
Conformational Selection Mechanism Provides Structural Insights into the Optimization of APC-Asef Inhibitors

Abstract
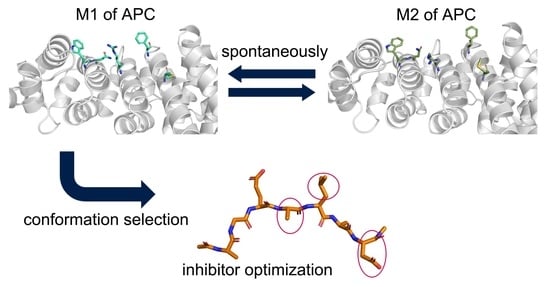
:
1. Introduction
2. Results
2.1. APC Covers a Wide Swath of Conformation Space Without a Ligand
2.2. Asef and ΔAsef Restrain APC Conformation in Ligand-Incompetent M2
2.3. MAI-108 Controls APC in Ligand-Competent M1 Conformation
2.4. The Fluctuation Analysis Confirms the Conformation Selection Mechanism
2.5. Dynamic Properties Provide Detailed Information for APC-Ligand Interactions
2.6. Binding Free Energy Analysis Provides Guidelines for Inhibitor Optimization
3. Discussion
4. Materials and Methods
4.1. MD Simulation Systems Setup
4.2. MD Simulations
4.3. Free Energy Landscape
4.4. Dynamic Network Analysis
4.5. Free Energy Calculations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability: Samples of the compounds are not available from the authors. |
References
- Fidler, I.J.; Kripke, M.L. The challenge of targeting metastasis. Cancer Metastasis Rev. 2015, 34, 635–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steeg, P.S. Targeting metastasis. Nat. Rev. Cancer 2016, 16, 201–218. [Google Scholar] [CrossRef]
- Nero, T.L.; Morton, C.J.; Holien, J.K.; Wielens, J.; Parker, M.W. Oncogenic protein interfaces: Small molecules, big challenges. Nat. Rev. Cancer 2014, 14, 248–262. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.A.; Khuri, F.R.; Fu, H. Targeting protein—protein interactions as an anticancer strategy. Trends Pharmacol. Sci. 2013, 34, 393–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schapira, M.; Tyers, M.; Torrent, M.; Arrowsmith, C.H. WD40 repeat domain proteins: A novel target class? Nat. Rev. Drug Discov. 2017, 16, 773–786. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xi, W.; Li, X.; Sun, H.; Li, Y. Advances in inhibition of protein-protein interactions targeting hypoxia-inducible factor-1 for cancer therapy. Bioorg. Med. Chem. 2019, 27, 1145–1158. [Google Scholar] [CrossRef]
- Kawasaki, Y.; Senda, T.; Ishidate, T.; Koyama, R.; Morishita, T.; Iwayama, Y.; Higuchi, O.; Akiyama, T. Asef, A Link between the Tumor Suppressor APC and G-Protein Signaling. Science 2000, 289, 1194–1197. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chen, L.; Gao, L.; Lin, K.; Zhu, L.; Lu, Y.; Shi, X.; Gao, Y.; Zhou, J.; Xu, P.; et al. Structural basis for the recognition of Asef by adenomatous polyposis coli. Cell Res. 2012, 22, 372–386. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Deng, R.; Yang, X.; Shang, J.; Lu, S.; Zhao, Y.; Song, K.; Liu, X.; Zhang, Q.; Chen, Y.; et al. Peptidomimetic inhibitors of APC–Asef interaction block colorectal cancer migration. Nat. Chem. Biol. 2017, 13, 994–1001. [Google Scholar] [CrossRef]
- Kawasaki, Y.; Sato, R.; Akiyama, T. Mutated APC and Asef are involved in the migration of colorectal tumour cells. Nat. Cell Biol. 2003, 5, 211–215. [Google Scholar] [CrossRef]
- Yang, X.; Zhong, J.; Zhang, Q.; Qian, J.; Song, K.; Ruan, C.; Xu, J.; Ding, K.; Zhang, J. Rational Design and Structure Validation of a Novel Peptide Inhibitor of the Adenomatous-Polyposis-Coli (APC)-Rho-Guanine-Nucleotide-Exchange-Factor-4 (Asef) Interaction. J. Med. Chem. 2018, 61, 8017–8028. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.-Q.; Wang, Z.-C.; Qi, P.-F.; Li, G.; Zhu, H.-L. Design, synthesis and biological evaluation of 2-H pyrazole derivatives containing morpholine moieties as highly potent small molecule inhibitors of APC-Asef interaction. Eur. J. Med. Chem. 2019, 177, 425–447. [Google Scholar] [CrossRef] [PubMed]
