
Development and Validation of an Automated Zone Fluidics-Based Sensor for In Vitro Dissolution Studies of Captopril Using Total Error Concept




Abstract
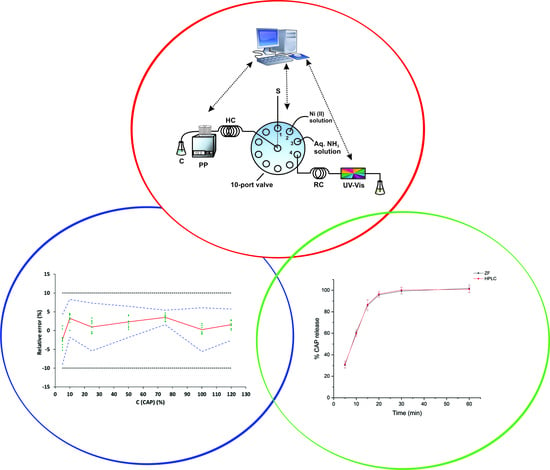
:
1. Introduction
2. Results and Discussion
2.1. Preliminary Experiments
2.2. Optimization of ZF Variables
2.3. Method Validation
2.3.1. Selectivity
2.3.2. Selection of the Response Function
2.3.3. Trueness, Precision and Accuracy
2.3.4. Linearity, LODs and LOQs
2.3.5. Robustness
2.4. Application in Dissolution Studies
3. Materials and Methods
3.1. Reagents, Solutions and Materials
3.2. Solutions for Method Validation
3.3. Instrumentation and Apparatus
3.4. ZF Procedure for the Determination of CAP
3.5. USP HPLC Conditions
3.6. Accuracy Profiles
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Musuc, A.M.; Anuta, V.; Atkinson, I.; Popa, V.T.; Sarbu, I.; Mircioiu, C.; Abdalrb, G.A.; Mitu, M.A.; Ozon, E.A. Development and Characterization of Orally Disintegrating Tablets Containing a Captopril-Cyclodextrin Complex. Pharmaceutics 2020, 12, 744. [Google Scholar] [CrossRef]
- Chikukwa, M.T.R.; Walker, R.B.; Khamanga, S.M.M. Formulation and Characterisation of a Combination Captopril and Hydrochlorothiazide Microparticulate Dosage Form. Pharmaceutics 2020, 12, 712. [Google Scholar] [CrossRef]
- Michalowski, C.B.; Arbo, M.D.; Altknecht, L.; Anciuti, A.N.; Abreu, A.S.G.; Alencar, L.M.R.; Pohlmann, A.R.; Garcia, S.C.; Guterres, S.S. Oral Treatment of Spontaneously Hypertensive Rats with Captopril-Surface Functionalized Furosemide-Loaded Multi-Wall Lipid-Core Nanocapsules. Pharmaceutics 2020, 12, 80. [Google Scholar] [CrossRef] [Green Version]
- Radivojev, S.; Zellnitz, S.; Paudel, A.; Fröhlich, E. Searching for physiologically relevant in vitro dissolution techniques for orally inhaled drugs. Int. J. Pharm. 2019, 556, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Dressman, J. Evolution of dissolution media over the last twenty years. Dissolution Technol. 2014, 21, 6–10. [Google Scholar] [CrossRef]
- Grignard, E.; Taylor, R.; McAllister, M.; Box, K.; Fotaki, N. Considerations for the development of in vitro dissolution tests to reduce or replace preclinical oral absorption studies. Eur. J. Pharm. Sci. 2017, 99, 193–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiwfo, K.; Wongwilai, W.; Sakai, T.; Teshima, N.; Grudpan, K. Determination of Albumin, Glucose, and Creatinine Employing a Single Sequential Injection Lab-at-Valve with Mono-Segmented Flow System Enabling In-Line Dilution, In-Line Single-Standard Calibration, and In-Line Standard Addition. Molecules 2020, 25, 1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzanavaras, P.D.; Papadimitriou, S.; Zacharis, C.K. Automated Stopped-Flow Fluorimetric Sensor for Biologically Active Adamantane Derivatives Based on Zone Fluidics. Molecules 2019, 24, 3975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motz, S.A.; Klimundová, J.; Schaefer, U.F.; Balbach, S.; Eichinger, T.; Solich, P.; Lehr, C.-M. Automated measurement of permeation and dissolution of propranolol HCl tablets using sequential injection analysis. Anal. Chim. Acta 2007, 581, 174–180. [Google Scholar] [CrossRef]
- Tzanavaras, P.D. Automated Determination of Captopril by Flow and Sequential Injection Analysis: A Review. Anal. Lett. 2011, 44, 560–576. [Google Scholar] [CrossRef]
- De Oliveira, D.M.; Suarez, W.T.; Júnior, B.R.A.; Gabriel, W.L.; dos Santos, V.B. Nitroprusside as a Novel Reagent for Flow Injection Spectrophotometric Determination of Captopril. Anal. Lett. 2016, 49, 200–207. [Google Scholar] [CrossRef]
