
Honey Phenolic Compound Profiling and Authenticity Assessment Using HRMS Targeted and Untargeted Metabolomics

 ,
,  , , , , , ,
, , , , , ,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Method Development and Validation
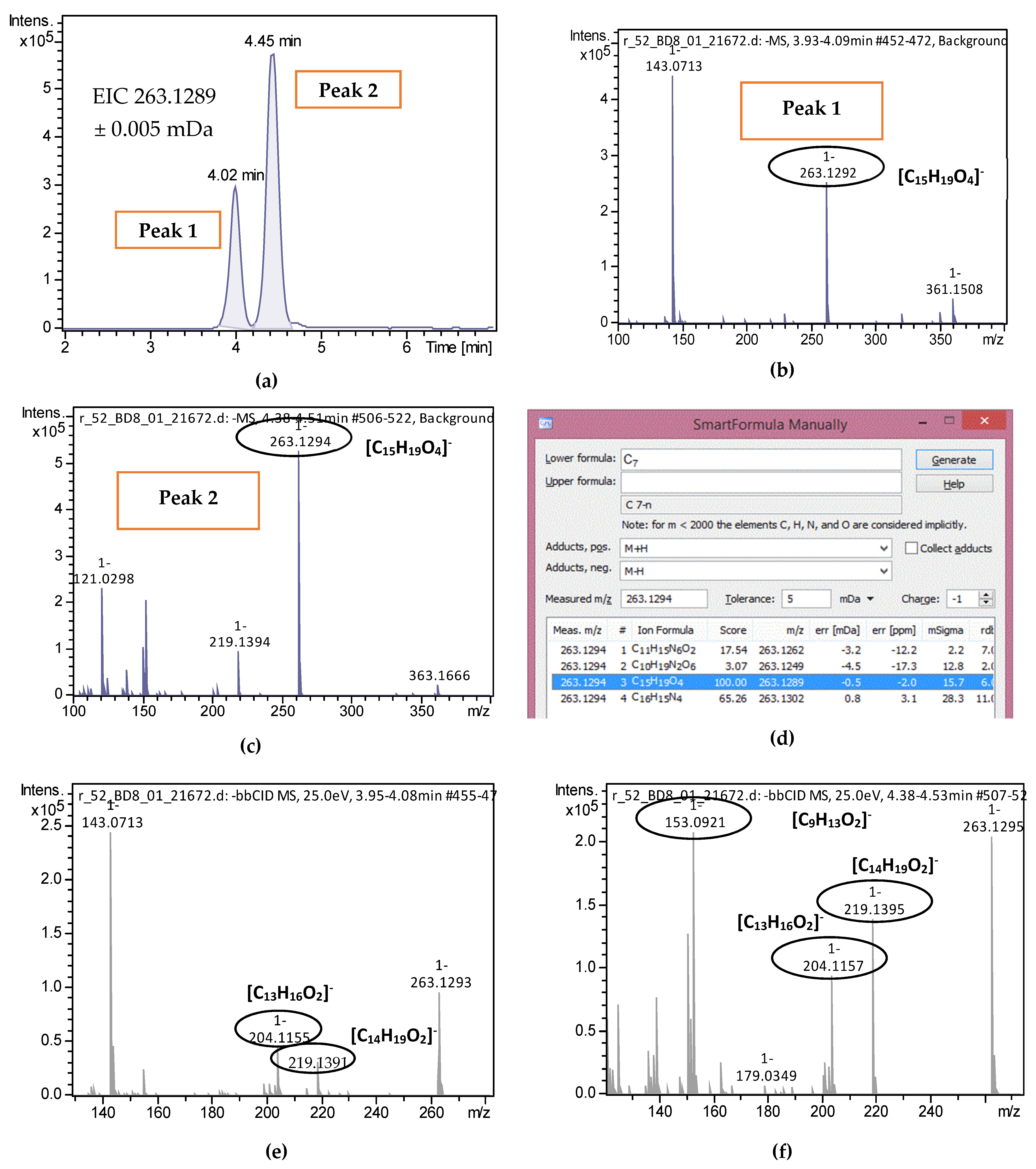
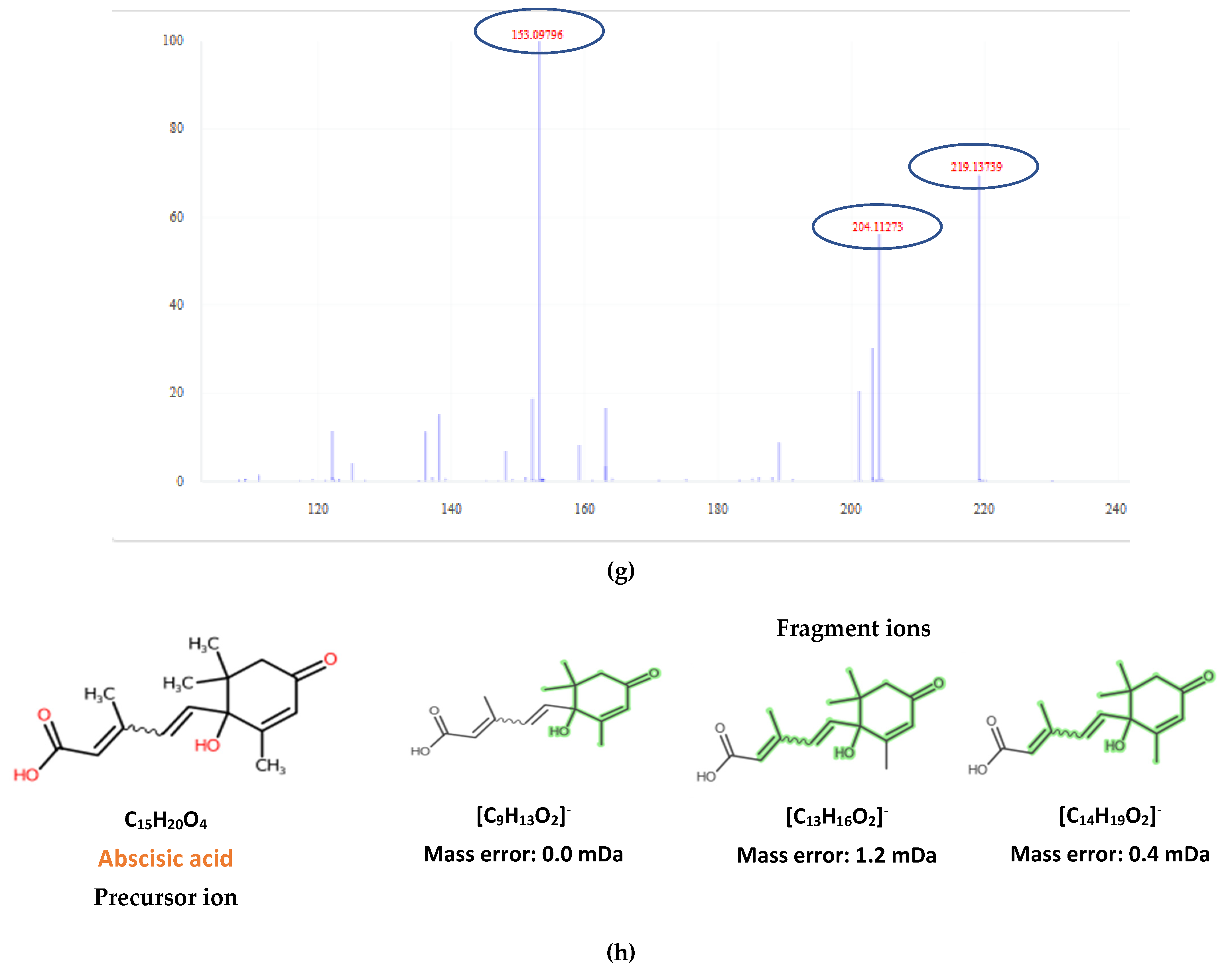
2.2. Target Screening
2.3. Suspect Screening
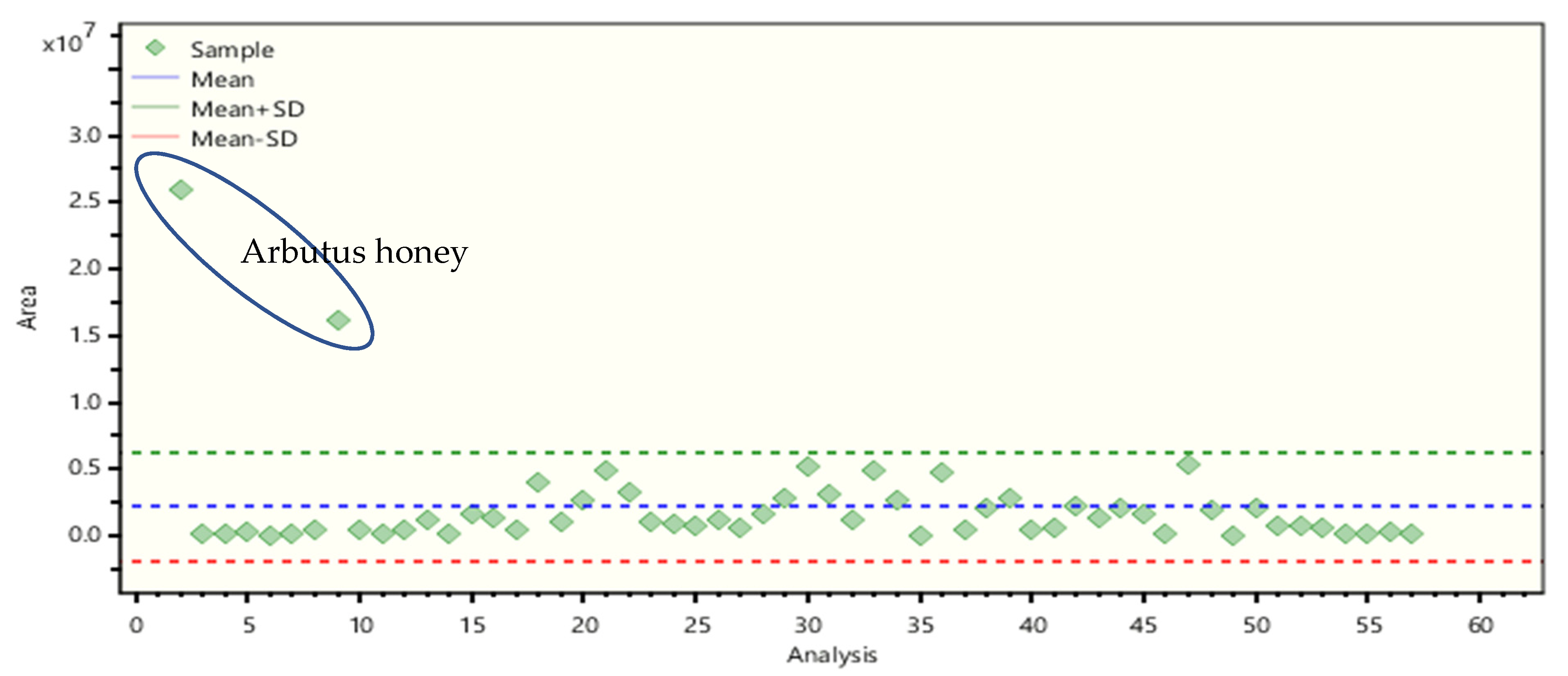
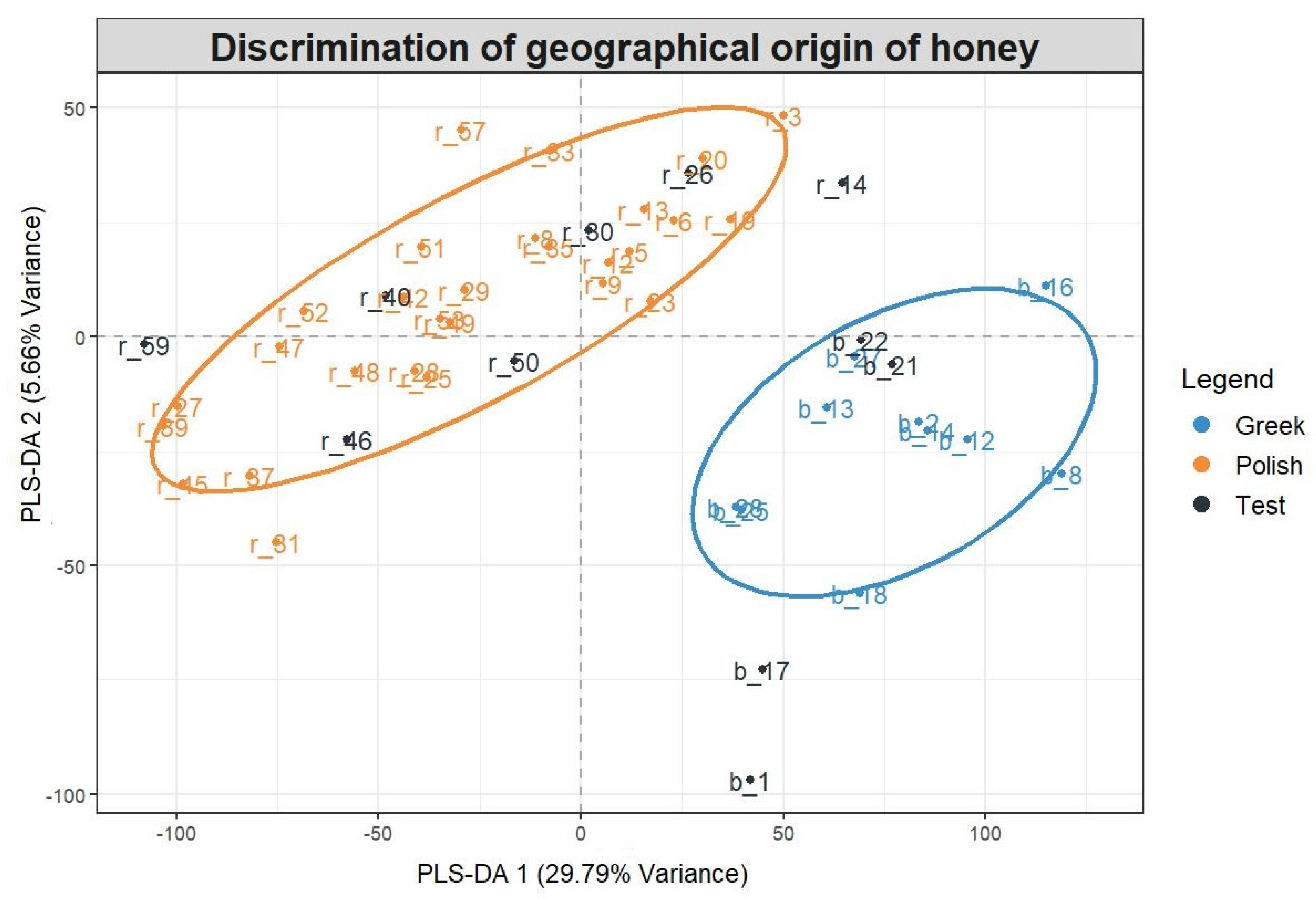
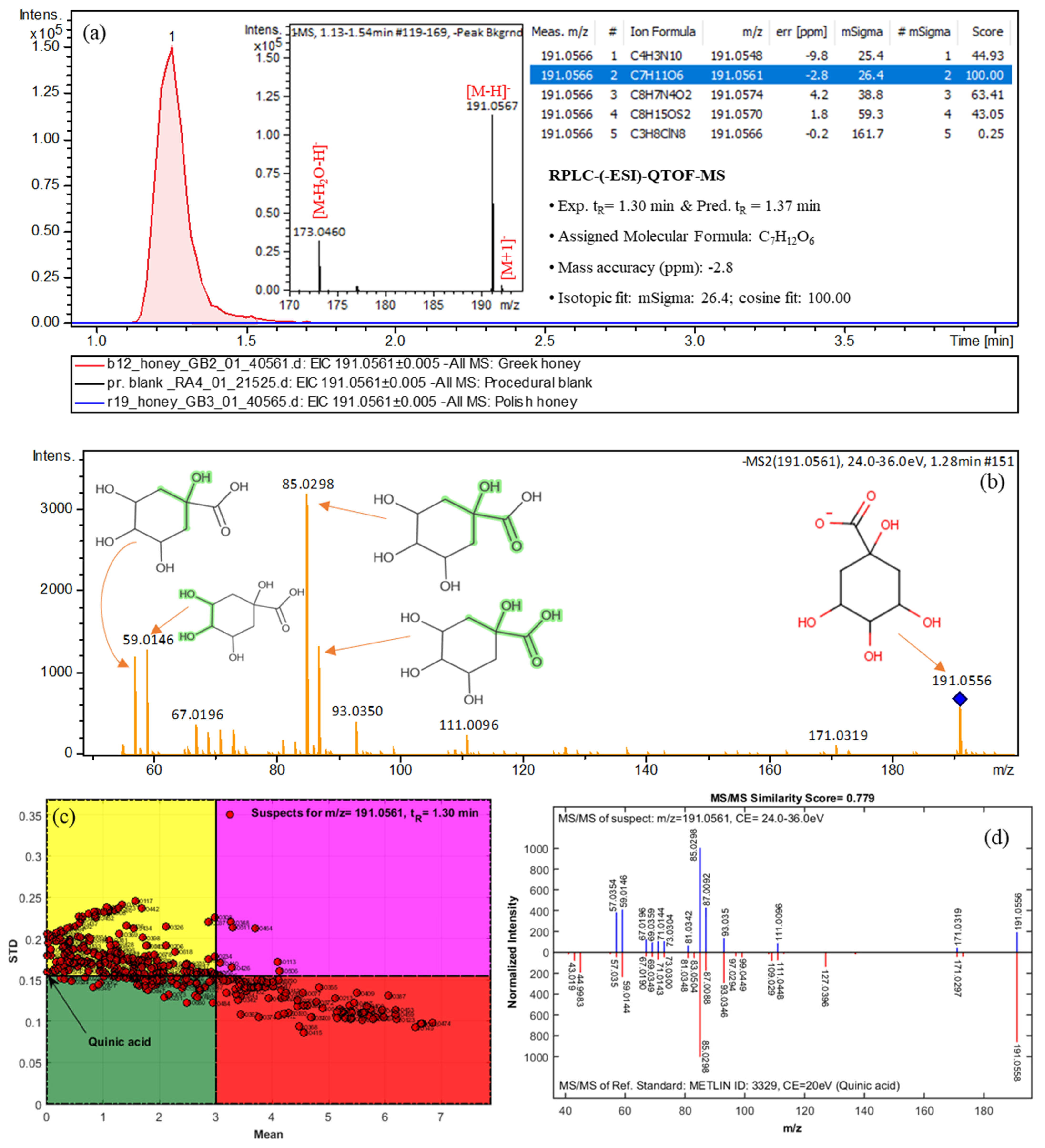
2.4. Non-Target Screening and Marker Identification
3. Materials and Methods
3.1. Chemicals
3.2. Preparation of Standards
3.3. Honey Samples
3.4. UPLC–QToF-MS Analysis
3.5. Sample Preparation Optimization
3.6. Validation
3.7. Screening Approaches
3.7.1. Target Screening Approach
3.7.2. Suspect Screening Approach
3.7.3. Non-Target Screening Approach
3.8. Chemometrics
Validation Criteria for Multivariate Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Danezis, G.P.; Tsagkaris, A.S.; Camin, F.; Brusic, V.; Georgiou, C.A. Food authentication: Techniques, trends & emerging approaches. TrAC Trends Anal. Chem. 2016, 85, 123–132. [Google Scholar]
- Alvarez-Suarez, J.M.; Giampieri, F.; Battino, M. Honey as a source of dietary antioxidants: Structures, bioavailability and evidence of protective effects against human chronic diseases. Curr. Med. Chem. 2013, 20, 621–638. [Google Scholar] [CrossRef]
- Pita-Calvo, C.; Vázquez, M. Differences between honeydew and blossom honeys: A review. Trends Food Sci. Technol. 2017, 59, 79–87. [Google Scholar] [CrossRef]
- Tsagkaris, A.S.; Koulis, G.A.; Danezis, G.P.; Martakos, I.; Dasenaki, M.; Georgiou, C.A.; Thomaidis, N.S. Honey authenticity: Analytical techniques, state of the art and challenges. RSC Adv. 2021, 11, 11273–11294. [Google Scholar] [CrossRef]
- Jamil Noor, M.; Ahmad, M.; Ashraf, M.A.; Zafar, M.; Sultana, S. A review of the pollen analysis of South Asian honey to identify the bee floras of the region. Palynology 2016, 40, 54–65. [Google Scholar] [CrossRef]
- Arvanitoyannis, I.S.; Chalhoub, C.; Gotsiou, P.; Lydakis-Simantiris, N.; Kefalas, P. Novel quality control methods in conjunction with chemometrics (multivariate analysis) for detecting honey authenticity. Crit. Rev. Food Sci. Nutr. 2005, 45, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Molan, P.C. The limitations of the methods of identifying the floral source of honeys. Bee World 1998, 79, 59–68. [Google Scholar] [CrossRef]
- Silvano, M.F.; Varela, M.S.; Palacio, M.A.; Ruffinengo, S.; Yamul, D.K. Physicochemical parameters and sensory properties of honeys from Buenos Aires region. Food Chem. 2014, 152, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Karabagias, I.K.; Badeka, A.; Kontakos, S.; Karabournioti, S.; Kontominas, M.G. Characterisation and classification of Greek pine honeys according to their geographical origin based on volatiles, physicochemical parameters and chemometrics. Food Chem. 2014, 146, 548–557. [Google Scholar] [CrossRef]
- Oroian, M.; Amariei, S.; Leahu, A.; Gutt, G. Multi-Element Composition of Honey as a Suitable Tool for Its Authenticity Analysis. Pol. J. Food Nutr. Sci. 2015, 65, 93–100. [Google Scholar] [CrossRef] [Green Version]
