
Application of Bar Adsorptive Microextraction for the Determination of Levels of Tricyclic Antidepressants in Urine Samples

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results and Discussion
2.1. LVI-GC-MS(SIM) Optimization
2.2. Optimization of the BAµE-µLD Efficiency
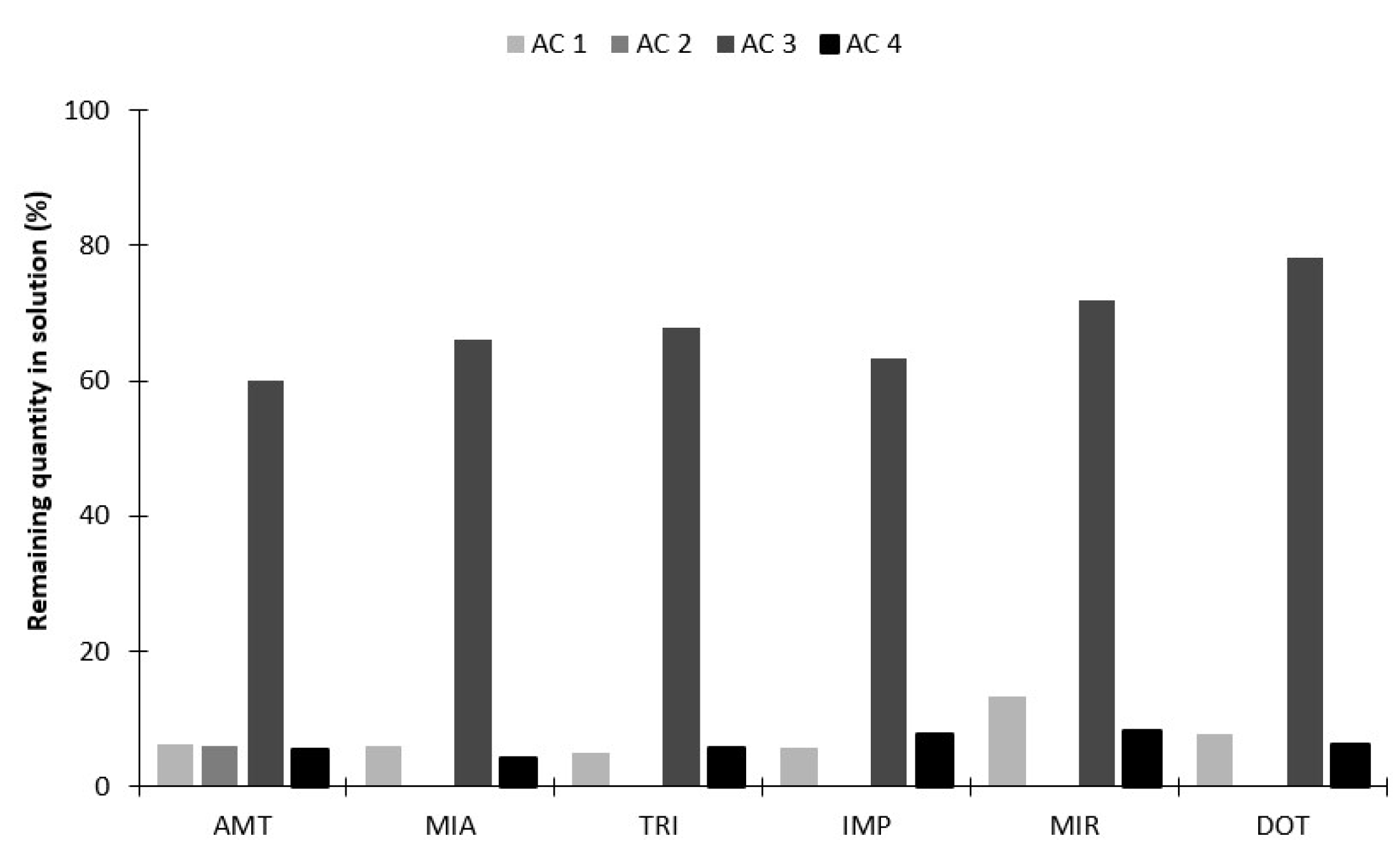
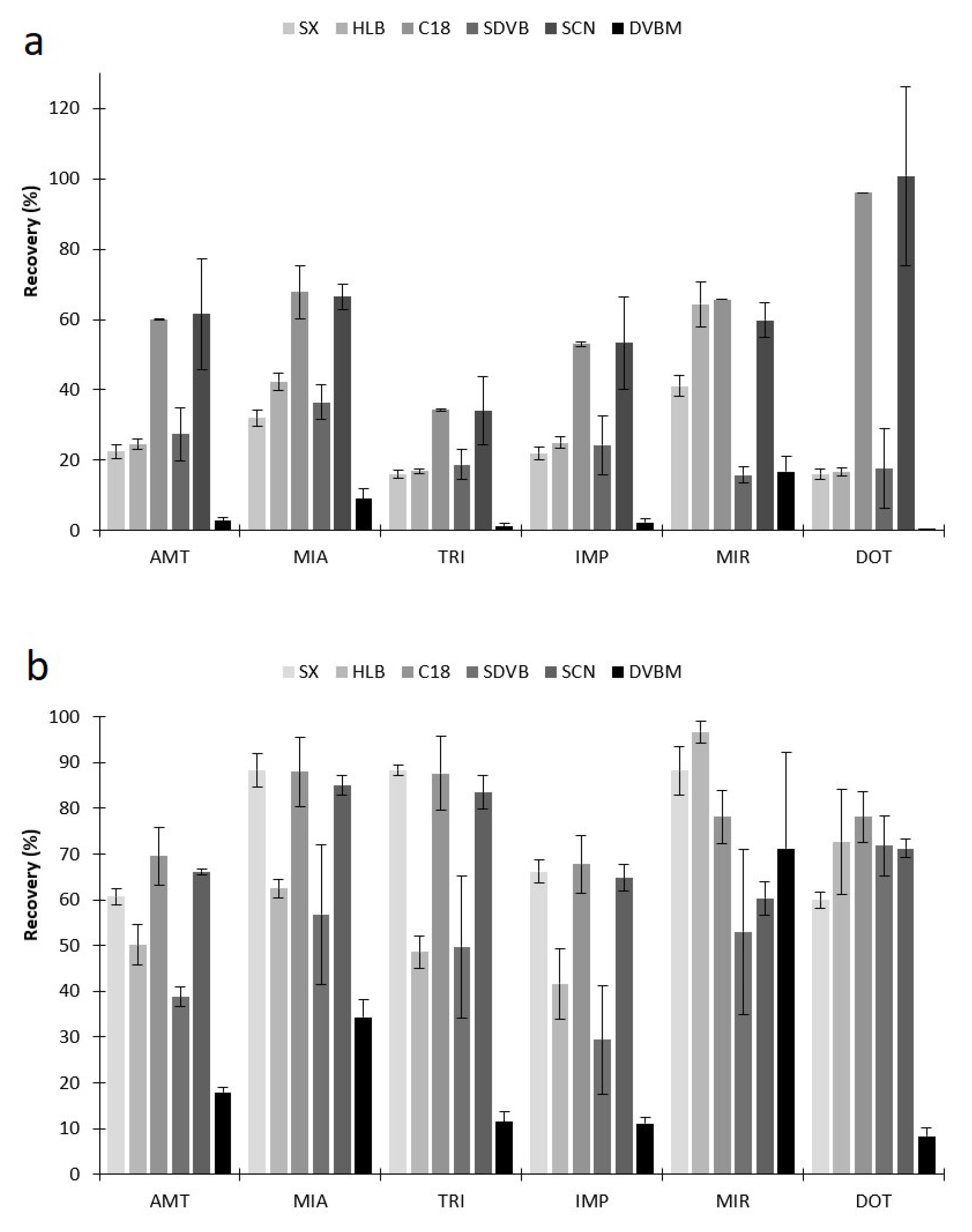
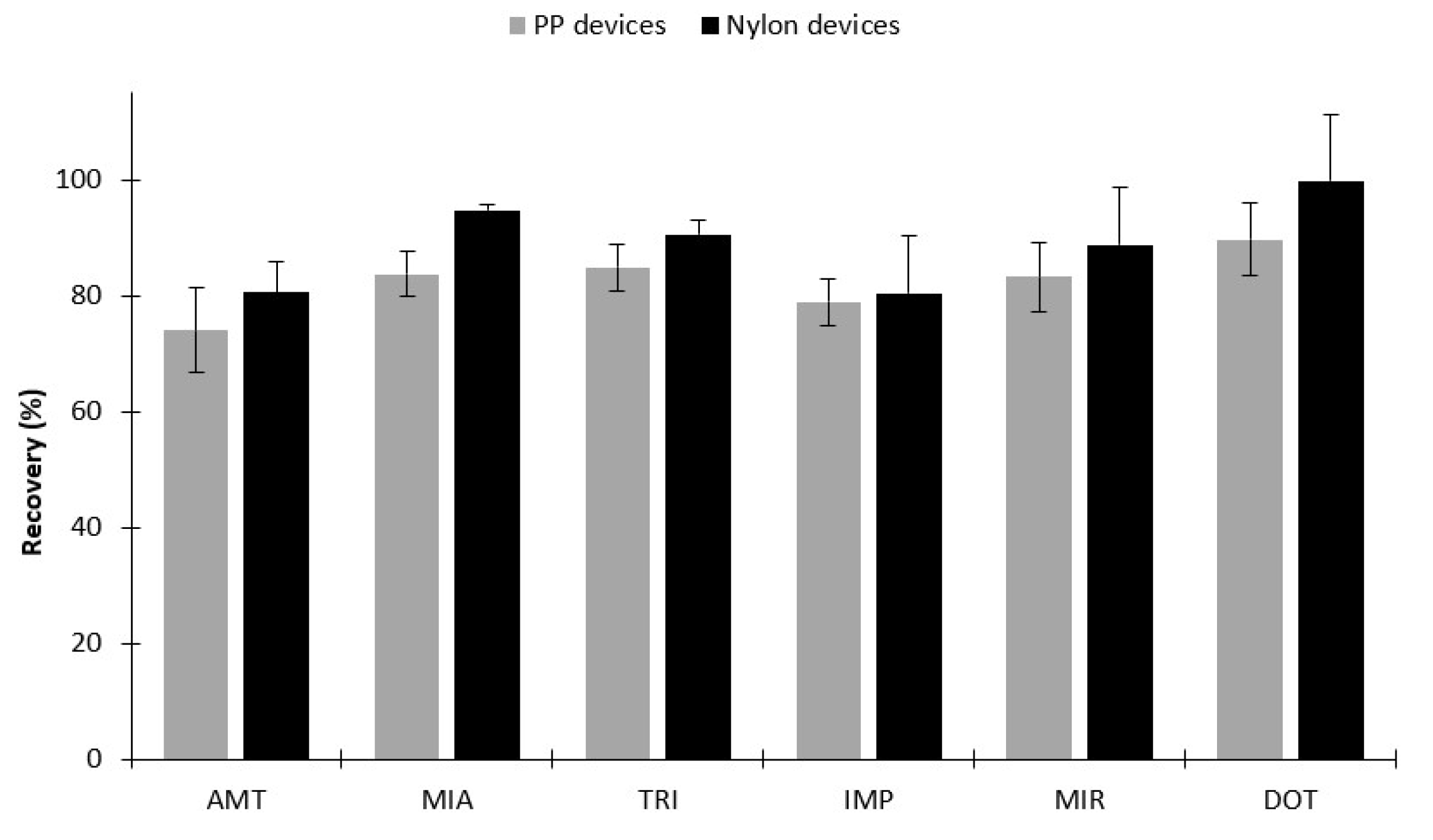
