
ChCl: Gly (DESs) Promote Environmentally Benign Synthesis of Xanthene Derivatives and Their Antitubercular Activity

 , , ,
, , , 
Abstract
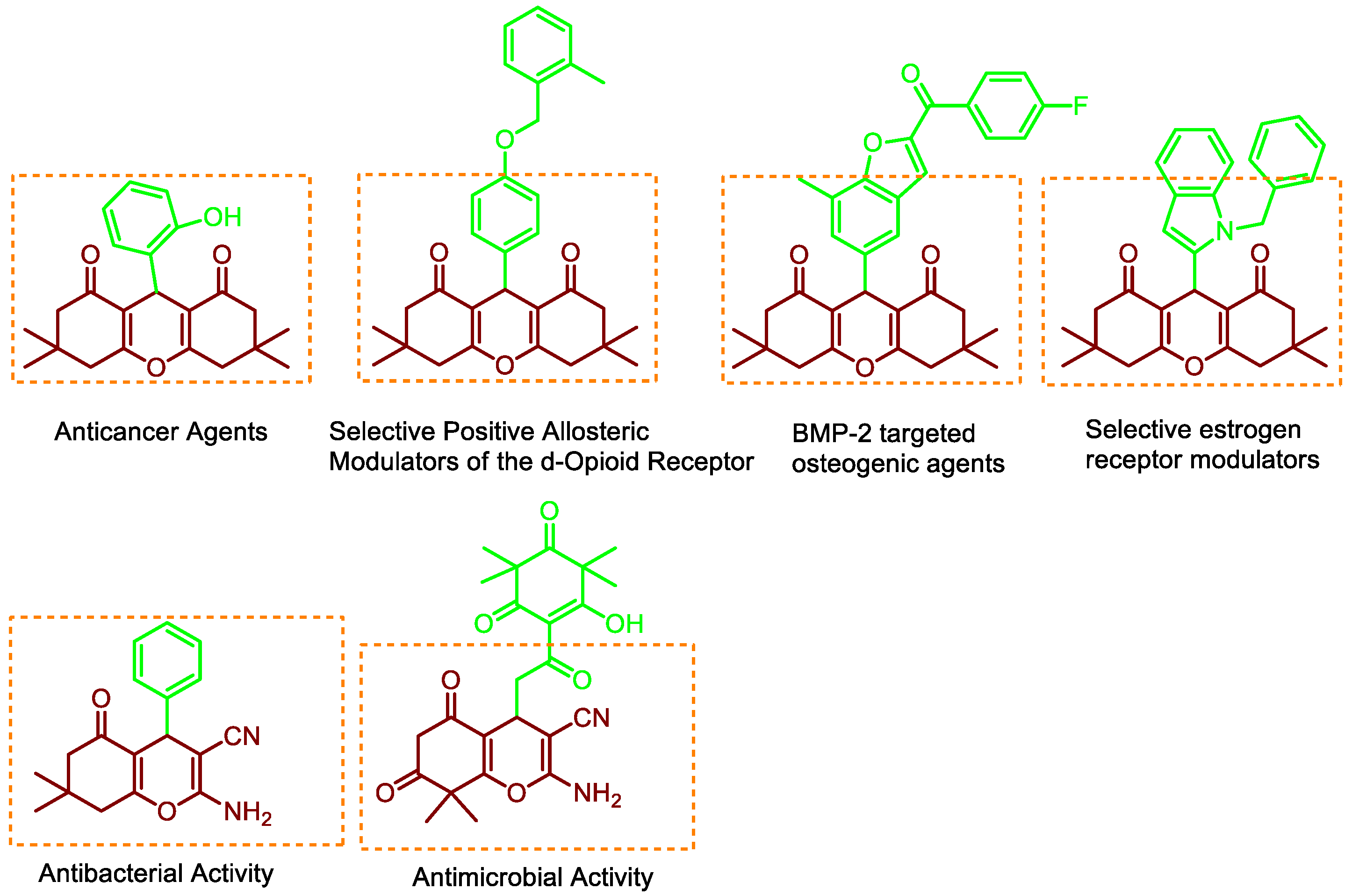
:1. Introduction
2. Materials and Methods
2.1. Preparation of ChCl: Gly
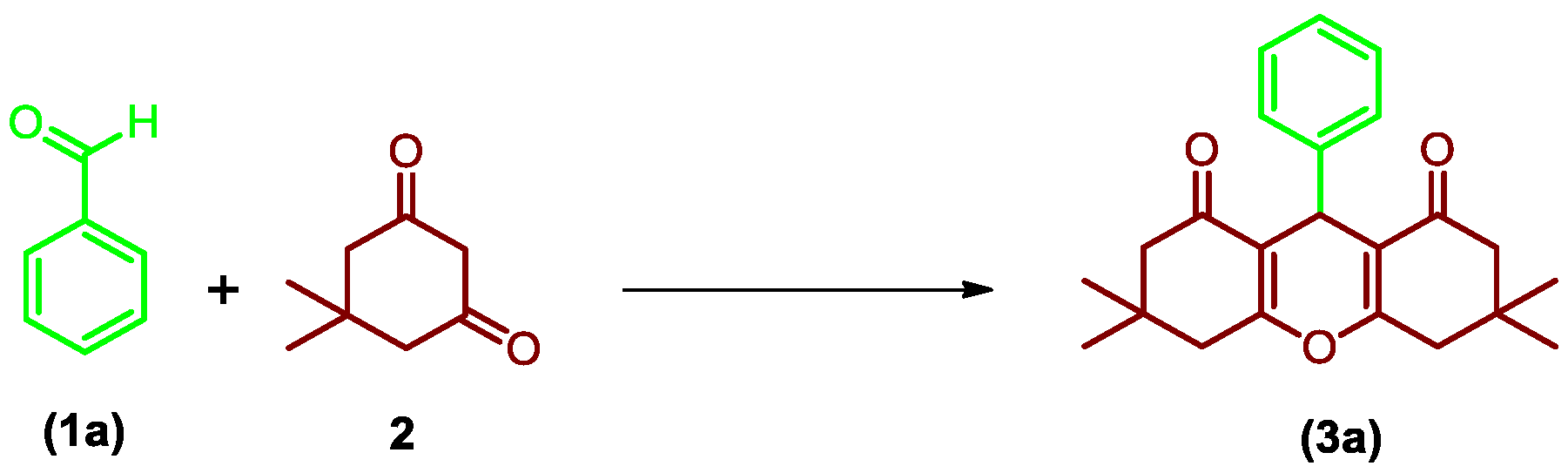
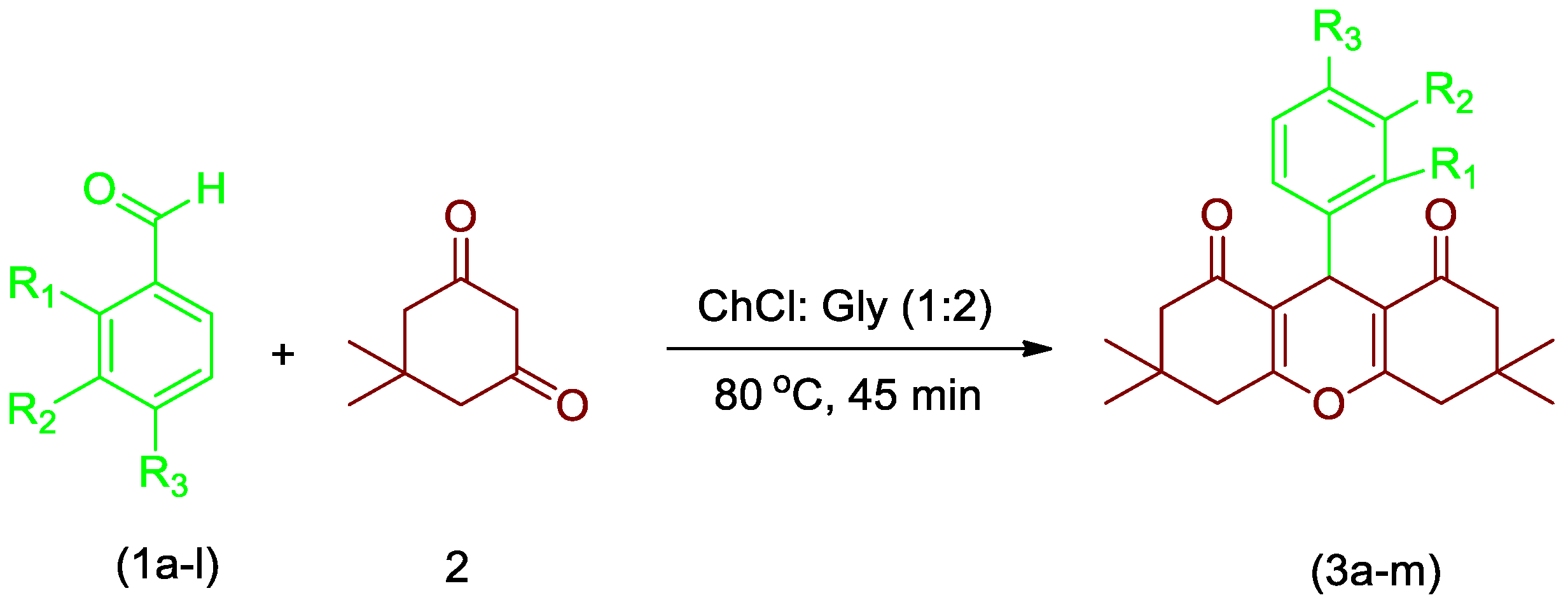
2.2. General Procedure for Synthesis of Xanthene Derivatives
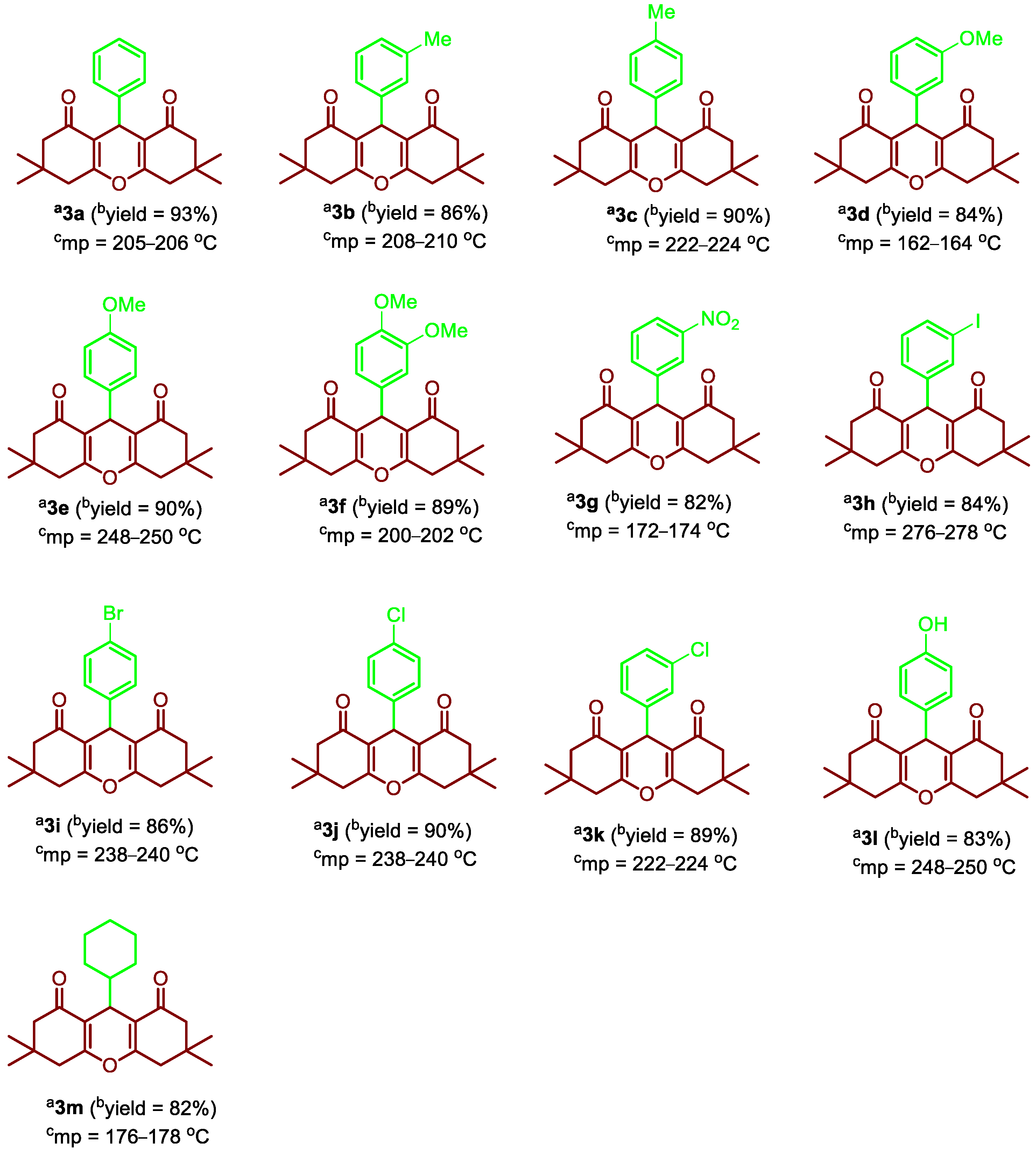
2.2.1. 3,3,6,6-Tetramethyl-9-phenyl-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (3a)
2.2.2. 3,3,6,6-Tetramethyl-9-(m-tolyl)-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (3b)
2.2.3. 3,3,6,6-Tetramethyl-9-(p-tolyl)-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (3c)
2.2.4. 9-(3-Methoxyphenyl)-3,3,6,6-tetramethyl-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (3d)
2.2.5. 9-(4-Methoxyphenyl)-3,3,6,6-tetramethyl-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (3e)
2.2.6. 9-(3,4-Dimethoxyphenyl)-3,3,6,6-tetramethyl-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (3f)
2.2.7. 3,3,6,6-Tetramethyl-9-(3-nitrophenyl)-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (3g)
2.2.8. 9-(3-Iodophenyl)-3,3,6,6-tetramethyl-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (3h)
2.2.9. 9-(4-Bromophenyl)-3,3,6,6-tetramethyl-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (3i)
2.2.10. 9-(4-Chlorophenyl)-3,3,6,6-tetramethyl-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (3j)
2.2.11. 9-(4-Hydroxyphenyl)-3,3,6,6-tetramethyl-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (3l)
2.2.12. 9-Cyclohexyl-3,3,6,6-tetramethyl-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (3m)
3. Results and Discussion
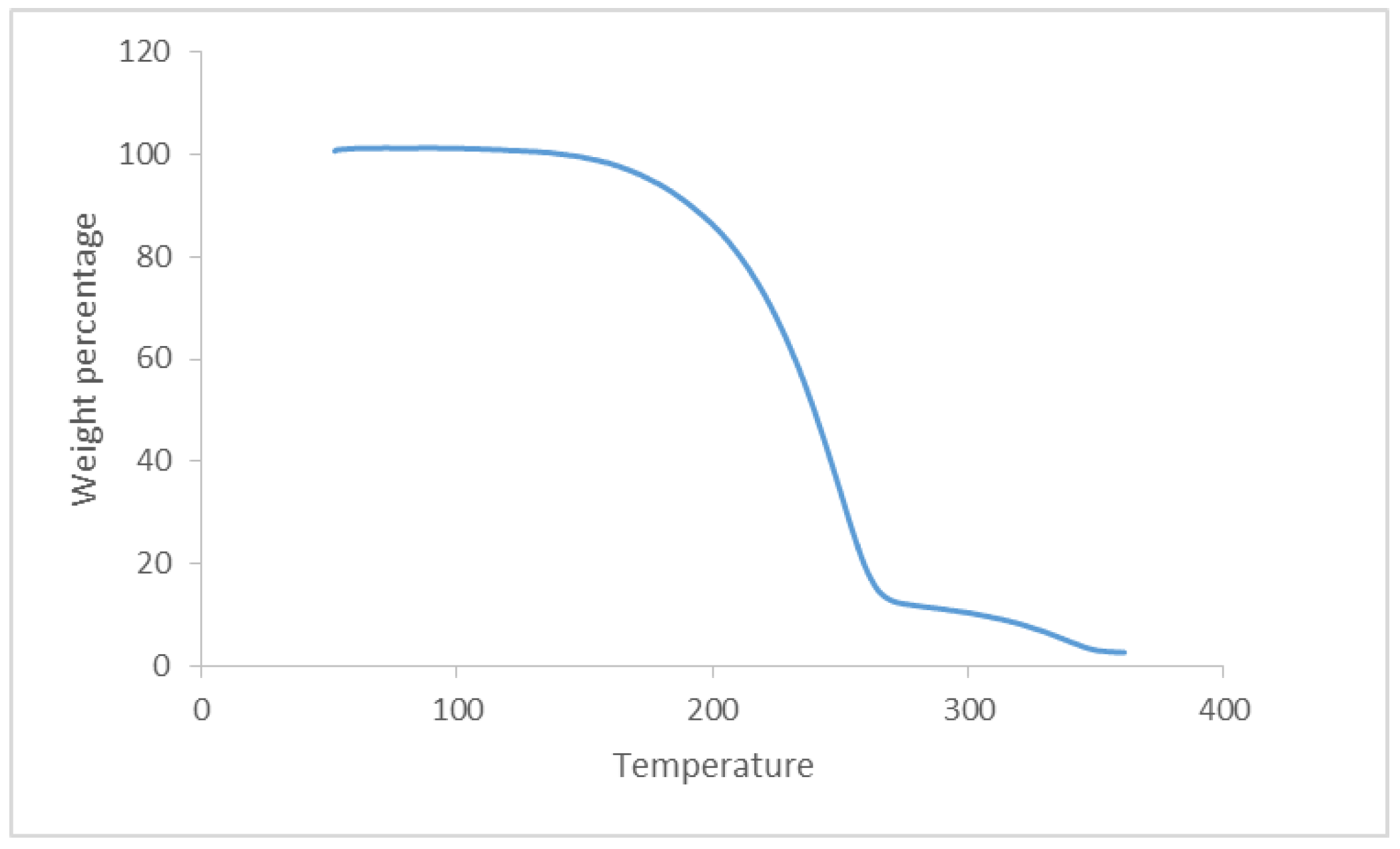
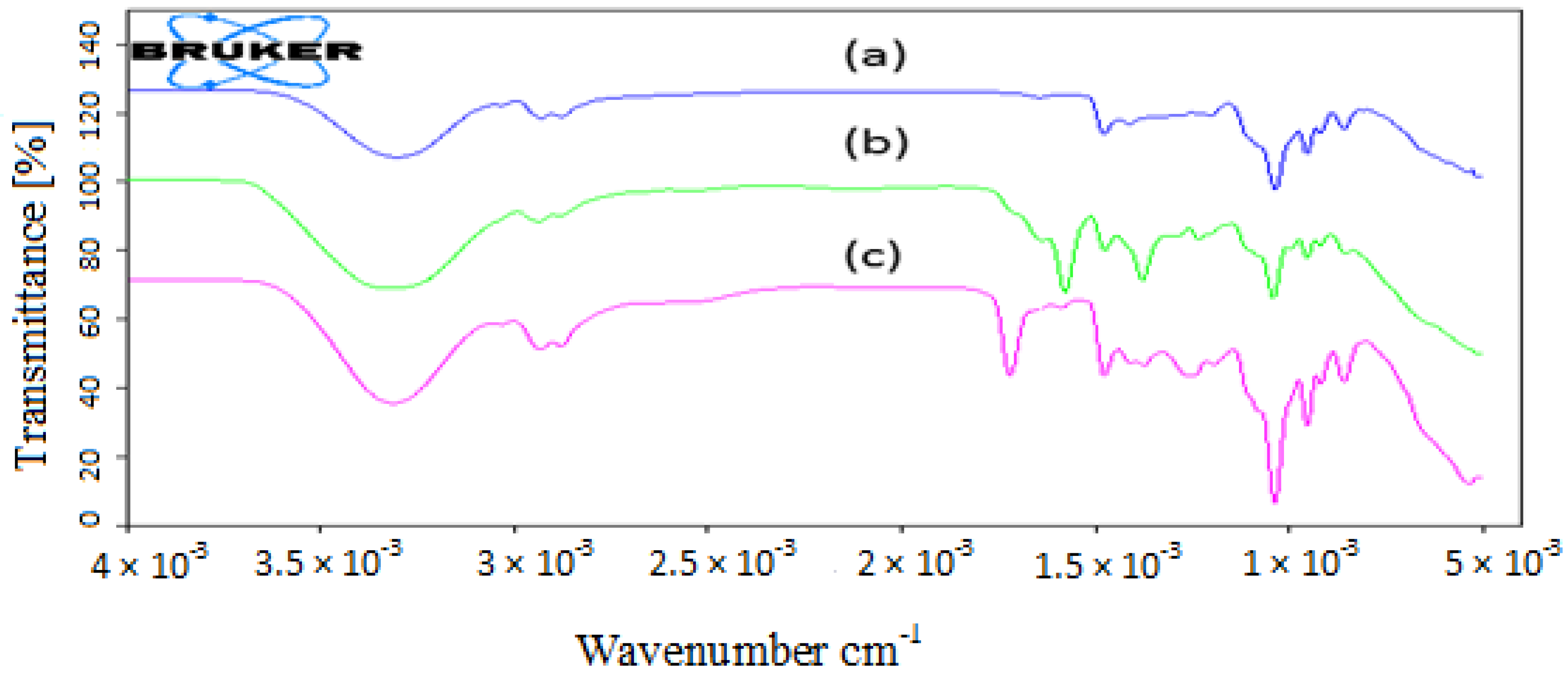
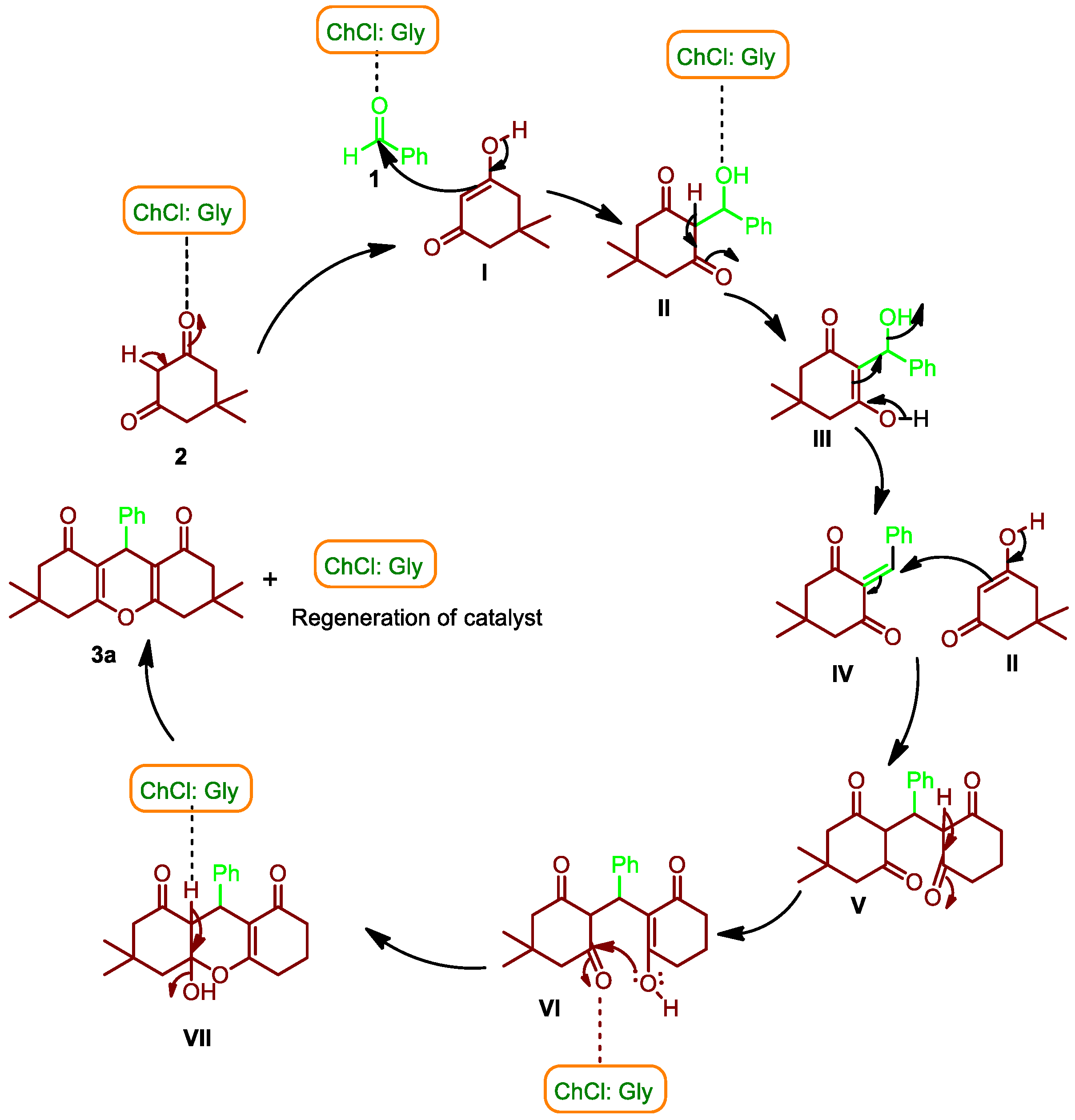
3.1. Chemistry
3.2. Biological Activity
3.2.1. Antitubercular Activity Screening
3.2.2. Structure–Activity Relationship (SAR)
3.3. Cytotoxicity
3.4. Selectivity Index (SI)
3.5. Antibacterial Activity
3.6. ADME Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Diana, I.S.P.R.; Duraes, F.; Maia, M.; Emilia, S.; Madalena, M.M.P. Recent advances in the synthesis of xanthones and azaxanthones. Org. Chem. Front. 2020, 7, 3027–3066. [Google Scholar] [CrossRef]
- Smith, E.L.; Abbott, A.P.; Ryder, K.S. Deep eutectic solvents (DESs) and their applications. Chem. Rev. 2014, 114, 11060–11082. [Google Scholar] [CrossRef] [Green Version]
- Abbott, A.P.; Boothby, D.; Capper, G.; Davies, D.L.; Rasheed, R.K. Deep eutectic solvents formed between choline chloride and carboxylic acids: Versatile alternatives to ionic liquids. J. Am. Chem. Soc. 2004, 9, 9142–9147. [Google Scholar] [CrossRef] [PubMed]