- Narayan, A.R.H.; Jiménez-Osés, G.; Podust, L.M.; Montgomery, J.; Houk, K.N.; Sherman, D.H.; Liu, P.; Negretti, S.; Zhao, W.; Gilbert, M.M.; et al. Enzymatic hydroxylation of an unactivated methylene C-H bond guided by molecular dynamics simulations. Nat. Chem. 2015, 7, 653–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaro, R.E.; Baudry, J.; Chodera, J.; Demir, Ö.; McCammon, J.A.; Miao, Y.; Smith, J.C. Ensemble docking in drug discovery. Biophys. J. 2018, 114, 2271–2278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; Deng, R.; Jiang, H.; Song, H.; Li, S.; Shen, Q.; Huang, W.; Nussinov, R.; Yu, J.; Zhang, J. The Mechanism of ATP-Dependent Allosteric Protection of Akt Kinase Phosphorylation. Structure 2015, 23, 1725–1734. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.; Chen, Y.; Wei, J.; Zhao, M.; Ni, D.; He, X.; Zhang, J. Mechanism of allosteric activation of SIRT6 revealed by the action of rationally designed activators. Acta Pharm. Sin. B 2020. [Google Scholar] [CrossRef]
- Ni, D.; Wei, J.; He, X.; Rehman, A.U.; Li, X.; Qiu, Y.; Pu, J.; Lu, S.; Zhang, J. Discovery of cryptic allosteric sites using reversed allosteric communication by a combined computational and experimental strategy. Chem. Sci. 2021, 12, 464–476. [Google Scholar] [CrossRef]
- Latorraca, N.R.; Venkatakrishnan, A.J.; Dror, R.O. GPCR Dynamics: Structures in Motion. Chem. Rev. 2016, 117, 139–155. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Rosell, M.; Fernández-Recio, J. Hot-spot analysis for drug discovery targeting protein-protein interactions. Expert Opin. Drug Discov. 2018, 13, 327–338. [Google Scholar] [CrossRef]
- Lu, S.; Jang, H.; Zhang, J.; Nussinov, R. Inhibitors of Ras-SOS Interactions. ChemMedChem 2016, 11, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Tsomaia, N. Peptide therapeutics: Targeting the undruggable space. Eur. J. Med. Chem. 2015, 94, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Nevola, L.; Giralt, E. Modulating protein–protein interactions: The potential of peptides. Chem. Commun. 2015, 51, 3302–3315. [Google Scholar] [CrossRef] [PubMed]
- Wójcik, P.; Berlicki, Ł. Peptide-based inhibitors of protein–protein interactions. Bioorg. Med. Chem. Lett. 2016, 26, 707–713. [Google Scholar] [CrossRef]
- Lu, S.; Shen, Q.; Zhang, J. Allosteric Methods and Their Applications: Facilitating the Discovery of Allosteric Drugs and the Investigation of Allosteric Mechanisms. Acc. Chem. Res. 2019, 52, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Ni, D.; Lu, S.; Zhang, J. Emerging roles of allosteric modulators in the regulation of protein-protein interactions (PPIs): A new paradigm for PPI drug discovery. Med. Res. Rev. 2019, 39, 2314–2342. [Google Scholar] [CrossRef]
- Lu, S.; He, X.; Ni, D.; Zhang, J. Allosteric Modulator Discovery: From Serendipity to Structure-Based Design. J. Med. Chem. 2019, 62, 6405–6421. [Google Scholar] [CrossRef]
- Lu, S.; Zhang, J. Small Molecule Allosteric Modulators of G-Protein-Coupled Receptors: Drug–Target Interactions. J. Med. Chem. 2019, 62, 24–45. [Google Scholar] [CrossRef]
- Leroux, A.E.; Biondi, R.M. Renaissance of Allostery to Disrupt Protein Kinase Interactions. Trends Biochem. Sci. 2020, 45, 27–41. [Google Scholar] [CrossRef]
- An, X.; Lu, S.; Song, K.; Shen, Q.; Huang, M.; Yao, X.; Liu, H.; Zhang, J. Are the Apo Proteins Suitable for the Rational Discovery of Allosteric Drugs? J. Chem. Inf. Model. 2019, 59, 597–604. [Google Scholar] [CrossRef] [Green Version]
- Rose, P.W.; Bi, C.; Bluhm, W.F.; Christie, C.H.; Dimitropoulos, D.; Dutta, S.; Green, R.K.; Goodsell, D.S.; Prlić, A.; Quesada, M.; et al. The RCSB Protein Data Bank: New resources for research and education. Nucleic Acids Res. 2012, 41, D475–D482. [Google Scholar] [CrossRef]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef]