- Spiteri, M.; Rogers, K.M.; Jamin, E.; Thomas, F.; Guyader, S.; Lees, M.; Rutledge, D.N. Combination of 1H NMR and chemometrics to discriminate manuka honey from other floral honey types from Oceania. Food Chem. 2017, 217, 766–772. [Google Scholar] [CrossRef]
- Kuballa, T.; Brunner, T.S.; Thongpanchang, T.; Walch, S.G.; Lachenmeier, D.W. Application of NMR for authentication of honey, beer and spices. Curr. Opin. Food Sci. 2018, 19, 57–62. [Google Scholar] [CrossRef]
- Strelec, I.; Brodar, L.; Flanjak, I.; Kenjerić, F.Č.; Kovač, T.; Kenjerić, D.Č.; Primorac, L. Characterization of Croatian honeys by right-angle fluorescence spectroscopy and chemometrics. Food Anal. Methods 2018, 11, 824–838. [Google Scholar] [CrossRef]
- Dinca, O.R.; Ionete, R.E.; Popescu, R.; Costinel, D.; Radu, G.L. Geographical and Botanical Origin Discrimination of Romanian Honey Using Complex Stable Isotope Data and Chemometrics. Food Anal. Methods 2015, 8, 401–412. [Google Scholar] [CrossRef]
- Seisonen, S.; Kivima, E.; Vene, K. Characterisation of the aroma profiles of different honeys and corresponding flowers using solid-phase microextraction and gas chromatography-mass spectrometry/olfactometry. Food Chem. 2015, 169, 34–40. [Google Scholar] [CrossRef]
- Gerhardt, N.; Birkenmeier, M.; Schwolow, S.; Rohn, S.; Weller, P. Volatile-Compound Fingerprinting by Headspace-Gas-Chromatography Ion-Mobility Spectrometry (HS-GC-IMS) as a Benchtop Alternative to 1 H NMR Profiling for Assessment of the Authenticity of Honey. Anal. Chem. 2018, 90, 1777–1785. [Google Scholar] [CrossRef] [PubMed]
- Aloglu, A.K.; Harrington, P.D.B.; Sahin, S.; Demir, C.; Gunes, M.E. Chemical profiling of floral and chestnut honey using high-performance liquid chromatography-ultraviolet detection. J. Food Compos. Anal. 2017, 62, 205–210. [Google Scholar] [CrossRef]
- Oroian, M.; Ropciuc, S. Honey authentication based on physicochemical parameters and phenolic compounds. Comput. Electron. Agric. 2017, 138, 148–156. [Google Scholar] [CrossRef]
- Gašić, U.M.; Milojković-Opsenica, D.M.; Tešić, Ž.L. Polyphenols as possible markers of botanical origin of honey. J. AOAC Int. 2017, 100, 852–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cianciosi, D.; Forbes-Hernández, T.Y.; Afrin, S.; Gasparrini, M.; Reboredo-Rodriguez, P.; Manna, P.P.; Zhang, J.; Lamas, L.B.; Flórez, S.M.; Toyos, P.A.; et al. Phenolic compounds in honey and their associated health benefits: A review. Molecules 2018, 23, 2322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciulu, M.; Spano, N.; Pilo, M.I.; Sanna, G. Recent Advances in the Analysis of Phenolic Compounds in Unifloral Honeys. Molecules 2016, 21, 451. [Google Scholar] [CrossRef]
- Jandrić, Z.; Frew, R.D.; Fernandez-Cedi, L.N.; Cannavan, A. An investigative study on discrimination of honey of various floral and geographical origins using UPLC-QToF MS and multivariate data analysis. Food Control. 2017, 72, 189–197. [Google Scholar] [CrossRef]
- Esslinger, S.; Riedl, J.; Fauhl-Hassek, C. Potential and limitations of non-targeted fingerprinting for authentication of food in official control. Food Res. Int. 2014, 60, 189–204. [Google Scholar] [CrossRef]