- Fei, H.; Li, P.; Gu, Y.; Li, G. Glycerol as a promoting medium for electrophilic activation of aldehydes: Catalyst-free synthesis of di(indolyl)methanes, xanthene-1,8(2H)-diones and 1-oxo-hexahydroxanthenes. Green Chem. 2009, 11, 1767–1773. [Google Scholar]
- Phadtare, S.B.; Shankarling, G.S. Greener coumarin synthesis by Knoevenagel condensation using biodegradable choline chloride. Environ. Chem. Lett. 2012, 10, 363–368. [Google Scholar] [CrossRef]
- Singh, A.S.; Shendage, S.S.; Nagarkar, J.M. Choline chloride based deep eutectic solvent as an efficient solvent for the benzylation of phenols. Tetrahedron Lett. 2014, 55, 7243–7246. [Google Scholar] [CrossRef]
- Azizi, N.; Dezfooli, S.; Mahmoudi, M. Greener synthesis of spiro oxindole in deep eutectic solvent. J. Mol. Liq. 2014, 194, 62–67. [Google Scholar] [CrossRef]
- Handy, S.; Wright, M. Organic synthesis in deep eutectic solvents: Paal-Knorr reactions. Tetrahedron Lett. 2014, 54, 4377–4379. [Google Scholar] [CrossRef]
- Wang, P.; Ma, F.; Zhang, Z. l-(+)-Tartaric acid and choline chloride based deep eutectic solvent: An efficient and reusable medium for synthesis of N-substituted pyrroles via Clauson-Kaas reaction. J. Mol. Liq. 2015, 198, 259–262. [Google Scholar] [CrossRef]
- Pawar, P.M.; Jarag, K.J.; Shankarling, G.S. Environmentally benign and energy efficient methodology for condensation: An interesting facet to the classical Perkin reaction. Green Chem. 2011, 13, 2130–2134. [Google Scholar] [CrossRef]
- Singh, B.; Lobo, H.; Shankarling, G. Selective N-Alkylation of Aromatic Primary Amines Catalyzed by Bio-catalyst or Deep Eutectic Solvent. Catal. Lett. 2011, 141, 178–182. [Google Scholar] [CrossRef]
- Shahbaz, K.; Mjalli, F.S.; Hashim, M.A.; Alnashef, I.M. Eutectic solvents for the removal of residual palm oil-based biodiesel catalyst. Sep. Purif. Technol. 2011, 81, 216–222. [Google Scholar] [CrossRef]
- Zahrina, I.; Nasikin, M.; Krisanti, E.; Mulia, K. Deacidification of palm oil using betaine monohydrate-based natural deep eutectic solvents. Food Chem. 2018, 240, 490–495. [Google Scholar] [CrossRef]
- Azizi, N.; Ahooie, T.S.; Hashemi, M.M. Multicomponent domino reactions in deep eutectic solvent: An efficient strategy to synthesize multisubstituted cyclohexa-1,3-dienamines. J. Mol. Liq. 2017, 246, 221–224. [Google Scholar] [CrossRef]
- Xing, W.; Xu, G.; Dong, J.; Han, R.; Ni, Y. Novel dihydrogen-bonding deep eutectic solvents: Pretreatment of rice straw for butanol fermentation featuring enzyme recycling and high solvent yield. Chem. Eng. J. 2018, 333, 712–720. [Google Scholar] [CrossRef]
- Gonzalez, C.G.; Mustafa, N.R.; Wilson, E.G.; Verpoorte, R.; Choi, Y.H. Application of natural deep eutectic solvents for the “green” extraction of vanillin from vanilla pods. Flavour Fragr. J. 2018, 33, 91–96. [Google Scholar] [CrossRef] [Green Version]