2.2.1. Selection of Sorbent Coatings and Back-Extraction Conditions
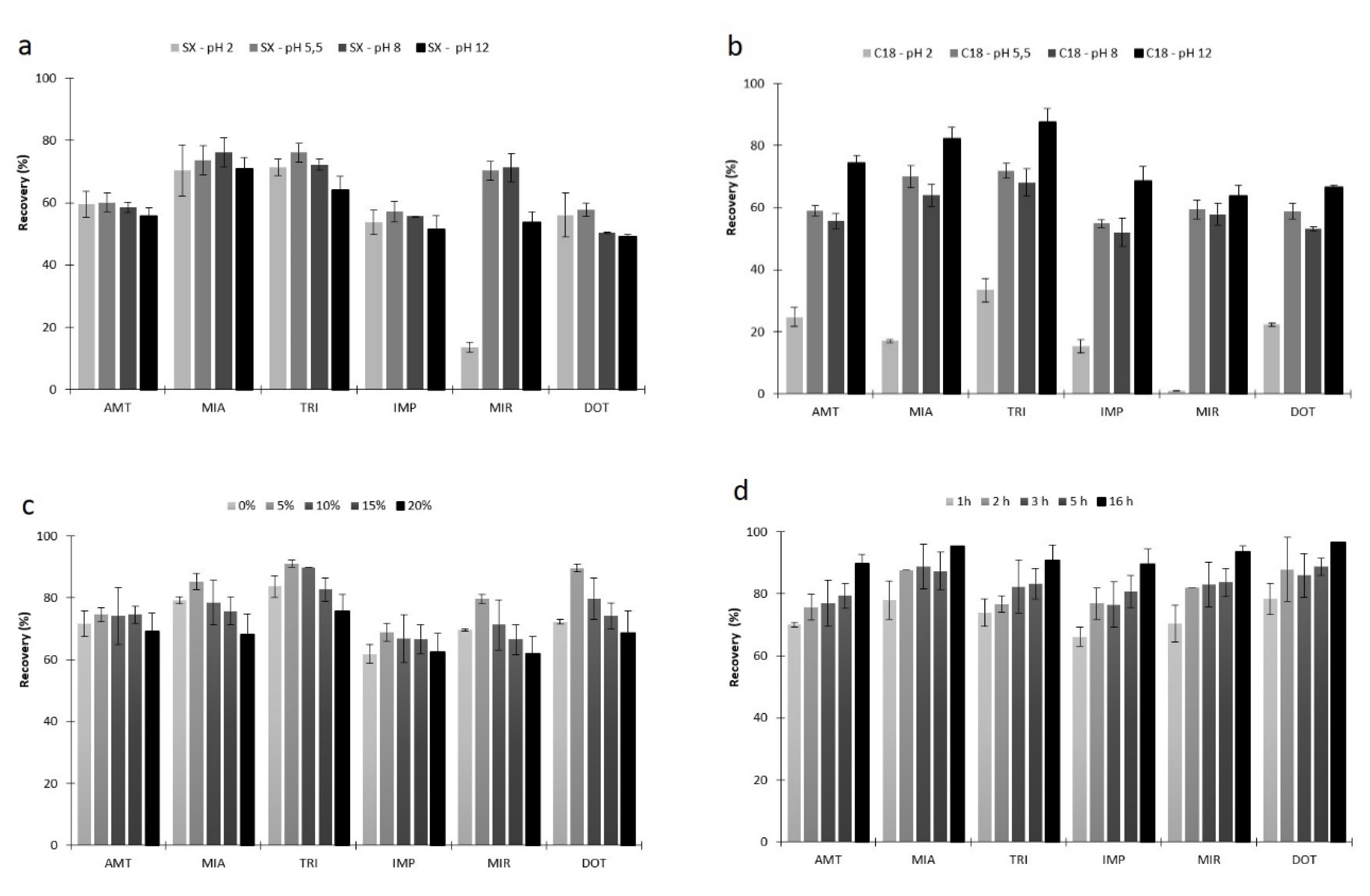
2.2.2. Microextraction Parameters
2.3. Validation of the Proposed Methodology
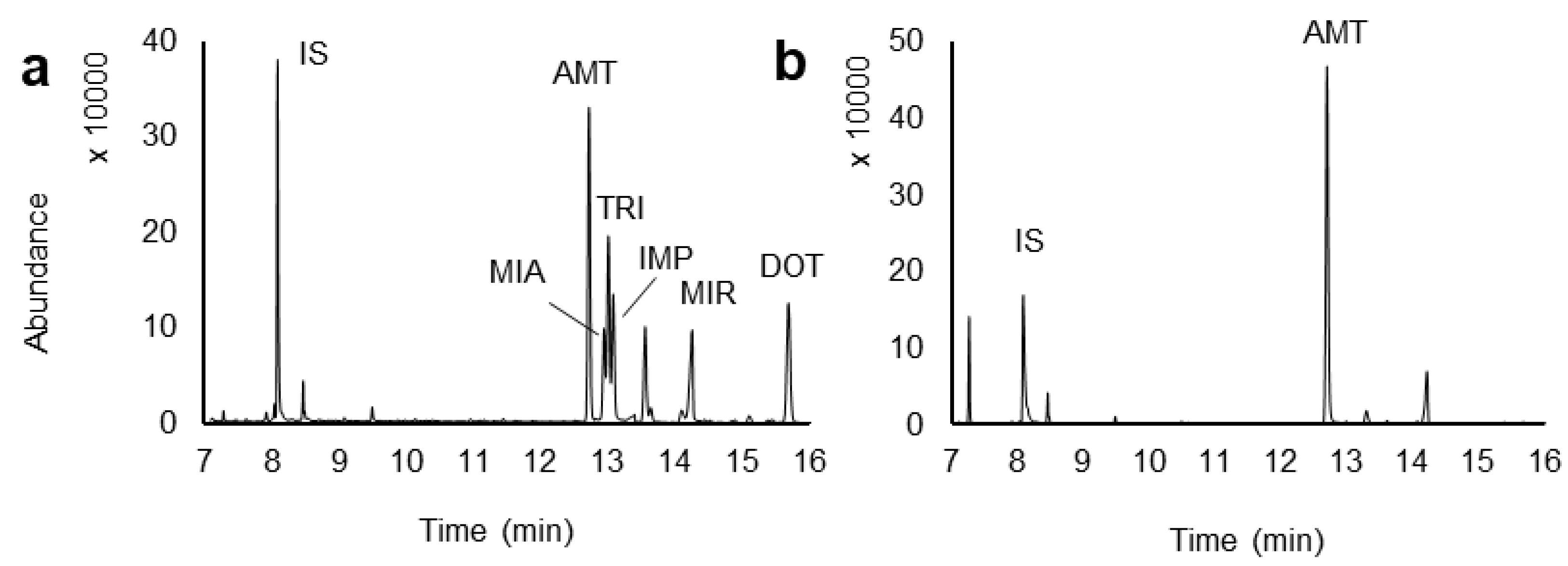
2.4. Application to Real Urine Samples
2.5. Performance Comparison with Other Microextraction Techniques
3. Materials and Methods
3.1. Chemicals and Standards
3.2. Sorbent Phases
3.3. Urine Matrices
3.4. Experimental Set-Up
3.4.1. Preparation of the BAμE Devices
3.4.2. Optimization Assays
3.4.3. Pre-Treatment of Biological Samples
3.4.4. Validation and Real Sample Assays
3.5. Instrumental Set-Up
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- World Health Organization. Mental Disorders. Available online: https://www.who.int/news-room/fact-sheets/detail/mental-disorders (accessed on 28 November 2019).