- Cavanna, D.; Righetti, L.; Elliott, C.; Suman, M. The scientific challenges in moving from targeted to non-targeted mass spectrometric methods for food fraud analysis: A proposed validation workflow to bring about a harmonized approach. Trends Food Sci. Technol. 2018, 80, 223–241. [Google Scholar] [CrossRef]
- Trautvetter, S.; Koelling-Speer, I.; Speer, K. Confirmation of phenolic acids and flavonoids in honeys by UPLC-MS. Apidologie 2009, 40, 140–150. [Google Scholar] [CrossRef] [Green Version]
- Kečkeš, S.; Gašić, U.; Veličković, T.Ć.; Milojković-Opsenica, D.; Natić, M.; Tešić, Ž. The determination of phenolic profiles of Serbian unifloral honeys using ultra-high-performance liquid chromatography/high resolution accurate mass spectrometry. Food Chem. 2013, 138, 32–40. [Google Scholar] [CrossRef]
- Gašić, U.; Kečkeš, S.; Dabić, D.; Trifković, J.; Milojković-Opsenica, D.; Natić, M.; Tešić, Z. Phenolic profile and antioxidant activity of Serbian polyfloral honeys. Food Chem. 2014, 145, 599–607. [Google Scholar] [CrossRef]
- Gašić, U.M.; Natić, M.M.; Mišić, D.M.; Lušić, D.V.; Milojković-Opsenica, D.M.; Tešić, Ž.L.; Lušić, D. Chemical markers for the authentication of unifloral Salvia officinalis L. honey. J. Food Compos. Anal. 2015, 44, 128–138. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Jin, Y.; Yang, S.; Zhang, W.; Zhang, J.; Zhao, W.; Chen, L.; Wen, Y.; Zhang, Y.; Lu, K.; et al. Strategy for comparative untargeted metabolomics reveals honey markers of different floral and geographic origins using ultrahigh-performance liquid chromatography-hybrid quadrupole-orbitrap mass spectrometry. J. Chromatogr. A 2017, 1499, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Jandrić, Z.; Haughey, S.A.; Frew, R.D.; McComb, K.; Galvin-King, P.; Elliott, C.T.; Cannavan, A. Discrimination of honey of different floral origins by a combination of various chemical parameters. Food Chem. 2015, 189, 52–59. [Google Scholar] [CrossRef]
- Spiteri, M.; Dubin, E.; Cotton, J.; Poirel, M.; Corman, B.; Jamin, E.; Lees, M.; Rutledge, D. Data fusion between high resolution 1 H-NMR and mass spectrometry: A synergetic approach to honey botanical origin characterization. Anal. Bioanal. Chem. 2016, 408, 4389–4401. [Google Scholar] [CrossRef]
- Galanakis, C.M.; Goulas, V.; Tsakona, S.; Manganaris, G.A.; Gekas, V. A knowledge base for the recovery of natural phenols with different solvents. Int. J. Food Prop. 2013, 16, 382–396. [Google Scholar] [CrossRef] [Green Version]
- Kaškoniene, V.; Venskutonis, P.R. Floral Markers in Honey of Various Botanical and Geographic Origins: A Review. Compr. Rev. Food Sci. Food Saf. 2010, 9, 620–634. [Google Scholar] [CrossRef] [PubMed]
- Silici, S.; Sarioglu, K.; Karaman, K. Determination of polyphenols of some turkish honeydew and nectar honeys using HPLC-DAD. J. Liq. Chromatogr. Relat. Technol. 2013, 36, 2330–2341. [Google Scholar] [CrossRef]
- Campillo, N.; Viñas, P.; Férez-Melgarejo, G.; Hernández-Córdoba, M. Dispersive liquid-liquid microextraction for the determination of flavonoid aglycone compounds in honey using liquid chromatography with diode array detection and time-of-flight mass spectrometry. Talanta 2015, 131, 185–191. [Google Scholar] [CrossRef]