- Ji, Y.; Hou, Y.; Ren, S.; Yao, C.; Wu, W. Highly efficient extraction of phenolic compounds from oil mixtures by trimethylamine-based dicationic ionic liquids via forming deep eutectic solvents. Fuel Process. Technol. 2018, 171, 183–191. [Google Scholar] [CrossRef]
- Rappuoli, R. Changing route: Aerosol vaccine against tuberculosis. Lancet Infect. Dis. 2014, 14, 901–902. [Google Scholar] [CrossRef] [Green Version]
- Global Tuberculosis Report. Available online: http://www.who.int/tb/publications/global_report/en/ (accessed on 12 January 2021).
- Kana, B.D.; Karakousis, P.C.; Parish, T.; Dick, T. Future target-based drug discovery for tuberculosis? Tuberculosis 2014, 94, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Rout, L.; Achary, L.S.K.; Dhaka, R.S.; Dash, P. Greener route for synthesis of aryl and alkyl-14H-dibenzo [a.j] xanthenes using graphene oxide-copper ferrite nanocomposite as a recyclable heterogeneous catalyst. Sci. Rep. 2017, 7, 42975. [Google Scholar] [CrossRef]
- Chibale, K.; Visser, M.; van Schalkwyk, D.; Smith, P.J.; Saravanamuthu, A.; Fairlamb, A.H. Exploring the potential of xanthene derivatives as trypanothionereductase inhibitors and chloroquine potentiating agents. Tetrahedron 2003, 59, 2289–2296. [Google Scholar] [CrossRef]
- Jin, T.S.; Liu, L.B.; Zhao, Y.; Li, T.S. Clean synthesis of compounds containing two 4H-pyrans or two tetra ketones in aqueous media. Synth. Commun. 2005, 35, 2379–2385. [Google Scholar] [CrossRef]
- Hafez, H.N.; Hegab, M.I.; Ahmed-Farag, I.S.; El-Gazzar, A.B.A. A facile regioselective synthesis of novel spiro-thio xanthene and spiro-xanthene-9′,2-[1,3,4]thiadiazole derivatives as potential analgesic and anti-inflammatory agents. Bioorg. Med. Chem. Lett. 2008, 18, 4538–4543. [Google Scholar] [CrossRef]
- Ilangovan, A.; Anandhan, K.; Prasad, K.M.; Vijayakumar, P.; Renganathan, R.; Ananth, D.A.; Sivasudha, T. Synthesis, DNA-binding study, and antioxidant activity of 14-aryl-14H-dibenzo [a,j] xanthene derivatives. Med. Chem. Res. 2015, 24, 344–355. [Google Scholar] [CrossRef]
- Spatafora, C.; Barresi, V.; Bhusainahalli, V.M.; Di Micco, S.; Musso, N.; Riccio, R.; Bifulco, G.; Condorelli, D.; Tringali, C. Bio-inspired benzo[k,l]xanthene lignans: Synthesis, DNA-interaction and antiproliferative properties. Org. Biomol. Chem. 2014, 12, 2686–2701. [Google Scholar] [CrossRef]
- Sashidhara, K.V.; Kumar, A.; Dodda, R.P.; Kumar, B.A. New iodine catalyzed regioselective synthesis of xanthene synthons. Tetrahedron Lett. 2012, 53, 3281–3283. [Google Scholar] [CrossRef]
- Khurana, J.M.; Magoo, D.; Aggarwal, K.; Aggarwal, N.; Kumar, R.; Srivastava, C. Synthesis of novel 12-aryl-8,9,10,12-tetrahydrobenzo[a]xanthene-11-thiones and evaluation of their biocidal effects. Eur. J. Med. Chem. 2012, 58, 470–477. [Google Scholar] [CrossRef]