- Lovatt, P. Tricyclic antidepressants: Pharmacological profile. Nurse Prescr. 2014, 9, 38–41. [Google Scholar] [CrossRef]
- Hecht, A. Understanding Drugs: Antidepressants and Antianxiety Drugs, 1st ed.; Chelsea House Publications: New York, NY, USA, 2011; ISBN 9781604135329. [Google Scholar]
- Santos, M.G.; Tavares, I.M.C.; Barbosa, A.F.; Bettini, J.; Figueiredo, E.C. Analysis of tricyclic antidepressants in human plasma using online-restricted access molecularly imprinted solid phase extraction followed by direct mass spectrometry identification/quantification. Talanta 2017, 163, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Mohebbi, A.; Yaripour, S.; Farajzadeh, M.A.; Afshar Mogaddam, M.R. Combination of dispersive solid phase extraction and deep eutectic solvent–based air–assisted liquid–liquid microextraction followed by gas chromatography–mass spectrometry as an efficient analytical method for the quantification of some tricyclic antidep. J. Chromatogr. A 2018, 1571, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Safari, M.; Shahlaei, M.; Yamini, Y.; Shakorian, M.; Arkan, E. Magnetic framework composite as sorbent for magnetic solid phase extraction coupled with high performance liquid chromatography for simultaneous extraction and determination of tricyclic antidepressants. Anal. Chim. Acta 2018, 1034, 204–213. [Google Scholar] [CrossRef]
- Nogueira, J.M.F. Stir-bar sorptive extraction: 15 years making sample preparation more environment-friendly. TrAC Trends Anal. Chem. 2015, 71, 214–223. [Google Scholar] [CrossRef]
- Prieto, A.; Basauri, O.; Rodil, R.; Usobiaga, A.; Fernández, L.A.; Etxebarria, N.; Zuloaga, O. Stir-bar sorptive extraction: A view on method optimisation, novel applications, limitations and potential solutions. J. Chromatogr. A 2010, 1217, 2642–2666. [Google Scholar] [CrossRef]
- Neng, N.R.; Silva, A.R.M.; Nogueira, J.M.F. Adsorptive micro-extraction techniques-novel analytical tools for trace levels of polar solutes in aqueous media. J. Chromatogr. A 2010, 1217, 7303–7310. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, J.M.F. Novel sorption-based methodologies for static microextraction analysis: A review on SBSE and related techniques. Anal. Chim. Acta 2012, 757, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kabir, A.; Locatelli, M.; Ulusoy, H. Recent Trends in Microextraction Techniques Employed in Analytical and Bioanalytical Sample Preparation. Separations 2017, 4, 36. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, K.; Podmore, I. Solid Phase Microextraction (SPME) Method Development in Analysis of Volatile Organic Compounds (VOCS) as Potential Biomarkers of Cancer. J. Mol. Biomark. Diagn. 2015, 6. [Google Scholar] [CrossRef]
- Nogueira, J.M.F. Extração Sortiva em Barra de Agitação (SBSE): Uma metodologia inovadora para microextração estática. Sci. Chromatogr. 2012, 4, 259–269. [Google Scholar] [CrossRef]
- Abujaber, F.; Ahmad, S.M.; Neng, N.R.; Rodríguez Martín-Doimeadios, R.C.; Guzmán Bernardo, F.J.; Nogueira, J.M.F. Bar adsorptive microextraction coated with multi-walled carbon nanotube phases-Application for trace analysis of pharmaceuticals in environmental waters. J. Chromatogr. A 2019, 1600, 17–22. [Google Scholar] [CrossRef]
- Ahmad, S.M.; Mestre, A.S.; Neng, N.R.; Ania, C.O.; Carvalho, A.P.; Nogueira, J.M.F. Carbon-Based Sorbent Coatings for the Determination of 2 Pharmaceutical Compounds by Bar Adsorptive Microextraction. ACS Appl. Bio Mater. 2020, 3, 2078–2091. [Google Scholar] [CrossRef]
- Mafra, G.; Oenning, A.L.; Dias, A.N.; Merib, J.; Budziak, D.; da Silveira, C.B.; Carasek, E. Low-cost approach to increase the analysis throughput of bar adsorptive microextraction (BAµE) combined with environmentally-friendly renewable sorbent phase of recycled diatomaceous earth. Talanta 2018, 178, 886–893. [Google Scholar] [CrossRef] [PubMed]
- De Souza, M.P.; Rizzetti, T.M.; Francesquett, J.Z.; Prestes, O.D.; Zanella, R. Bar adsorptive microextraction (BAμE) with a polymeric sorbent for the determination of emerging contaminants in water samples by ultra-high performance liquid chromatography with tandem mass spectrometry. Anal. Methods 2018, 10, 697–705. [Google Scholar] [CrossRef]
- Ahmad, S.M.; Nogueira, J.M.F. High throughput bar adsorptive microextraction: A simple and effective analytical approach for the determination of nicotine and cotinine in urine samples. J. Chromatogr. A 2019, 1615, 460750. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.M.; Gomes, M.I.; Ide, A.H.; Neng, N.R.; Nogueira, J.M.F. Monitoring traces of organochlorine pesticides in herbal matrices by bar adsorptive microextraction–Application to black tea and tobacco. Int. J. Environ. Anal. Chem. 2019, 1–15. [Google Scholar] [CrossRef]
- Oenning, A.L.; Morés, L.; Dias, A.N.; Carasek, E. A new configuration for bar adsorptive microextraction (BAμE) for the quantification of biomarkers (hexanal and heptanal) in human urine by HPLC providing an alternative for early lung cancer diagnosis. Anal. Chim. Acta 2017, 965, 54–62. [Google Scholar] [CrossRef]