- Dimitrova, B.; Gevrenova, R.; Anklam, E. Analysis of phenolic acids in honeys of different floral origin by solid-phase extraction and high-performance liquid chromatography. Phytochem. Anal. 2007, 18, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Can, Z.; Yildiz, O.; Sahin, H.; Akyuz Turumtay, E.; Silici, S.; Kolayli, S. An investigation of Turkish honeys: Their physico-chemical properties, antioxidant capacities and phenolic profiles. Food Chem. 2015, 180, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Biesaga, M.; Pyrzynska, K. Liquid chromatography/tandem mass spectrometry studies of the phenolic compounds in honey. J. Chromatogr. A 2009, 1216, 6620–6626. [Google Scholar] [CrossRef] [PubMed]
- Aalizadeh, R.; Nika, M.-C.; Thomaidis, N.S. Development and application of retention time prediction models in the suspect and non-target screening of emerging contaminants. J. Hazard. Mater. 2019, 363, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Ruttkies, C.; Schymanski, E.L.; Wolf, S.; Hollender, J.; Neumann, S. MetFrag relaunched: Incorporating strategies beyond in silico fragmentation. J. Cheminform. 2016, 8, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Tuberoso, C.I.G.; Bifulco, E.; Caboni, P.; Cottiglia, F.; Cabras, P.; Floris, I. Floral markers of strawberry tree (Arbutus unedo L.) honey. J. Agric. Food Chem. 2010, 58, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Baliño, P.; Gómez-Cadenas, A.; López-Malo, D.; Romero, F.J.; Muriach, M. Is there a role for abscisic acid, a proven anti-inflammatory agent, in the treatment of ischemic retinopathies? Antioxidants 2019, 8, 104. [Google Scholar] [CrossRef] [Green Version]
- Bertoncelj, J.; Polak, T.; Kropf, U.; Korošec, M.; Golob, T. LC-DAD-ESI/MS analysis of flavonoids and abscisic acid with chemometric approach for the classification of Slovenian honey. Food Chem. 2011, 127, 296–302. [Google Scholar] [CrossRef]
- Silva, T.M.; dos Santos, F.P.; Evangelista-Rodrigues, A.; da Silva, E.M.; da Silva, G.S.; de Novais, J.S.; dos Santos, F.D.; Camara, C.A. Phenolic compounds, melissopalynological, physicochemical analysis and antioxidant activity of jandaíra (Melipona subnitida) honey. J. Food Compos. Anal. 2013, 29, 10–18. [Google Scholar] [CrossRef] [Green Version]
- Jerković, I.; Kuś, P.M.; Tuberoso, C.I.G.; Šarolić, M. Phytochemical and physical-chemical analysis of Polish willow (Salix spp.) honey: Identification of the marker compounds. Food Chem. 2014, 145, 8–14. [Google Scholar] [CrossRef]
- Campone, L.; Piccinelli, A.L.; Pagano, I.; Carabetta, S.; Di Sanzo, R.; Russo, M.; Rastrelli, L. Determination of phenolic compounds in honey using dispersive liquid-liquid microextraction. J. Chromatogr. A 2014, 1334, 9–15. [Google Scholar] [CrossRef]
- Deiana, V.; Tuberoso, C.; Satta, A.; Pinna, C.; Camarda, I.; Spano, N.; Ciulu, M.; Floris, I. Relationship between markers of botanical origin in nectar and honey of the strawberry tree (Arbutus unedo) throughout flowering periods in different years and in different geographical areas. J. Apic. Res. 2015, 54, 342–349. [Google Scholar] [CrossRef]
- Tomas-Barberan, F.A.; Martos, I.; Ferreres, F.; Radovic, B.S.; Anklam, E. HPLC flavonoid profiles as markers for the botanical origin of European unifloral honeys. J. Sci. Food Agric. 2001, 81, 485–496. [Google Scholar] [CrossRef]