- Kushwaha, P.; Tripathi, A.K.; Gupta, S.; Kothari, P.; Upadhyay, A.; Ahmad, N.; Sharma, T.; Siddiqi, M.I.; Trivedi, R.; Sashidhara, K.V. Synthesis and study of benzofuran-pyran analogs as BMP-2 targeted osteogenic agents. Eur. J. Med. Chem. 2018, 156, 103–117. [Google Scholar] [CrossRef]
- Burford, N.T.; Livingston, K.E.; Canals, M.; Ryan, M.R.; Budenholzer, L.M.L.; Han, Y.; Shang, Y.; Herbst, J.J.; O’Connell, J.; Banks, M.; et al. Discovery, synthesis, and molecular pharmacology of selective positive allosteric modulators of the δ-opioid receptor. J. Med. Chem. 2015, 58, 4220–4229. [Google Scholar] [CrossRef]
- Ramit, S.; Gupta, K.B.; Upadhyay, S.; Dhiman, M.; Jaitak, V. Design, synthesis and biological evaluation of novel indole-xanthendione hybrids as selective estrogen receptor modulators. Bioorg. Med. Chem. 2018, 26, 266–277. [Google Scholar]
- Kantevari, S.; Bantu, R.; Nagarapu, L. HClO4-SiO2 and PPASiO2 catalyzed efficient one pot Knoevenagel condensation, Michael addition and cyclo-dehydration of dimedone and aldehydes in acetonitrile, aqueous and solvent free conditions: Scope and limitations. J. Mol. Catal. A Chem. 2007, 269, 53–57. [Google Scholar] [CrossRef]
- Rajitha, B.; Sunil Kumar, B.; Thirupathi Reddy, Y.; Narsimha Reddy, P.; Sreenivasulu, N. Sulfamic acid: A novel and efficient catalyst for the synthesis of aryl-14h-dibenzo [a.j] xanthenes under conventional heating and microwave irradiation. Tetrahedron Lett. 2005, 46, 8691–8693. [Google Scholar] [CrossRef]
- Prasad, D.; Preetam, A.; Nath, M. Microwave-assisted green synthesis of dibenzo[a,j]xanthenes using p-dodecylbenzenesulfonic acid as an efficient Bronsted acid catalyst under solvent-free conditions. Comptus Rendus Chim. 2012, 15, 675–678. [Google Scholar] [CrossRef]
- Karimi-Jaberi, Z.; Keshavarzi, M. Efficient one-pot synthesis of 14-substituted-14H-dibenzo[a,j]xanthenes using boric acid under solvent-free conditions. Chin. Chem. Lett. 2010, 21, 547–549. [Google Scholar] [CrossRef]
- Khosropour, A.; Khodaei, M.; Moghannian, H. A facile, simple and convenient method for the synthesis of 14-alkyl or aryl-14H-dibenzo[a,j]xanthenes Catalyzed by pTSA in Solution and Solvent Free Conditions. Synlett 2005, 6, 955–958. [Google Scholar] [CrossRef]
- Das, B.; Thirupathi, P.; Reddy, K.R.; Ravikanth, B.; Nagarapu, L. An efficient synthesis of 1,8-dioxo-octahydroxanthenes using heterogeneous catalysts. Catal. Commun. 2007, 8, 535–538. [Google Scholar] [CrossRef]
- Jin, T.S.; Zhang, J.S.; Wang, A.Q.; Li, T.S. Solid-state condensation reactions between aldehydes and 5,5-dimethyl-1,3-cyclohexanedione by grinding at room temperature. Synth. Commun. 2005, 35, 2339–2345. [Google Scholar] [CrossRef]