- Vicentini, F.C.; Suarez, W.T.; Cavalheiro, É.T.G.; Fatibello-Filho, O. Flow-injection spectrophotometric determination of captopril in pharmaceutical formulations using a new solid-phase reactor containing AgSCN immobilized in a polyurethane resin. Brazilian J. Pharm. Sci. 2012, 48, 325–333. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, S.S.M.; Santos, J.L.M. Chemiluminometric determination of captopril in a multi-pumping flow system. Talanta 2012, 96, 210–215. [Google Scholar] [CrossRef]
- Karakosta, T.D.; Tzanavaras, P.D.; Themelis, D.G. Automated determination of total captopril in urine by liquid chromatography with post-column derivatization coupled to on-line solid phase extraction in a sequential injection manifold. Talanta 2012, 88, 561–566. [Google Scholar] [CrossRef]
- Wei, X.; Du, L.; Li, D.; Gong, Q.; Wang, L.; Lin, Y. Spectral characteristic investigation on complex of Ni (II) with captopril and its analytical application. Spectrochim. Acta Part. A Mol. Biomol. Spectrosc. 2012, 94, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Refat, M.S.; Alghool, S.; de Farias, R.F. Synthesis, Spectral, and Thermal Studies of Cu(II), Ni(II), Fe(III), and Sn(II) Complexes with Captopril Drug. Synth. React. Inorganic, Met. Nano-Metal. Chem. 2010, 40, 585–591. [Google Scholar] [CrossRef]
- IFPMA. ICH Harmonised Tripartite Guideline, Validation of Analytical Procedures: Text and Methodology, Q2 (R1). In International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use; IFPMA: Geneva, Switzerland, 2005. [Google Scholar]
- USP-38 <1902>: The dissolution procedure: Development and validation. United States Pharmacop. Natl. Formul. 2015.
- Hubert, P.; Nguyen-Huu, J.-J.; Boulanger, B.; Chapuzet, E.; Chiap, P.; Cohen, N.; Compagnon, P.-A.; Dewé, W.; Feinberg, M.; Lallier, M.; et al. Harmonization of strategies for the validation of quantitative analytical procedures A SFSTP proposal–Part I. J. Pharm. Biomed. Anal. 2004, 36, 579–586. [Google Scholar] [CrossRef]
- Hubert, P.; Nguyen-Huu, J.J.; Boulanger, B.; Chapuzet, E.; Chiap, P.; Cohen, N.; Compagnon, P.A.; Dewe, W.; Feinberg, M.; Lallier, M.; et al. Validation of quantitative analytical procedure, harmonization of approaches. S.T.P. Pharma Prat. 2003, 13, 101–138. [Google Scholar]
- U.S. Pharmacopeia XXIX. 2005; 1430–1431.
- Feinberg, M. Validation of analytical methods based on accuracy profiles. J. Chromatogr. A 2007, 1158, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Hubert, P.; Nguyen-Huu, J.-J.; Boulanger, B.; Chapuzet, E.; Cohen, N.; Compagnon, P.-A.; Dewé, W.; Feinberg, M.; Laurentie, M.; Mercier, N.; et al. Harmonization of strategies for the validation of quantitative analytical procedures A SFSTP proposal–Part II. J. Pharm. Biomed. Anal. 2007, 45, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Hubert, P.; Nguyen-Huu, J.-J.; Boulanger, B.; Chapuzet, E.; Cohen, N.; Compagnon, P.-A.; Dewé, W.; Feinberg, M.; Laurentie, M.; Mercier, N.; et al. Harmonization of strategies for the validation of quantitative analytical procedures: A SFSTP proposal-Part III. J. Pharm. Biomed. Anal. 2007, 45, 82–96. [Google Scholar] [CrossRef] [PubMed]
- Hubert, P.; Nguyen-Huu, J.-J.; Boulanger, B.; Chapuzet, E.; Cohen, N.; Compagnon, P.-A.; Dewé, W.; Feinberg, M.; Laurentie, M.; Mercier, N.; et al. Harmonization of strategies for the validation of quantitative analytical procedures: A SFSTP proposal-Part IV. J. Pharm. Biomed. Anal. 2008, 48, 760–771. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Validation Criteria | |||