- Bobis, O.; Mărghitaş, A.l.L.; Bonta, V.; Dezmirean, D.; Maghear, O. Free phenolic acids, flavonoids and abscisic acid related to HPLC sugar profile in acacia honey. Bull. Univ. Agric. Sci. Vet. Med. Cluj-Napoca Anim. Sci. Biotechnol. 2007, 64, 179–185. [Google Scholar]
- Martos, I.; Ferreres, F.; Tomás-Barberán, F.A. Identification of flavonoid markers for the botanical origin of Eucalyptus honey. J. Agric. Food Chem. 2000, 48, 1498–1502. [Google Scholar] [CrossRef] [PubMed]
- Zieliński, Ł.; Deja, S.; Jasicka-Misiak, I.; Kafarski, P. Chemometrics as a tool of origin determination of polish monofloral and multifloral honeys. J. Agric. Food Chem. 2014, 62, 2973–2981. [Google Scholar] [CrossRef]
- Tuberoso, C.I.G.; Bifulco, E.; Caboni, P.; Sarais, G.; Cottiglia, F.; Floris, I. Lumichrome and phenyllactic acid as chemical markers of thistle (Galactites tomentosa Moench) honey. J. Agric. Food Chem. 2011, 59, 364–369. [Google Scholar] [CrossRef]
- Oelschlaegel, S.; Gruner, M.; Wang, P.N.; Boettcher, A.; Koelling-Speer, I.; Speer, K. Classification and characterization of manuka honeys based on phenolic compounds and methylglyoxal. J. Agric. Food Chem. 2012, 60, 7229–7237. [Google Scholar] [CrossRef]
- Beitlich, N.; Koelling-Speer, I.; Oelschlaegel, S.; Speer, K. Differentiation of manuka honey from kanuka honey and from jelly bush honey using HS-SPME-GC/MS and UHPLC-PDA-MS/MS. J. Agric. Food Chem. 2014, 62, 6435–6444. [Google Scholar] [CrossRef]
- Cabras, P.; Angioni, A.; Tuberoso, C.; Floris, I.; Reniero, F.; Guillou, C.; Ghelli, S. Homogentisic acid: A phenolic acid as a marker of strawberry-tree (Arbutus unedo) honey. J. Agric. Food Chem. 1999, 47, 4064–4067. [Google Scholar] [CrossRef]
- Scanu, R.; Spano, N.; Panzanelli, A.; Pilo, M.I.; Piu, P.C.; Sanna, G.; Tapparo, A. Direct chromatographic methods for the rapid determination of homogentisic acid in strawberry tree (Arbutus unedo L.) honey. J. Chromatogr. A 2005, 1090, 76–80. [Google Scholar] [CrossRef]
- Andrade, P.; Ferreres, F.; Gil, M.I.; Tomás-Barberán, F.A. Determination of phenolic compounds in honeys with different floral origin by capillary zone electrophoresis. Food Chem. 1997, 60, 79–84. [Google Scholar] [CrossRef]
- Tuberoso, C.I.G.; Bifulco, E.; Jerkovic, I.; Caboni, P.; Cabras, P.; Floris, I. Methyl syringate: A chemical marker of asphodel (asphodelus microcarpus salzm. et viv.) monofloral honey. J. Agric. Food Chem. 2009, 57, 3895–3900. [Google Scholar] [CrossRef] [PubMed]
- Joshi, R.; Gangabhagirathi, R.; Venu, S.; Adhikari, S.; Mukherjee, T. Antioxidant activity and free radical scavenging reactions of gentisic acid: In-vitro and pulse radiolysis studies. Free Radic. Res. 2012, 46, 11–20. [Google Scholar] [CrossRef]
- Gómez-Caravaca, A.M.; Gómez-Romero, M.; Arráez-Román, D.; Segura-Carretero, A.; Fernández-Gutiérrez, A. Advances in the analysis of phenolic compounds in products derived from bees. J. Pharm. Biomed. Anal. 2006, 41, 1220–1234. [Google Scholar] [CrossRef]
- Krauss, M.; Singer, H.; Hollender, J. LC-high resolution MS in environmental analysis: From target screening to the identification of unknowns. Anal. Bioanal. Chem. 2010, 397, 943–951. [Google Scholar] [CrossRef] [Green Version]