- Pasha, M.A.; Jayashankara, V.P. Molecular iodine catalyzed synthesis of aryl-14H-dibenzo[a,j]xanthenes under solvent-free condition. Bioorg. Med. Chem. Lett. 2007, 17, 621–623. [Google Scholar] [CrossRef] [Green Version]
- Song, G.; Wang, B.; Luo, H.; Yang, L. Fe3+-montmorilloniteas a cost-effective and recyclable solid acidic catalyst for the synthesis of xanthene diones. Catal. Commun. 2007, 8, 673–676. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Tao, X.Y. 2,4,6-Trichloro-1,3,5-triazinepromoted synthesis of 1,8-dioxo-octahydroxanthenes under solvent-free conditions. Aust. J. Chem. 2008, 61, 77–79. [Google Scholar] [CrossRef]
- Pourian, E.; Javanshir, S.; Dolatkhah, Z.; Molaei, S.; Maleki, A. Ultrasonic-assisted preparation, characterization, and use of novel biocompatible core/shell Fe3O4@GA@isinglass in the synthesis of 1,4-dihydropyridine and 4H-pyran derivatives. ACS Omega 2018, 3, 5012–5020. [Google Scholar] [CrossRef]
- Bhat, M.A.; Al-Omar, M.A.; Naglah, A.M.; Khan, A.A. [Et3NH][HSO4]-mediated efficient synthesis of novel xanthene derivatives and their biological evaluation. J. Saudi Chem. Soc. 2020, 24, 425–433. [Google Scholar] [CrossRef]
- Abbott, A.P.; Harris, R.C.; Ryder, K.S.; D’Agostino, C.; Gladden, L.F.; Mantle, M.D. Glycerol eutectics as sustainable solvent systems. Green Chem. 2011, 13, 82–90. [Google Scholar] [CrossRef]
- Liu, P.; Hao, J.W.; Mo, L.P.; Zhang, Z.H. Recent advances in the application of deep eutectic solvents as sustainable media as well as catalysts in organic reactions. RSC Adv. 2015, 5, 48675–48704. [Google Scholar] [CrossRef]
- Zhao, H.; Gary, A.B.; Holmes, S. Protease activation in glycerol-based deep eutectic solvents. J. Mol. Catal. B Enzym. 2011, 72, 163–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kokkirala, S.; Sabbavarapu, N.M.; Yadavalli, V.D.N. β-Cyclodextrin mediated synthesis of 1,8-dioxooctahydroxanthenes in water. Eur. J. Chem. 2011, 2, 272–275. [Google Scholar] [CrossRef] [Green Version]
- Niknam, K.; Panahi, F.; Saberi, D.; Mohagheghnejad, M. Silica-bonded S-sulfonic acid as recyclable catalyst for the synthesis of 1,8-dioxo-decahydroacridines and 1,8-dioxo-octahydroxanthenes. J. Heterocycl. Chem. 2010, 47, 292–300. [Google Scholar]
- Fan, X.S.; Li, Y.Z.; Zhang, X.Y.; Hu, X.Y.; Wang, J.J. FeCl3.6H2O Catalyzed Reaction of Aromatic Aldehydes with 5, 5-Dimethyl-1, 3-cyclohexandione in Ionic Liquids. Chin. Chem. Lett. 2005, 16, 897–899. [Google Scholar]
- Das, B.; Thirupathi, P.; Mahender, I.; Reddy, V.S.; Rao, Y.K. Amberlyst-15: An efficient reusable heterogeneous catalyst for the synthesis of 1, 8-dioxo-octahydroxanthenes and 1,8-dioxo-decahydroacridines. J. Mol. Catal. A Chem. 2006, 247, 233. [Google Scholar] [CrossRef]