- Dias, A.N.; da Silva, A.C.; Simão, V.; Merib, J.; Carasek, E. A novel approach to bar adsorptive microextraction: Cork as extractor phase for determination of benzophenone, triclocarban and parabens in aqueous samples. Anal. Chim. Acta 2015, 888, 59–66. [Google Scholar] [CrossRef]
- Almeida, C.; Nogueira, J.M.F. Determination of steroid sex hormones in real matrices by bar adsorptive microextraction (BAμE). Talanta 2015, 136, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Ide, A.H.; Nogueira, J.M.F. New-generation bar adsorptive microextraction (BAμE) devices for a better eco-user-friendly analytical approach–Application for the determination of antidepressant pharmaceuticals in biological fluids. J. Pharm. Biomed. Anal. 2018, 153, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Ferrari Júnior, E.; Caldas, E.D. Simultaneous determination of drugs and pesticides in postmortem blood using dispersive solid-phase extraction and large volume injection-programmed temperature vaporization-gas chromatography–mass spectrometry. Forensic Sci. Int. 2018, 290, 318–326. [Google Scholar] [CrossRef]
- Wille, S.M.R.; Van Hee, P.; Neels, H.M.; Van Peteghem, C.H.; Lambert, W.E. Comparison of electron and chemical ionization modes by validation of a quantitative gas chromatographic-mass spectrometric assay of new generation antidepressants and their active metabolites in plasma. J. Chromatogr. A 2007, 1176, 236–245. [Google Scholar] [CrossRef]
- Gunnar, T.; Mykkänen, S.; Ariniemi, K.; Lillsunde, P. Validated semiquantitative/quantitative screening of 51 drugs in whole blood as silylated derivatives by gas chromatography-selected ion monitoring mass spectrometry and gas chromatography electron capture detection. J. Chromatogr. B 2004, 806, 205–219. [Google Scholar] [CrossRef]
- Rosado, T.; Gonçalves, A.; Martinho, A.; Alves, G.; Duarte, A.P.; Domingues, F.; Silvestre, S.; Breitenfeld, L.; Víctor, G.; Carlos, O.; et al. Simultaneous Quantification of Antidepressants and Metabolites in Urine and Plasma Samples by GC–MS for Therapeutic Drug Monitoring. Chromatographia 2017, 80, 301–328. [Google Scholar] [CrossRef]
- Camino-Sánchez, F.J.; Rodríguez-Gómez, R.; Zafra-Gómez, A.; Santos-Fandila, A.; Vílchez, J.L. Stir bar sorptive extraction: Recent applications, limitations and future trends. Talanta 2014, 130, 388–399. [Google Scholar] [CrossRef]
- Almeida, C.; Ahmad, S.M.; Nogueira, J.M.F. Bar adsorptive microextraction technique-application for the determination of pharmaceuticals in real matrices. Anal. Bioanal. Chem. 2017, 409, 2093–2106. [Google Scholar] [CrossRef]
- Ahmad, S.M.; Almeida, C.; Neng, N.R.; Nogueira, J.M.F. Bar adsorptive microextraction (BAμE) coated with mixed sorbent phases-Enhanced selectivity for the determination of non-steroidal anti-inflammatory drugs in real matrices in combination with capillary electrophoresis. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2016, 1008, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.M.; Almeida, C.; Neng, N.R.; Nogueira, J.M.F. Application of bar adsorptive microextraction (BAμE) for anti-doping control screening of anabolic steroids in urine matrices. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2014, 969. [Google Scholar] [CrossRef]
- Neng, N.R.; Ahmad, S.M.; Gaspar, H.; Nogueira, J.M.F. Determination of mitragynine in urine matrices by bar adsorptive microextraction and HPLC analysis. Talanta 2015, 144, 105–109. [Google Scholar] [CrossRef]
- Ide, A.H.; Ahmad, S.M.; Neng, N.R.; Nogueira, J.M.F. Enhancement for trace analysis of sulfonamide antibiotics in water matrices using bar adsorptive microextraction (BAμE). J. Pharm. Biomed. Anal. 2016, 129, 593–599. [Google Scholar] [CrossRef]
- Ahmad, S.M.; Ide, A.H.; Neng, N.R.; Nogueira, J.M.F. Application of bar adsorptive microextraction to determine trace organic micro-pollutants in environmental water matrices. Int. J. Environ. Anal. Chem. 2017, 97, 484–498. [Google Scholar] [CrossRef]
- Neng, N.R.; Nogueira, J.M.F. Monitoring trace levels of hydroxy aromatic compounds in urine matrices by bar adsorptive microextraction (BAμE). Anal. Methods 2017, 9, 5260–5265. [Google Scholar] [CrossRef]
- Ide, A.H.; Nogueira, J.M.F. Hollow fiber microextraction: A new hybrid microextraction technique for trace analysis. Anal. Bioanal. Chem. 2018, 410, 2911–2920. [Google Scholar] [CrossRef]
- Quintana, J.B.; Rodil, R.; Muniategui-Lorenzo, S.; López-Mahía, P.; Prada-Rodríguez, D. Multiresidue analysis of acidic and polar organic contaminants in water samples by stir-bar sorptive extraction–liquid desorption–gas chromatography–mass spectrometry. J. Chromatogr. A 2007, 1174, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.K.; Krause, L.C.; Carvalho, A.P.; Nogueira, J.M.F.; Caramão, E.B. Adsorption Properties of Activated Biochars Produced from Agro-industrial Residual Biomass. Int. J. Adv. Eng. Res. Sci. 2020, 7, 121–129. [Google Scholar] [CrossRef]
- Almeida, C.; Strzelczyk, R.; Nogueira, J.M.F. Improvements on bar adsorptive microextraction (BAμE) technique-Application for the determination of insecticide repellents in environmental water matrices. Talanta 2014, 120, 126–134. [Google Scholar] [CrossRef]
- MarvinSketch 20.10, version 20.10.0, ChemAxon Marvin 6.2.2 2014.