- Gago-Ferrero, P.; Schymanski, E.L.; Bletsou, A.A.; Aalizadeh, R.; Hollender, J.; Thomaidis, N.S. Extended Suspect and Non-Target Strategies to Characterize Emerging Polar Organic Contaminants in Raw Wastewater with LC-HRMS/MS. Environ. Sci. Technol. 2015, 49, 12333–12341. [Google Scholar] [CrossRef] [Green Version]
- Truchado, P.; Ferreres, F.; Bortolotti, L.; Sabatini, A.G.; Tomás-Barberán, F.A. Nectar flavonol rhamnosides are floral markers of acacia (Robinia pseudacacia) honey. J. Agric. Food Chem. 2008, 56, 8815–8824. [Google Scholar] [CrossRef]
- Beretta, G.; Granata, P.; Ferrero, M.; Orioli, M.; Facino, R.M. Standardization of antioxidant properties of honey by a combination of spectrophotometric/fluorimetric assays and chemometrics. Anal. Chim. Acta 2005, 533, 185–191. [Google Scholar] [CrossRef]
- da Silva, I.A.A.; da Silva, T.M.S.; Camara, C.A.; Queiroz, N.; Magnani, M.; de Novais, J.S.; Soledade, L.E.B.; Lima, E.D.E.O.; de Souza, A.L.; de Souza, A.G. Phenolic profile, antioxidant activity and palynological analysis of stingless bee honey from Amazonas, Northern Brazil. Food Chem. 2013, 141, 3552–3558. [Google Scholar] [CrossRef] [Green Version]
- Cherchi, A.; Spanedda, L.; Tuberoso, C.; Cabras, P. Solid-phase extraction and high-performance liquid chromatographic determination of organic acids in honey. J. Chromatogr. A 1994, 669, 59–64. [Google Scholar] [CrossRef]
- Stephens, J.M.; Schlothauer, R.C.; Morris, B.D.; Yang, D.; Fearnley, L.; Greenwood, D.R.; Loomes, K.M. Phenolic compounds and methylglyoxal in some New Zealand manuka and kanuka honeys. Food Chem. 2010, 120, 78–86. [Google Scholar] [CrossRef]
- Hadjmohammadi, M.R.; Nazari, S.; Kamel, K. Determination of Flavonoid Markers in Honey with SPE and LC using Experimental Design. Chromatographia 2009, 69, 1291–1297. [Google Scholar] [CrossRef]
- Yaoa, L.; Jiang, Y.; Singanusong, R.; Datta, N.; Raymont, K. Phenolic acids in Australian Melaleuca, Guioa, Lophostemon, Banksia and Helianthus honeys and their potential for floral authentication. Food Res. Int. 2005, 38, 651–658. [Google Scholar] [CrossRef]
- Andrade, P.; Ferreres, F.; Teresa Amaral, M. Analysis of honey phenolic acids by HPLC, its application to honey botanical characterization. J. Liq. Chromatogr. Relat. Technol. 1997, 20, 2281–2288. [Google Scholar] [CrossRef]
- Lianda, R.L.P.; D’Oliveira Sant’Ana, L.; Echevarria, A.; Castro, R.N. Antioxidant activity and phenolic composition of brazilian honeys and their extracts. J. Braz. Chem. Soc. 2012, 23, 618–627. [Google Scholar] [CrossRef] [Green Version]
- Lachman, J.; Orsák, M.; Hejtmánková, A.; Kovářová, E. Evaluation of antioxidant activity and total phenolics of selected Czech honeys. LWT Food Sci. Technol. 2010, 43, 52–58. [Google Scholar] [CrossRef]
- Viuda-Martos, M.; Ruiz-Navajas, Y.; Fernández-López, J.; Pérez-Álvarez, J.A. Functional properties of honey, propolis, and royal jelly. J. Food Sci. 2008, 73, 117–124. [Google Scholar] [CrossRef]
- Kalogiouri, N.P.; Alygizakis, N.A.; Aalizadeh, R.; Thomaidis, N.S. Olive oil authenticity studies by target and nontarget LC–QTOF-MS combined with advanced chemometric techniques. Anal. Bioanal. Chem. 2016, 408, 7955–7970. [Google Scholar] [CrossRef] [PubMed]
- FiehnLab. MassBank of North America. 2019. Available online: http://mona.fiehnlab.ucdavis.edu/ (accessed on 9 March 2021).