- Oskooie, H.A.; Tahershamsi, L.; Heravi, M.M.; Baghernejad, B. Cellulose sulfonic acid: An efficient heterogeneous catalyst for the synthesis of 1, 8-Dioxo-octahydroxanthenes. Eur. J. Chem. 2010, 7, 717. [Google Scholar] [CrossRef]
- Niknam, K.; Damya, M.J. 1-Butyl-3-methylimidazolium Hydrogen Sulfate [Bmim]HSO4: An Efficient Reusable Acidic Ionic Liquid for the Synthesis of 1,8-Dioxo-Octahydroxanthenes. Chin. Chem. Soc. 2009, 56, 659–665. [Google Scholar] [CrossRef]
- Ghassamipour, S.; Ghashghaei, R. Zirconium dodecyl phosphonate promoted synthesis of xanthene derivatives by condensation reaction of aldehydes and β-naphthol or dimedone in green media. Monatsh. Chem. 2015, 146, 159–163. [Google Scholar] [CrossRef]
- Maghsoodlou, M.T.; Habibi-Khorassani, S.M.; Shahkarami, Z.; Maleki, N.; Rostamizadeh, M. An efficient synthesis of 2,2′-arylmethylene bis(3-hydroxy-5,5-dimethyl-2-cyclohexene-1-one) and 1,8-dioxooctahydroxanthenes using ZnO and ZnO-acetyl chloride. Chin. Chem. Lett. 2010, 21, 686–689. [Google Scholar] [CrossRef]
- Pardini, E.; Gagliardi, M.; Colone, M.C.; Stringaro, M.; Teloni, A.R.; Brunori, R.; Nisini, L.; Fattorini, R.; Giannoni, L. Infection of human THP-1 cells with dormant Mycobacterium tuberculosis. Microbes Infect. 2012, 14, 959–967. [Google Scholar]
- Van de Loosdrecht, A.A.; Beelen, R.H.; Ossenkoppele, G.J.; Broekhoven, M.G.; Langenhuijsen, M.M. A tetrazolium-based colorimetric MTT assay to quantitate human monocyte mediated cytotoxicity against leukemic cells from cell lines and patients with acute myeloid. J. Immunol. Methods 1994, 174, 311–320. [Google Scholar] [CrossRef]
- Hartkoorn, R.C.; Chandler, B.; Owen, A.; Ward, S.A.; Squire, S.B.; Back, D.J.; Khoo, S.H. Differential drug susceptibility of intracellular and extracellular tuberculosis, and the impact of P-glycoprotein. Tuberculosis 2007, 87, 248–255. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, L.; Dominy, B.W.; Freeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Molinspiration Chemoinformatics Brastislava, Slovak Republic. Available online: http://www.molinspiration.com/cgi-bin/properties (accessed on 12 January 2021).
- Zhao, Y.H.; Abraham, M.H.; Lee, J.; Hersey, A.; Luscombe, N.C.; Beck, G.; Sherborne, B.; Cooper, I. Rate-Limited Steps of Human Oral Absorption and QSAR Studies. Pharm. Res. 2002, 19, 1446–1457. [Google Scholar] [CrossRef] [PubMed]
- Drug-Likeness and Molecular Property Prediction. Available online: http://www.molsoft.com/mprop/ (accessed on 12 January 2021).
- Ertl, P.; Rohde, B.; Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
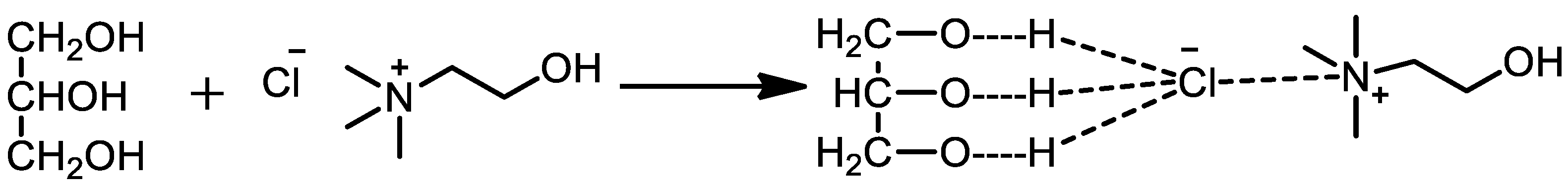{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Solvent | Temperature (°C) | Time (min) | Yield (%) b |
|---|---|---|---|---|