- Jafari, M.T.; Saraji, M.; Sherafatmand, H. Electrospray ionization-ion mobility spectrometry as a detection system for three-phase hollow fiber microextraction technique and simultaneous determination of trimipramine and desipramine in urine and plasma samples. Anal. Bioanal. Chem. 2011, 399, 3555–3564. [Google Scholar] [CrossRef]
- Erik, B.; Pat, S.; Frank, D.; Carel, C. Stir bar sorptive extraction (SBSE), a novel extraction technique for aqueous samples: Theory and principles. J. Microcolumn Sep. 1999, 11, 737–747. [Google Scholar] [CrossRef]
- Baltussen, E.; Cramers, C.A.; Sandra, P.J.F. Sorptive sample preparation—A review. Anal Bioanal Chem 2002, 373, 3–22. [Google Scholar] [CrossRef] [Green Version]
- David, F.; Sandra, P. Stir bar sorptive extraction for trace analysis. J. Chromatogr. A 2007, 1152, 54–69. [Google Scholar] [CrossRef]
- Dos Santos, M.F.; Ferri, C.C.; Seulin, S.C.; Leyton, V.; Pasqualucci, C.A.G.; Muñoz, D.R.; Yonamine, M. Determination of antidepressants in whole blood using hollow-fiber liquid-phase microextraction and gas chromatography-mass spectrometry. Forensic Toxicol. 2014, 32, 214–224. [Google Scholar] [CrossRef]
- Mansilha, C.; Melo, A.; Rebelo, H.; Ferreira, I.M.P.L.V.O.; Pinho, O.; Domingues, V.; Pinho, C.; Gameiro, P. Quantification of endocrine disruptors and pesticides in water by gas chromatography-tandem mass spectrometry. Method validation using weighted linear regression schemes. J. Chromatogr. A 2010, 1217, 6681–6691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, F.I.C.C.; Barbosa, P.G.A.; Zocolo, G.J.; do Nascimento, R.F. Method Validation Using Normal and Weighted Linear Regression Models for Quantification of Pesticides in Mango (Mangifera indica L.) Samples. Chromatographia 2018, 81, 677–688. [Google Scholar] [CrossRef]
- Da Silva, C.P.; Emídio, E.S.; De Marchi, M.R.R. Method validation using weighted linear regression models for quantification of UV filters in water samples. Talanta 2015, 131, 221–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutkowska, E.; Lozowicka, B.; Kaczynski, P. Three approaches to minimize matrix effects in residue analysis of multiclass pesticides in dried complex matrices using gas chromatography tandem mass spectrometry. Food Chem. 2019, 279, 20–29. [Google Scholar] [CrossRef]
- Montenarh, D.; Wernet, M.P.; Hopf, M.; Maurer, H.H.; Schmidt, P.H.; Ewald, A.H. Quantification of 33 antidepressants by LC-MS/MS-Comparative validation in whole blood, plasma, and serum. Anal. Bioanal. Chem. 2014, 406, 5939–5953. [Google Scholar] [CrossRef] [PubMed]
- Amaratunga, P.; Thomas, C.; Lemberg, B.L.; Lemberg, D. Quantitative Measurement of XLR11 and UR-144 in Oral Fluid by LC–MS-MS. J. Anal. Toxicol. 2014, 38, 315–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohebbi, A.; Farajzadeh, M.A.; Yaripour, S.; Afshar, M.R. Determination of Tricyclic Antidepressants in Human Urine Samples by the Three–Step Sample Pretreatment Followed by HPLC–UV Analysis: An Efficent Analytical Method for Further Pharmcokinetic and Forensic Studies. EXCLI J. 2018, 17, 952–963. [Google Scholar]