- Smith, C.A.; O’Maille, G.; Want, E.J.; Qin, C.; Trauger, S.A.; Brandon, T.R.; Custodio, D.E.; Abagyan, R.; Siuzdak, G. METLIN: A metabolite mass spectral database. Ther. Drug Monit. 2005, 27, 747–751. [Google Scholar] [CrossRef]
- Wolf, S.; Schmidt, S.; Müller-Hannemann, M.; Neumann, S. In silico fragmentation for computer assisted identification of metabolite mass spectra. BMC Bioinform. 2010, 11, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aalizadeh, R.; Thomaidis, N.S.; Bletsou, A.A.; Gago-Ferrero, P. Quantitative Structure-Retention Relationship Models to Support Nontarget High-Resolution Mass Spectrometric Screening of Emerging Contaminants in Environmental Samples. J. Chem. Inf. Model. 2016, 56, 1384–1398. [Google Scholar] [CrossRef]
- Polykarpos, B.; Nikolaos, S.; Thomaidis, R.A. Minimizing analytical procedural mass spectral features as false positive peaks in untargeted liquid chromatography—High resolutin mass spectrometry. In Proceedings of the 11th Aegean Analytical Chemistry Days, Crete, Greece, 25–29 September 2018; p. 70. [Google Scholar]
- Schymanski, E.L.; Williams, A.J. Open Science for Identifying “Known Unknown” Chemicals. Environ. Sci. Technol. 2017, 51, 5357–5359. [Google Scholar] [CrossRef]
- Schymanski, E.L.; Jeon, J.; Gulde, R.; Fenner, K.; Ruff, M.; Singer, H.P.; Hollender, J. Identifying small molecules via high resolution mass spectrometry: Communicating confidence. Environ. Sci. Technol. 2014, 48, 2097–2098. [Google Scholar] [CrossRef] [PubMed]
- Aalizadeh, R.; Thomaidis, N.S. AutoSuspect: An R package to Perform Automatic Suspect Screening based on Regulatory Databases. In Proceedings of the 11th Aegean Analytical Chemistry Days, Crete, Greece, 25–29 September 2018; p. 32. [Google Scholar]
- Keun, H.C.; Ebbels, T.M.D.; Antti, H.; Bollard, M.E.; Beckonert, O.; Holmes, E.; Lindon, J.C.; Nicholson, J.K. Improved analysis of multivariate data by variable stability scaling: Application to NMR-based metabolic profiling. Anal. Chim. Acta 2003, 490, 265–276. [Google Scholar] [CrossRef]
- van den Berg, R.A.; Hoefsloot, H.C.J.; Westerhuis, J.A.; Smilde, A.K.; van der Werf, M.J. Centering, scaling, and transformations: Improving the biological information content of metabolomics data. BMC Genom. 2006, 7, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barker, M.; Rayens, W. Partial least squares for discrimination. J. Chemom. 2003, 17, 166–173. [Google Scholar] [CrossRef]
- Chong, I.G.; Jun, C.H. Performance of some variable selection methods when multicollinearity is present. Chemom. Intell. Lab. Syst. 2005, 78, 103–112. [Google Scholar] [CrossRef]
- Taylor, P.; Kennard, R.W.; Stone, L.A. Technometrics Computer Aided Design of Experiments. Technometric 1969, 11, 137–148. [Google Scholar]
- Kalogiouri, N.P.; Aalizadeh, R.; Thomaidis, N.S. Application of an advanced and wide scope non-target screening workflow with LC-ESI-QTOF-MS and chemometrics for the classification of the Greek olive oil varieties. Food Chem. 2018, 256, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Ballabio, D.; Consonni, V. Classification tools in chemistry. Part 1: Linear models. PLS-DA. Anal. Methods 2013, 5, 3790–3798. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Median Concentrations (mg/kg) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Acacia (n = 6) | Arbutus (n = 2) | Blossom (n = 2) | Buckwheat (n = 8) | Chestnut (n = 2) | Fir (n = 2) | Heather (n = 10) | Linden (n = 9) | Rape (n = 7) | Thyme (n = 3) | |
| Apigenin | 0.14 | <LOD | <LOD | 0.13 | <LOD | <LOD | 0.36 | 0.13 | <LOD | <LOD |
| Cinnamic acid | 0.061 | 0.050 | 0.54 | 0.21 | 0.61 | <LOD | 2.1 | 0.10 | 0.052 | 0.049 |
| Ferulic acid | 0.83 | 0.051 | 0.14 | 0.18 | 0.18 | 0.032 | 0.15 | 0.24 | 0.36 | 0.061 |
| Luteolin | <LOD | <LOD | 0.18 | <LOD | <LOD | <LOD | <LOD | 0.11 | ND | 0.083 |
| p-Coumaric acid | 0.22 | <LOD | 1.0 | 4.2 | 1.7 | 0.45 | 0.29 | 1.5 | 1.1 | 1.3 |
| Quercetin | <LOD | 0.19 | <LOD | 0.13 | <LOD | 5.0 | 0.18 | 0.15 | <LOD | 0.13 |
| Salicylic acid | 0.40 | <LOD | 1.2 | 1.8 | ND | 0.56 | ND | 0.88 | 0.74 | <LOD |
| Taxifolin | ND | ND | 0.51 | <LOD | 0.15 | 0.61 | <LOD | ND | <LOD | 0.16 |
| Vanillin | 0.27 | <LOD | 0.54 | 0.37 | ND | 0.11 | 0.25 | 0.20 | 0.26 | 0.089 |
| 3,4-Dihydroxybenzoic acid | 0.13 | 0.57 | 0.50 | 0.41 | 2.9 | 2.5 | 0.28 | 0.54 | 0.11 | 0.47 |
| 4-Hydroxybenzoic acid | 0.50 | 2.9 | 0.94 | 15 | 1.3 | 1.3 | 2.1 | 1.1 | 1.0 | 0.60 |