| 1 | - | 70 | 520 | - |
| 2 | ChCl:Urea(1:2) | 70 | 80 | 60 |
| 3 | ChCl:Thiourea(1:2) | 70 | 80 | 58 |
| 5 | ChCl:Adipic Acid (1:1) | 70 | 80 | 28 |
| 4 | ChCl:Oxalic Acid(1:1) | 70 | 80 | 63 |
| 7 | ChCl:Malonic Acid(1:1) | 70 | 80 | 38 |
| 6 | ChCl:Succinic Acid(1:1) | 70 | 80 | 46 |
| 9 | ChCl:ZnCl2 (1:2) | 70 | 85 | 30 |
| 8 | ChCl:FeCl3 (1:2) | 70 | 85 | 35 |
| 10 | ChCl:Gly (1:1) | 70 | 80 | 76 |
| 11 | ChCl:Gly (1:2) | 70 | 60 | 82 |
| 12 | ChCl:Gly (1:3) | 70 | 60 | 83 |
| 13 | ChCl:Gly (1:2) | 80 | 45 | 93 |
| 14 | ChCl:Gly (1:2) | 90 | 45 | 93 |
| 15 | Gly | 80 | 80 | 62 |
| 16 | ChCl | 80 | 520 | 25 |
| Entry | ChCl: Gly (g) | Time (min) | Temp (°C) | % Yield b |
|---|---|---|---|---|
| 1 | 0.5 | 45 | 80 | 28 |
| 2 | 1 | 45 | 80 | 58 |
| 3 | 1.5 | 45 | 80 | 78 |
| 4 | 2 | 45 | 80 | 93 |
| 5 | 2.5 | 45 | 80 | 94 |
| Entry | Run | Time (min) | % Yield b |
|---|---|---|---|
| 1 | Fresh | 45 | 93 |
| 2 | 1 | 45 | 93 |
| 3 | 2 | 45 | 91 |
| 4 | 3 | 45 | 89 |
| 5 | 4 | 45 | 85 |
| 6 | 5 | 45 | 82 |
| Entry | Catalyst | Amount of Catalyst | Time (min) | Yield (%) | Solvent/Condition | Ref. |
|---|---|---|---|---|---|---|
| 1 | β-CD (1 mmol) | 1 mmol | 10 h | 96 | EtOH/60 °C | [48] |
| 2 | SBSSA | 30 mg | 10 h | 98 | EtOH/reflux | [49] |
| 3 | FeCl3.6H2O (10 mol%) | 10 mol% | 6 h | 92 | [bmim][BF4]/80 °C | [50] |
| 4 | Amberlyst-15 | 200 mg | 5 h | 92 | CH3CN/Reflux | [51] |
| 5 | Cellulose sulfonic acid | 50 mg | 5 h | 94 | Solvent free/110 °C | [52] |
| 6 | [bmim][HSO4] (42 mol%) | 42 mol% | 3 h | 85 | Solvent free/100 °C | [40] |
| 7 | Fe3+ montmorillonite | 85 mg | 6 h | 94 | EtOH/reflux | [53] |
| 8 | Zr(DP)2 | 10 mol % | 24 h | 98 | EtOH/80 °C | [54] |
| 9 | ZnO-CH3COCl | 30 mol % | 5 h | 86 | CH3CN/reflux | [55] |
| 10 | ChCl:Gly | 2.0 g | 45 min | 93 | ChCl:Gly act as a solvent | Present work |
| Compound | MTB H37Ra | M. bovis BCG | ||
|---|---|---|---|---|
| a MIC | a IC50 | a MIC | a IC50 | |
| 3a | >30 | >30 | >30 | >30 |
| 3b | >30 | >30 | >30 | >30 |
| 3c | >30 | >30 | >30 | >30 |
| 3d | 15.10 | 3.20 | 14.92 | 3.40 |
| 3e | 4.20 | 0.74 | 0.26 | 0.28 |
| 3f | 2.5 | 0.79 | 1.22 | 0.32 |
| 3g | >30 | >30 | >30 | >30 |
| 3h | >30 | >30 | >30 | >30 |
| 3i | >30 | >30 | >30 | >30 |
| 3j | 4.74 | 0.50 | 2.58 | 0.6 |
| 3k | 7.8 | 2.3 | 9.4 | 5.8 |
| 3l | >30 | >30 | >30 | >30 |
| 3m | >30 | >30 | >30 | >30 |
| bRifampicin | 0.045 | 0.0017 | 0.017 | 0.0015 |
| Compound | MCF-7 | A549 | HCT 116 | THP-1 |
|---|---|---|---|---|
| 3d | >100 | >100 | >100 | >100 |
| 3e | >100 | >100 | >100 | >100 |
| 3f | >100 | >100 | >100 | >100 |
| 3j | >100 | >100 | >100 | >100 |
| 3k | >100 | >100 | >100 | >100 |
| Rifampicin | >100 | >100 | >100 | >100 |
| Paclitaxel | 0.0048 | 0.0035 | 0.0260 | 0.1374 |