- Wang, E.; Weng, G.; Sun, H.; Du, H.; Zhu, F.; Chen, F.; Wang, Z.; Hou, T. Assessing the performance of the MM/PBSA and MM/GBSA methods. 10. Impacts of enhanced sampling and variable dielectric model on protein–protein Interactions. Phys. Chem. Chem. Phys. 2019, 21, 18958–18969. [Google Scholar] [CrossRef]
- Fluitt, A.M.; de Pablo, J.J. An Analysis of Biomolecular Force Fields for Simulations of Polyglutamine in Solution. Biophys. J. 2015, 109, 1009–1018. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Adelman, S.A.; Doll, J.D. Generalized Langevin equation approach for atom/solid-surface scattering: General formulation for classical scattering off harmonic solids. J. Chem. Phys. 1976, 64, 2375–2388. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cerutti, D.; Cheateham, T.; Darden, T.; Duke, R.; Giese, T.; Gohlke, H.; Goetz, A.W.; Greene, D.; Homeyer, N. AMBER16 package; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Swaminathan, S.; Harte, W.E., Jr.; Beveridge, D.L. Investigation of domain structure in proteins via molecular dynamics simulation: Application to HIV-1 protease dimer. J. Am. Chem. Soc. 1991, 113, 2717–2721. [Google Scholar] [CrossRef]
- Miller, B.R., III; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Duan, L.; Chen, F.; Liu, H.; Wang, Z.; Pan, P.; Zhu, F.; Zhang, J.Z.H.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 7. Entropy effects on the performance of end-point binding free energy calculation approaches. Phys. Chem. Chem. Phys. 2018, 20, 14450–14460. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Dai, J.; Ni, D.; He, X.; Zhang, H.; Zhang, J.; Fu, Q.; Liu, Y.; Lu, S. Insight into the mechanism of allosteric activation of PI3Kα by oncoprotein K-Ras4B. Int. J. Biol. Macromol. 2020, 144, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ye, M.; Wang, Y.; Qiu, M.; Fu, T.; Zhang, J.; Zhou, B.; Lu, S. How Parkinson’s disease-related mutations disrupt the dimerization of WD40 domain in LRRK2: A comparative molecular dynamics simulation study. Phys. Chem. Chem. Phys. 2020, 22, 20421–20433. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Residue Number | RMSF of APC | RMSF of APC-108 |
|---|---|---|
| R549 | 1.10 | 0.73 |
| F458 | 1.31 | 1.08 |
| R463 | 1.06 | 1.17 |
| F510 | 1.16 | 0.79 |
| Energy Items | MAI-108 | ΔAsef | Asef |
|---|---|---|---|
| ΔEvdw | −62.26 ± 3.97 | −50.67 ± 3.67 | −158.43 ± 9.58 |
| ΔEele | −185.94 ± 28.51 | −139.41 ± 37.90 | −999.05 ± 62.78 |
| ΔGP | 190.25 ± 24.27 | 161.53 ± 35.03 | 1083.73 ± 60.40 |
| ΔGnp | −9.00 ± 0.45 | −6.64 ± 0.66 | −21.77 ± 1.45 |
| ΔEMM | −248.21 ± 28.40 | −190.08 ± 38.72 | −1157.48 ± 64.48 |
| ΔGsolv | 181.25 ± 24.09 | 154.90 ± 34.51 | 1061.96 ± 59.79 |
| ΔGbinding | −66.95 ± 6.94 | −35.18 ± 7.52 | −95.52 ± 12.00 |
| System Name | Molecule Occupying ARM Domain | PDB ID | PME Grid Sizes (Å) |
|---|---|---|---|
| APC | None | 3NMZ | 108 × 108 × 108 |
| APC-108 | MAI-108 (AGEALAD) | 5IZ8 | 108 × 108 × 108 |
| APC-Asef | Asef | 3NMZ | 112 × 112 × 112 |
| APC-ΔAsef | Residue 181–187 of Asef (GGEQLAI) | 3NMZ | 112 × 112 × 112 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, X.; Huang, N.; Qiu, Y.; Zhang, J.; Liu, Y.; Yin, X.-L.; Lu, S. Conformational Selection Mechanism Provides Structural Insights into the Optimization of APC-Asef Inhibitors. Molecules 2021, 26, 962. https://doi.org/10.3390/molecules26040962
He X, Huang N, Qiu Y, Zhang J, Liu Y, Yin X-L, Lu S. Conformational Selection Mechanism Provides Structural Insights into the Optimization of APC-Asef Inhibitors. Molecules. 2021; 26(4):962. https://doi.org/10.3390/molecules26040962
Chicago/Turabian StyleHe, Xinheng, Ning Huang, Yuran Qiu, Jian Zhang, Yaqin Liu, Xiao-Lan Yin, and Shaoyong Lu. 2021. "Conformational Selection Mechanism Provides Structural Insights into the Optimization of APC-Asef Inhibitors" Molecules 26, no. 4: 962. https://doi.org/10.3390/molecules26040962