- Shamsipur, M.; Mirmohammadi, M. High performance liquid chromatographic determination of ultra-traces of two tricyclic antidepressant drugs imipramine and trimipramine in urine samples after their dispersive liquid-liquid microextraction coupled with response surface optimization. J. Pharm. Biomed. Anal. 2014, 100, 271–278. [Google Scholar] [CrossRef]
- Esrafili, A.; Yamini, Y.; Shariati, S. Hollow fiber-based liquid phase microextraction combined with high-performance liquid chromatography for extraction and determination of some antidepressant drugs in biological fluids. Anal. Bioanal. Chem. 2007, 604, 127–133. [Google Scholar] [CrossRef]
- Ito, R.; Ushiro, M.; Takahashi, Y.; Saito, K.; Ookubo, T.; Iwasaki, Y. Improvement and validation the method using dispersive liquid–liquid microextraction with in situ derivatization followed by gas chromatography–mass spectrometry for determination of tricyclic antidepressants in human urine samples. J. Chromatogr. B. 2011, 879, 3714–3720. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.N.; Samanidou, V.F.; Papadoyannis, I.N. Development and Validation of an HPLC Method for the Determination of Six 1,4-Benzodiazepines in Pharmaceuticals and Human Biological Fluids. J. Liq. Chromatogr. Relat. Technol. 2008, 31, 1258–1282. [Google Scholar] [CrossRef]
- Ahmad, S.M.; Nogueira, J.M.F. High throughput bar adsorptive microextraction: A novel cost-effective tool for monitoring benzodiazepines in large number of biological samples. Talanta 2019, 199, 195–202. [Google Scholar] [CrossRef] [PubMed]







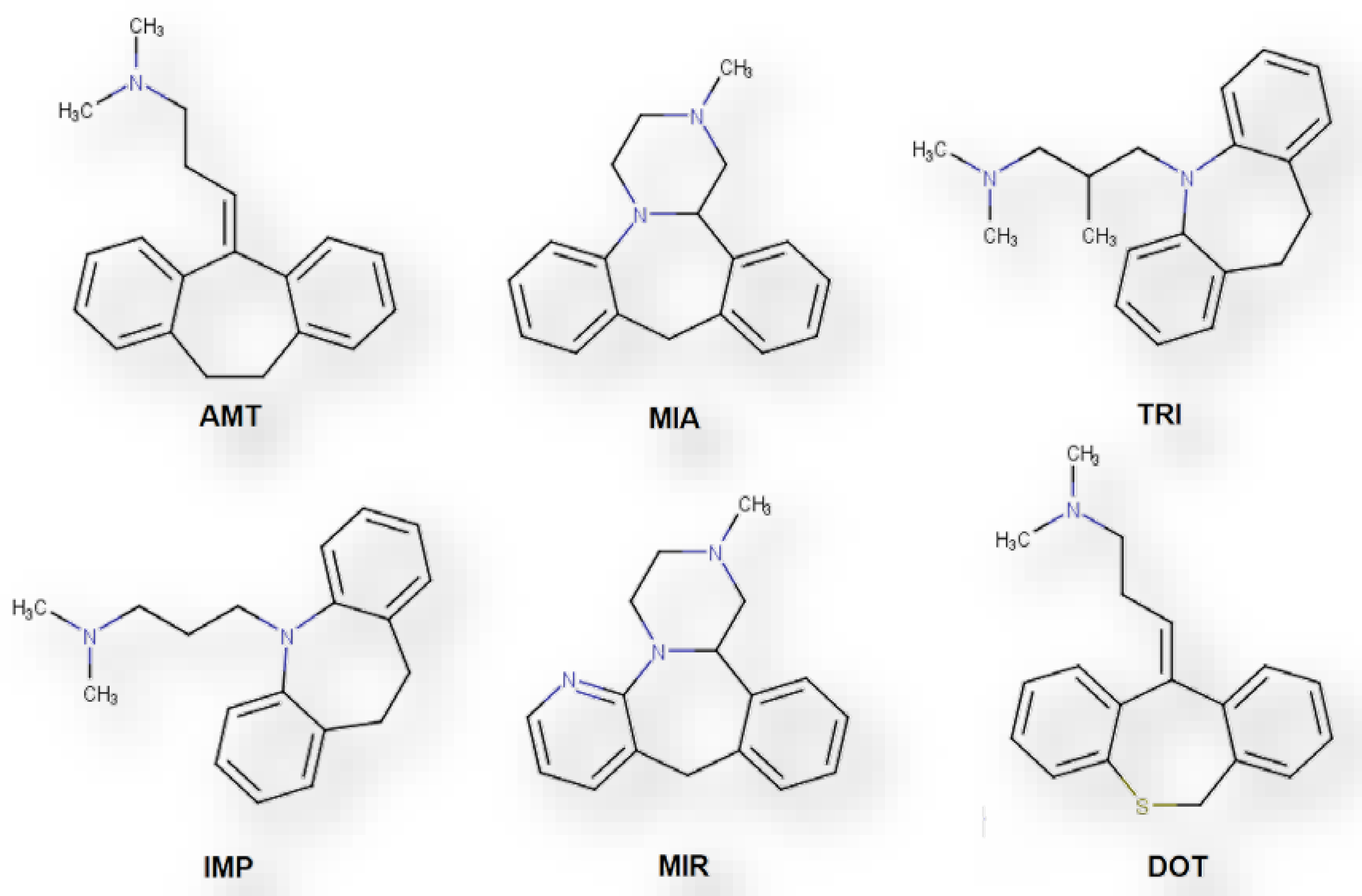{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TCAs | Ions (m/z) | Retention Time (min) |
|---|---|---|
| AMT | 58/202/215 | 12.91 |
| MIA | 193/220/264 | 13.13 |
| TRI | 58/249/294 | 13.22 |
| IMP | 58/234/280 | 13.28 |
| MIR | 195/208/265 | 13.77 |
| DOT | 58/202/295 | 15.96 |
| TCAs | LODs (µg L−1) | LLOQs (µg L−1) | Calibration Equations | r2 |
|---|---|---|---|---|
| AMT | 0.20 | 10.00 | y = 34.4780 x − 0.0174 | 0.9974 |
| MIA | y = 7.3701 x − 0.0026 | 0.9974 | ||
| TRI | y = 13.0250 x − 0.0033 | 0.9988 | ||
| IMP | y = 3.5938 x − 0.0027 | 0.9960 | ||
| MIR | 0.39 | y = 11.2264 x − 0.0026 | 0.9982 | |
| DOT | 1.56 | y = 29.1992 x − 0.0094 | 0.9978 |
| TCAs | Spiking Level (μg L−1) | Intraday | Interday | ||||
|---|---|---|---|---|---|---|---|
| Accuracy (%) ± Precision (%) | Accuracy (%) ± Precision (%) | ||||||
| AMT | 10.0 | 5.1 | ± | 8.2 | 20.0 | ± | 15.0 |
| 50.0 | −5.5 | ± | 4.6 | −5.0 | ± | 8.9 | |
| 500.0 | 1.7 | ± | 4.8 | 2.9 | ± | 12.2 | |
| 1000.0 | 4.8 | ± | 8.8 | −4.4 | ± | 11.8 | |
| MIA | 10.0 | −6.6 | ± | 4.3 | −2.8 | ± | 16.7 |
| 50.0 | −0.5 | ± | 11.1 | 0.1 | ± | 13.2 | |
| 500.0 | 4.8 | ± | 8.6 | 1.9 | ± | 11.8 | |
| 1000.0 | −0.1 | ± | 8.9 | 2.5 | ± | 13.2 | |
| TRI | 10.0 | 0.9 | ± | 6.8 | 13.1 | ± | 18.1 |
| 50.0 | −1.7 | ± | 6.1 | 11.0 | ± | 13.8 | |
| 500.0 | 5.7 | ± | 4.8 | 13.0 | ± | 9.2 | |
| 1000.0 | −4.1 | ± | 13.7 | 5.8 | ± | 11.7 | |
| IMP | 10.0 | −8.4 | ± | 9.0 | 14.8 | ± | 17.6 |
| 50.0 | 6.7 | ± | 12.8 | 2.4 | ± | 14.5 | |
| 500.0 | −1.2 | ± | 6.3 | 14.2 | ± | 14.9 | |
| 1000.0 | −0.5 | ± | 11.2 | −4.3 | ± | 10.1 | |
| MIR | 10.0 | 1.3 | ± | 15.8 | 15.9 | ± | 19.9 |