| Compound | MCF-7 | A549 | HCT 116 | THP-1 | ||||
|---|---|---|---|---|---|---|---|---|
| MTB | BCG | MTB | BCG | MTB | BCG | MTB | BCG | |
| 3d | 7 | 7 | 7 | 7 | 7 | 7 | 7 | 7 |
| 3e | 20 | 330 | 20 | 333 | 20 | 330 | 20 | 330 |
| 3f | 44 | 82 | 42 | 82 | 42 | 82 | 44 | 82 |
| 3j | 18 | 40 | 18 | 40 | 18 | 40 | 18 | 40 |
| 3k | 14 | 12 | 14 | 12 | 14 | 12 | 14 | 12 |
| Rifampicin | 196 | 222 | 196 | 222 | 196 | 222 | 196 | 222 |
| Compound | E. coli | P. fluorescens | S. aureus | B. subtilis |
|---|---|---|---|---|
| 3d | >30 | >30 | >30 | >30 |
| 3e | >30 | >30 | >30 | >30 |
| 3f | >30 | >30 | >30 | >30 |
| 3j | >30 | >30 | >30 | >30 |
| 3k | >30 | >30 | >30 | >30 |
| Kanamycin | 3.34 ± 0.41 | 1.01 ± 0.09 | >61.92 | 2.78 ± 0.85 |
| Ampicillin | 4.17 ± 1.04 | 12.47 ± 1.28 | 2.86 ± 0.78 | 29.53 ± 1.88 |
| Com | % ABS | TPSA (A2) | n-ROTB | MV | MW | miLogP | n-ON | n-OHNH | Lipinski Violation | Drug Likeness Model Score |
|---|---|---|---|---|---|---|---|---|---|---|
| Rule | - | - | - | - | <500 | ≤5 | <10 | <5 | ≤1 | |
| 3a | 94.03 | 43.38 | 1 | 336.59 | 350.46 | 5.20 | 3 | 0 | 1 | −1.31 |
| 3b | 94.03 | 43.38 | 1 | 353.15 | 364.49 | 5.62 | 3 | 0 | 1 | −0.98 |
| 3c | 94.03 | 43.38 | 2 | 353.15 | 364.49 | 5.65 | 3 | 0 | 1 | −1.27 |
| 3d | 90.84 | 52.61 | 2 | 362.13 | 380.48 | 5.23 | 4 | 0 | 2 | −0.89 |
| 3e | 90.84 | 52.61 | 3 | 362.13 | 380.48 | 5.25 | 4 | 0 | 2 | −1.09 |
| 3f | 87.66 | 61.84 | 2 | 387.8 | 410.51 | 4.84 | 5 | 0 | 3 | 0.41 |
| 3g | 78.22 | 89.20 | 1 | 359.92 | 395.45 | 5.13 | 6 | 0 | 1 | −0.85 |
| 3h | 94.03 | 43.38 | 1 | 360.58 | 476.35 | 6.26 | 3 | 0 | 1 | −0.87 |
| 3i | 94.03 | 43.38 | 1 | 354.48 | 429.35 | 6.01 | 3 | 0 | 1 | −1.16 |
| 3j | 94.03 | 43.38 | 1 | 350.12 | 384.90 | 5.88 | 3 | 0 | 1 | −0.77 |
| 3k | 87.05 | 63.60 | 1 | 344.61 | 366.46 | 4.72 | 4 | 0 | 1 | −0.72 |
| 3l | 94.03 | 43.38 | 1 | 355.18 | 356.51 | 5.79 | 3 | 1 | 1 | −1.32 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhat, M.A.; Naglah, A.M.; Akber Ansari, S.; Al-Tuwajiria, H.M.; Al-Dhfyan, A. ChCl: Gly (DESs) Promote Environmentally Benign Synthesis of Xanthene Derivatives and Their Antitubercular Activity. Molecules 2021, 26, 3667. https://doi.org/10.3390/molecules26123667
Bhat MA, Naglah AM, Akber Ansari S, Al-Tuwajiria HM, Al-Dhfyan A. ChCl: Gly (DESs) Promote Environmentally Benign Synthesis of Xanthene Derivatives and Their Antitubercular Activity. Molecules. 2021; 26(12):3667. https://doi.org/10.3390/molecules26123667
Chicago/Turabian StyleBhat, Mashooq A., Ahmed M. Naglah, Siddique Akber Ansari, Hanaa M. Al-Tuwajiria, and Abdullah Al-Dhfyan. 2021. "ChCl: Gly (DESs) Promote Environmentally Benign Synthesis of Xanthene Derivatives and Their Antitubercular Activity" Molecules 26, no. 12: 3667. https://doi.org/10.3390/molecules26123667