| Compound | Molecular Formula | m/z (Pseudo-Molecular Ion) | Mass Error (mDa) | tR Experimental (min) | tR Predicted (min) | Isotopic Fitting (mSigma) | m/z (Fragment Ions) | No of Samples Detected (in Total 51 Samples) |
|---|---|---|---|---|---|---|---|---|
| 2-trans,4-trans-Abscisic acid | C15H20O4 | 263.1289 | 0.3 | 4.02 | 4.44 | 15 | 219.1396; 204.1159 | 51 |
| 2-cis,4-trans-Abscisic acid | C15H20O4 | 263.1289 | 0.5 | 4.45 | 4.44 | 15.7 | 153.0922; 219.1396; 204.1159 | 51 |
| Acacetin | C16H12O5 | 283.0612 | 0.3 | 9.87 | 8.09 | 17.5 | 268.038 | 42 |
| Chrysin | C15H10O4 | 253.0506 | 0.1 | 9.14 | 8.15 | 14.3 | 143.0502; 209.0611 | 51 |
| Dehydrovomifoliol | C13H18O3 | 221.1183 | 0.3 | 5.07 | 5.42 | 17.6 | 206.0948; 179.1078; 161.0972; 108.0217; 191.0714 | 38 |
| Galangin | C15H10O5 | 269.0455 | 0.1 | 10.02 | 7.48 | 16.5 | 239.0349; 227.0349; 223.0404 | 51 |
| Homogentisic acid | C8H8O4 | 167.0350 | 0.6 | 1.68 | 3.29 | 20.8 | 122.0383; 108.0221 | 14 |
| Isorhamnetin | C16H12O7 | 315.0510 | 0.2 | 7.96 | 7.63 | 24.6 | 300.0277 | 51 |
| Kaempferol | C15H10O6 | 285.0405 | 0.9 | 8.17 | 7.26 | 7.4 | 229.051 | 45 |
| Lumichrome | C12H9N4O2 | 241.0731 | 0.4 | 6.42 | 5.85 | 22.3 | 198.0675; 170.0739 | 40 |
| Methyl Syringate | C10H12O5 | 211.0612 | 0.4 | 5.95 | 5.77 | 21.6 | 181.0144; 153.0194 | 43 |
| Phenyllactic acid | C8H8O2 | 165.0557 | 0.6 | 3.13 | 3.65 | 5.8 | 119.0506; 147.0455; 72.9935; 103.0558 | 47 |
| Pinobanksin | C15H12O5 | 271.0612 | 0.4 | 7.15 | 7.35 | 4.3 | 253.0511; 125.0244; 197.0608 | 51 |
| Pinocembrin | C15H12O4 | 255.0663 | 0.9 | 9.13 | 8.14 | 5.7 | 213.0562; 151.0037; 107.0139 | 51 |
| Rosmarinic acid | C18H16O8 | 359.0772 | 0.3 | 4.09 | 5.49 | 34.1 | 161.0242; 197.0449; 179.0346 | 1 |
| Sakuranetin | C16H14O5 | 285.0768 | 0.4 | 9.25 | 8.06 | 11.2 | 119.0500; 165.0191 | 51 |
| Compound Name | Measured m/z | Molecular Formula | Exp. tR (Pred. tR) min | Marker in Greek/Polish Honey | Level of Identification Confidence | MS/MS Fragments (5 Most Abundant Fragments) in UPLC–QTOF-MS |
|---|---|---|---|---|---|---|
| Quinic acid | 191.0566 | C7H12O6 | 1.30 (1.37) | Greek | 2a | 171.0319, 191.0556, 85.0298, 93.0350, 59.0146 |
| Gentisic acid | 153.0198 | C7H6O4 | 1.78 (2.41) | Greek | 2a | 108.0214, 109.0295, 91.0189, 81.0350, 110.0326 |
| Unknown | 239.1291 | C13H20O4 | 4.21 | Greek | 4 | 57.0355, 58.0392, 59.0151, 61.9911, 69.0351 |
| Unknown | 313.1803 | C20H26O3 | 10.75 | Greek | 4 | 255.1954, 313.1810, 315.2538 |
| 3-(2,5-Dimethoxyphenyl) propanoic acid | 209.0822 | C11H14O4 | 3.49 (4.94) | Greek | 3 | 91.0556, 72.994, 135.047, 119.0493, 147.0464 |
| 3-[[3-[(3S,4R,5S,6R)-3,4,5-Trihydroxy-6-(hydroxymethyl)oxan-2-yl]phenoxy]methyl]benzoic acid | 389.1243 | C20H22O8 | 3.71 (4.34) | Greek | 3 | 165.0561, 134.0375, 193.0511, 150.0321, 178.0276 |
| Chrysin | 253.0512 | C15H10O4 | 9.66 (8.05) | Polish | 2a | 253.0512, 143.0500, 209.0626, 63.0249, 119.0513 |
| Acacetin | 283.0611 | C16H12O5 | 10.15 (8.03) | Polish | 2a | 268.0383, 239.0353, 211.0404, 283.0611, 167.0505 |
| Sebacic acid | 201.1135 | C10H18O4 | 3.94 (4.21) | Polish | 2a | 139.1127, 59.0148, 165.0188, 183.1011, 121.0289 |
| Isorhamnetin | 315.0522 | C16H12O7 | 7.86 (7.56) | Polish | 2a | 300.0275, 315.0511, 165.9912 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koulis, G.A.; Tsagkaris, A.S.; Aalizadeh, R.; Dasenaki, M.E.; Panagopoulou, E.I.; Drivelos, S.; Halagarda, M.; Georgiou, C.A.; Proestos, C.; Thomaidis, N.S. Honey Phenolic Compound Profiling and Authenticity Assessment Using HRMS Targeted and Untargeted Metabolomics. Molecules 2021, 26, 2769. https://doi.org/10.3390/molecules26092769
Koulis GA, Tsagkaris AS, Aalizadeh R, Dasenaki ME, Panagopoulou EI, Drivelos S, Halagarda M, Georgiou CA, Proestos C, Thomaidis NS. Honey Phenolic Compound Profiling and Authenticity Assessment Using HRMS Targeted and Untargeted Metabolomics. Molecules. 2021; 26(9):2769. https://doi.org/10.3390/molecules26092769
Chicago/Turabian StyleKoulis, Georgios A., Aristeidis S. Tsagkaris, Reza Aalizadeh, Marilena E. Dasenaki, Eleni I. Panagopoulou, Spyros Drivelos, Michał Halagarda, Constantinos A. Georgiou, Charalampos Proestos, and Nikolaos S. Thomaidis. 2021. "Honey Phenolic Compound Profiling and Authenticity Assessment Using HRMS Targeted and Untargeted Metabolomics" Molecules 26, no. 9: 2769. https://doi.org/10.3390/molecules26092769