| 50.0 | 13.3 | ± | 0.9 | 14.5 | ± | 2.1 | |
| 500.0 | 6.5 | ± | 6.0 | 13.0 | ± | 12.1 | |
| 1000.0 | −0.7 | ± | 7.3 | 11.1 | ± | 13.8 | |
| DOT | 10.0 | 8.1 | ± | 0.4 | 14.1 | ± | 9.4 |
| 50.0 | 14.2 | ± | 2.5 | 1.0 | ± | 13.5 | |
| 500.0 | −8.2 | ± | 10.3 | −4.5 | ± | 14.5 | |
| 1000.0 | −12.0 | ± | 7.2 | −8.2 | ± | 10.1 | |
| TCAs | Spiking Level (μg L−1) | Matrix Effects (%) ± RSD (%) | Recovery Yields (%) ± RSD (%) | Process Efficiency (%) ± RSD (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| AMT | 25.0 | 90.2 | ± | 6.6 | 95.3 | ± | 9.6 | 86.0 | ± | 7.5 |
| 750.0 | 93.7 | ± | 12.5 | 107.9 | ± | 6.9 | 101.1 | ± | 12.9 | |
| MIA | 25.0 | 100.1 | ± | 9.8 | 95.3 | ± | 7.2 | 95.4 | ± | 8.6 |
| 750.0 | 111.5 | ± | 11.2 | 111.5 | ± | 7.7 | 124.3 | ± | 11.7 | |
| TRI | 25.0 | 102.1 | ± | 8.4 | 103.3 | ± | 9.3 | 105.5 | ± | 12.0 |
| 750.0 | 99.3 | ± | 8.2 | 109.0 | ± | 5.0 | 108.2 | ± | 7.9 | |
| IMP | 25.0 | 91.9 | ± | 4.1 | 92.3 | ± | 11.1 | 84.9 | ± | 10.4 |
| 750.0 | 109.3 | ± | 13.9 | 106.5 | ± | 10.0 | 116.5 | ± | 12.6 | |
| MIR | 25.0 | 95.6 | ± | 10.9 | 108.5 | ± | 12.3 | 103.6 | ± | 12.0 |
| 750.0 | 112.9 | ± | 14.4 | 99.0 | ± | 9.9 | 111.7 | ± | 13.9 | |
| DOT | 25.0 | 91.3 | ± | 4.4 | 99.6 | ± | 8.9 | 90.9 | ± | 8.4 |
| 750.0 | 99.4 | ± | 11.6 | 103.7 | ± | 7.5 | 103.1 | ± | 13.9 | |
| Analytical Method | TCAs | Recovery Yields (%) | RSD (%) | LOD (µg L−1) | LOQ (µg L−1) | Linear Range (µg L−1) | r2 | Ref. |
|---|---|---|---|---|---|---|---|---|
| BAμE-μLD/LVI-GC-MS(SIM) | AMT, MIA, TRI, IMP, MIR, DOT | ~100.0 | <9.6 | 0.20 | 10.0 (LLOQ) | 10.0–1000.0 | 0.9974 | This study |
| MSPE/HPLC-UV | AMT, IMP | 98.5–99.5 (RR) | [6] | |||||
| SPE/GC-MS | AMT, MIA, TRI, MIR, DOT | 64.4–99.8 | 6.0–20.6 | 1.0–2.5 | - | 1.0–320.0 | 0.9963–0.9996 | [27] |
| SI-HLLE-DSPE-DLLME-SFO/HPLC-UV | AMT, IMP | 69.0–84.0 | 3.0–4.0 | 0.2–0.3 | 0.7–1.1 (LOQ) | 0.7–1000.0 | 0.9960–0.9970 | [52] |
| DLLME/HPLC-UV | TRI | 112.0 | 6.1 | 0.6 | - | 2.0–100.0 | 0.9946 | [53] |
| HF-LPME/HPLC-UV | AMT, IMP, | - | 6.8 | - | - | - | [54] | |
| DLLME/GC-MS | AMT, IMP | 88.2–103.6 | 7.4–7.9 | 0.5 | 2.0 | 2.0–100.0 | 0.9990 | [55] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliveira, M.N.; Gonçalves, O.C.; Ahmad, S.M.; Schneider, J.K.; Krause, L.C.; Neng, N.R.; Caramão, E.B.; Nogueira, J.M.F. Application of Bar Adsorptive Microextraction for the Determination of Levels of Tricyclic Antidepressants in Urine Samples. Molecules 2021, 26, 3101. https://doi.org/10.3390/molecules26113101
Oliveira MN, Gonçalves OC, Ahmad SM, Schneider JK, Krause LC, Neng NR, Caramão EB, Nogueira JMF. Application of Bar Adsorptive Microextraction for the Determination of Levels of Tricyclic Antidepressants in Urine Samples. Molecules. 2021; 26(11):3101. https://doi.org/10.3390/molecules26113101
Chicago/Turabian StyleOliveira, Mariana N., Oriana C. Gonçalves, Samir M. Ahmad, Jaderson K. Schneider, Laiza C. Krause, Nuno R. Neng, Elina B. Caramão, and José M. F. Nogueira. 2021. "Application of Bar Adsorptive Microextraction for the Determination of Levels of Tricyclic Antidepressants in Urine Samples" Molecules 26, no. 11: 3101. https://doi.org/10.3390/molecules26113101