|---|---|---|---|
| Response function (linear regression) | Slope | Intercept (×103) | r2 |
| (k a = 3; m = 7; n = 3) (5–120%) | |||
| Day 1 | 3.5007 | −1.3337 | 0.9986 |
| Day 2 | 3.5055 | 1.257 | 0.9979 |
| Day 3 | 3.4988 | 1.9391 | 0.9978 |
| Precision (k = 3; n = 3) | |||
| C (%) | sr (%) b | sR (%) c | |
| 5 | 1.7 | 2.3 | |
| 10 | 1.1 | 1.6 | |
| 25 | 1.0 | 1.8 | |
| 50 | 1.3 | 1.5 | |
| 75 | 0.8 | 2.1 | |
| 100 | 0.74 | 1.4 | |
| 120 | 0.6 | 1.2 | |
| Trueness (k = 3; n = 3) | |||
| C (%) | Relative bias (%) | ||
| 5 | −2.3 | ||
| 10 | +3.3 | ||
| 25 | +1.0 | ||
| 50 | +2.3 | ||
| 75 | +3.5 | ||
| 100 | +0.3 | ||
| 120 | +1.6 | ||
| Accuracy (k = 5; n = 3) | |||
| C (%) | Relative β-ΕΤΙ (%) | ||
| 5 | (−9.00, 4.33) | ||
| 10 | (−1.77, 8.29) | ||
| 25 | (−5.42, 7.34) | ||
| 50 | (−1.81, 6.47) | ||
| 75 | (−1.60, 5.41) | ||
| 100 | (−5.59, 6.09) | ||
| 120 | (−2.62, 5.74) | ||
| Linearity (k = 3; n = 3; m = 7) (5–120%) | |||
| Slope | 1.026 | ||
| Intercept | 1.026 | ||
| r2 | 0.9999 | ||
| LOD (%) | 1 | ||
| LLOQ/ULOQ (%) | 5/120 | ||
| Time (min) | % CAP Release (±SD) (Brand A) | % CAP Release (±SD) (Brand B) | ||||
|---|---|---|---|---|---|---|
| ZF | HPLC | p-Value | ZF | HPLC | p-Value | |
| 5 | 30.4 (±2.9) | 31.2 (±3.2) | 0.769 | 27.3 (±3.0) | 26.5 (±2.5) | 0.803 |
| 10 | 60.8 (±2.7) | 59.6 (±2.9) | 0.636 | 63.7 (±5.8) | 61.8 (±4.1) | 0.675 |
| 15 | 85.9 (±5.1) | 86.7 (±4.6) | 0.853 | 88.9 (±5.7) | 87.1 (±4.9) | 0.706 |
| 20 | 95.7 (±2.3) | 96.5 (±2.7) | 0.722 | 92.6 (±4.9) | 94.1 (±4.1) | 0.712 |
| 30 | 99.3 (±3.1) | 100.2 (±2.9) | 0.738 | 100.5 (±3.5) | 99.7 (±4.0) | 0.811 |
| 60 | 101.7 (±3.4) | 100.9 (±3.2) | 0.786 | 98.6 (±2.3) | 99.1 (±2.9) | 0.830 |
| a/a | Time (s) | Valve Position | Pump Action | Flow Rate (mL min−1) | Volume (μL) | Action Description |
|---|---|---|---|---|---|---|
| 1 | 1 | 2 | Off | — | — | Selection of Ni(II) solution port |
| 2 | 5 | 2 | Aspirate | 0.6 | 50 | Aspiration of Ni(II) solution in the HC |
| 3 | 1 | 1 | Off | — | — | Selection of sample port |
| 4 | 10 | 1 | Aspirate | 0.6 | 100 | Aspiration of sample in the HC |
| 5 | 1 | 3 | Off | — | — | Selection of aqueous NH3 solution port |
| 6 | 5 | 3 | Aspirate | 0.6 | 50 | Aspiration of aqueous NH3 solution in the HC |
| 7 | 1 | 4 | Off | — | — | Selection of UV-Vis detector port |
| 8 | 120 | 4 | Deliver | 0.9 | 1800 | Deliver of the reaction mixture to the UV-Vis detector |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karakosta, T.D.; Tzanavaras, P.D.; Zacharis, C.K. Development and Validation of an Automated Zone Fluidics-Based Sensor for In Vitro Dissolution Studies of Captopril Using Total Error Concept. Molecules 2021, 26, 824. https://doi.org/10.3390/molecules26040824
Karakosta TD, Tzanavaras PD, Zacharis CK. Development and Validation of an Automated Zone Fluidics-Based Sensor for In Vitro Dissolution Studies of Captopril Using Total Error Concept. Molecules. 2021; 26(4):824. https://doi.org/10.3390/molecules26040824
Chicago/Turabian StyleKarakosta, Theano D., Paraskevas D. Tzanavaras, and Constantinos K. Zacharis. 2021. "Development and Validation of an Automated Zone Fluidics-Based Sensor for In Vitro Dissolution Studies of Captopril Using Total Error Concept" Molecules 26, no. 4: 824. https://doi.org/10.3390/molecules26040824