—Calc) | Assignment (b) |
|---|---|---|---|---|
| 2781 | 2780 | 1.1 | 1 | δasymCH3 + (δring, δCH) |
| 2759 | 2750 | 1.9 | 9 | (δring, δCH) + δCH |
| 2747 | 2743 | 3.6 | 4 | 2δsymCH3 |
| 2727 | 2737 | 1.6 | −10 | (δring, δCH) + δCH |
| 2711 | 2723 | 1.6 | −12 | δsymCH3 + (δring, δCH) |
| 2702 | 2713 | 2.1 | −11 | (δring, δCH) + δCH |
| 2680 | 2687 | 4.4 | −7 | (δring, δCH) + (δring, δCH) |
| 2655 | 2661 | 3.4 | −6 | (δring, ρCH3) + (δring, νC=O) |
| 2625 | 2652 | 5.5 | −27 | 2δring |
| 2600 | 2633 | 1.5 | −33 | (δring, δCH) + (δring, δCH) |
| 2586 | 2616 | 2.4 | −30 | (δring, δCH) + δring |
| 2564 | 2603 | 2.8 | −39 | (δring, δCH) + (δring, δCH) |
| 2550 | 2580 | 3.4 | −30 | 2(δring, δCH) |
| 2536 | 2547 | 2.3 | −11 | (δring, δCH) + δsymCH3 |
| 2505 | 2506 | 1.6 | −1 | (δring, δCH) + (δring, δCH) |
| 2489 | 2495 | 2.7 | −6 | (δring, δCH) + (δring, δCH) |
| 2456 | 2462 | 1.2 | −6 | ρCH3 + δasymCH3 |
| 2443 | 2451 | 2.3 | −8 | (δring, ρCH3) + δasymCH3 |
| 2414 | 2432 | 2.2 | −18 | (δring, δCH) + (δring, δCH) |
| 2396 | 2418 | 1.6 | −22 | ρCH3 + δsymCH3 |
| 2378 | 2381 | 2.1 | −3 | (δring, δCH) + (δring, δCH) |
| 2371 | 2370 | 4.1 | 1 | (δring, δCH) + (δring, δCH) |
| 2330 | 2333 | 4.1 | −3 | 2(δring, δCH) |
| 2318 | 2323 | 2.9 | −5 | 2(δring, δCH); (δring, δCH) + (δring, δCH) |
| 2292 | 2298 | 1.3 | −6 | δring (breath) + (δring, δCH) |
| 2275 | 2282 | 2.1 | −7 | δring + (δring, δCH) |
| 2270 | 2275 | 2.1 | −5 | (δring, δCH) + (δring, δCH) |
| 2250 | 2255 | 2.2 | −5 | (δring, δCH) + (δring, δCH) |
| 2223 | 2217 | 1.8 | 6 | 2(δring, δCH) |
| 2203 | 2204 | 2.8 | −1 | δring + (δring, δCH) |
| 2188 | 2176 | 2.3 | 12 | δring + (δring, δCH) |
| 2172 | 2163 | 1.8 | 9 | δring + (δring, νC=O) |
| 2143 | 2140 | 3.1 | 3 | (δring, ρCH3) + (δring, δCH) |
| 2111 | 2123 | 1.6 | −12 | (δring, ρCH3) + (δring, δCH) |
| 2114 | 2110 | 1.2 | 4 | (δring, ρCH3) + (δring, δCH) |
| 2098 | 2089 | 2.3 | 9 | (δring, ρCH3) + (δring, δCH) |
| 2093 | 2080 | 1.5 | 13 | 2ρCH3 |
| 2074 | 2057 | 1.9 | 17 | δring + (δring, δCH) |
| 2058 | 2052 | 1.6 | 6 | δring + (δring, νC=O) |
| 2029 | 2030 | 1.4 | −1 | (δring, ρCH3) + (δring, δCH) |
| 2008 | 2009; 1997 | 4.6/9.0 | −1; 11 | δring + (δring, δCH); 2γCH |
| 1981 | 1977 | 12.0 | 4 | γCH + γCH |
| 1954 | 1959 | 3.5 | −5 | 2γCH |
| 1946 | 1943 | 2.2 | 3 | δring + (δring, δCH) |
| 1903 | 1913; 1905 | 5.0/6.8 | −10; −2 | γCH + γCH |
| 1877 | 1892; 1884 | 6.5/9.1 | −15; −7 | γCH + γCH |
| 1840 | 1860 | 1.9 | −20 | δring + (δring, δCH) |
| 1825 | 1824 | 5.6 | 1 | 2γCH |
| 1810 | 1817; 1807 | 7.8/9.0 | −7; 3 | γCH + γCH; δring + (δring, δCH) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beć, K.B.; Grabska, J.; Huck, C.W.; Mazurek, S.; Czarnecki, M.A. Anharmonicity and Spectra–Structure Correlations in MIR and NIR Spectra of Crystalline Menadione (Vitamin K3). Molecules 2021, 26, 6779. https://doi.org/10.3390/molecules26226779
Beć KB, Grabska J, Huck CW, Mazurek S, Czarnecki MA. Anharmonicity and Spectra–Structure Correlations in MIR and NIR Spectra of Crystalline Menadione (Vitamin K3). Molecules. 2021; 26(22):6779. https://doi.org/10.3390/molecules26226779
Chicago/Turabian StyleBeć, Krzysztof B., Justyna Grabska, Christian W. Huck, Sylwester Mazurek, and Mirosław A. Czarnecki. 2021. "Anharmonicity and Spectra–Structure Correlations in MIR and NIR Spectra of Crystalline Menadione (Vitamin K3)" Molecules 26, no. 22: 6779. https://doi.org/10.3390/molecules26226779