Antiangiogenic Activity and in Silico Cereblon Binding Analysis of Novel Thalidomide Analogs

 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
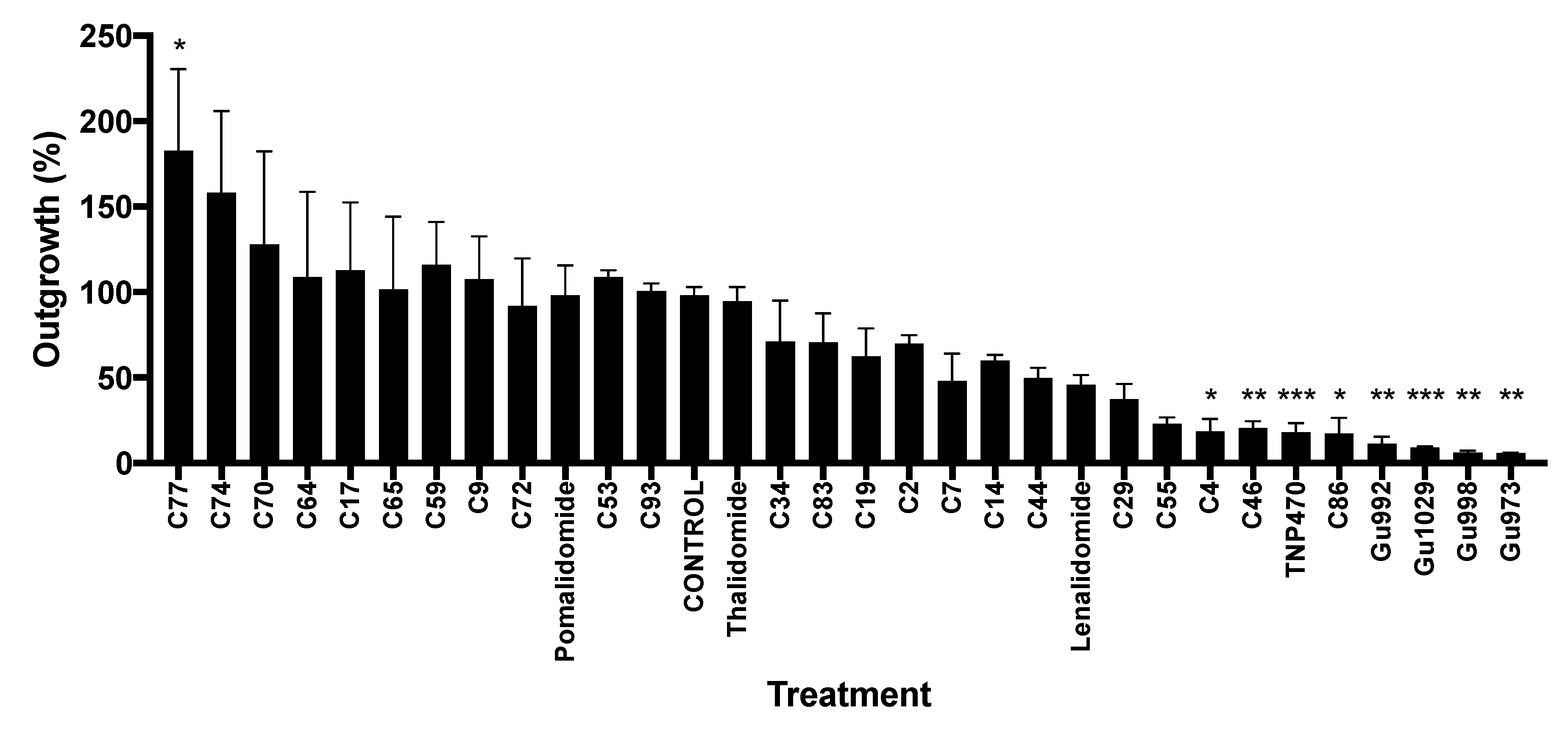
2.1. Biological Testing of Thalidomide Analogs
2.2. In Silico Protein Docking Simulation
3. Material and Methods
3.1. Thalidomide Analogs
3.2. Rat Aorta Ring (RAR) Assay of Angiogenesis
3.3. Molecular Modeling
3.4. Induced-Fit Docking
3.5. Pharmacophore Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Aragon-Ching, J.B.; Li, H.; Gardner, E.R.; Figg, W.D. Thalidomide Analogues as Anticancer Drugs. Recent Pat. Anticancer Drug Discov. 2007, 2, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Millrine, D.; Kishimoto, T. A Brighter Side to Thalidomide: Its Potential Use in Immunological Disorders. Trends. Mol. Med. 2017, 23, 348–361. [Google Scholar] [CrossRef]
- Sherbet, G.V. Therapeutic Potential of Thalidomide and Its Analogues in the Treatment of Cancer. Anticancer Res. 2015, 35, 5767–5772. [Google Scholar]
- Lepper, E.R.; Smith, N.F.; Cox, M.C.; Scripture, C.D.; Figg, W.D. Thalidomide Metabolism and Hydrolysis: Mechanisms and Implications. Curr. Drug Metab. 2006, 7, 677–685. [Google Scholar] [CrossRef]
- Paravar, T.; Lee, D.J. Thalidomide: Mechanisms of Action. Int. Rev. Immunol. 2008, 27, 111–135. [Google Scholar] [CrossRef]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a Primary Target of Thalidomide Teratogenicity. Science 2010, 327, 1345–1350. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Handa, H. Cereblon and Its Downstream Substrates as Molecular Targets of Immunomodulatory Drugs. Int. J. Hematol. 2016, 104, 293–299. [Google Scholar] [CrossRef] [Green Version]
- Kronke, J.; Fink, E.C.; Hollenbach, P.W.; MacBeth, K.J.; Hurst, S.N.; Udeshi, N.D.; Chamberlain, P.P.; Mani, D.R.; Man, H.W.; Gandhi, A.K.; et al. Lenalidomide Induces Ubiquitination and Degradation of Ck1alpha in Del(5q) Mds. Nature 2015, 523, 183–188. [Google Scholar] [CrossRef]
- Matyskiela, M.E.; Couto, S.; Zheng, X.; Lu, G.; Hui, J.; Stamp, K.; Drew, C.; Ren, Y.; Wang, M.; Carpenter, A.; et al. Sall4 Mediates Teratogenicity as a Thalidomide-Dependent Cereblon Substrate. Nat. Chem. Biol. 2018, 14, 981–987. [Google Scholar] [CrossRef]
- Chamberlain, P.P.; Lopez-Girona, A.; Miller, K.; Carmel, G.; Pagarigan, B.; Chie-Leon, B.; Rychak, E.; Corral, L.G.; Ren, Y.J.; Wang, M.; et al. Structure of the Human Cereblon-Ddb1-Lenalidomide Complex Reveals Basis for Responsiveness to Thalidomide Analogs. Nat. Struct. Mol. Biol. 2014, 21, 803–809. [Google Scholar] [CrossRef]
- Fischer, E.S.; Bohm, K.; Lydeard, J.R.; Yang, H.; Stadler, M.B.; Cavadini, S.; Nagel, J.; Serluca, F.; Acker, V.; Lingaraju, G.M.; et al. Structure of the Ddb1-Crbn E3 Ubiquitin Ligase in Complex with Thalidomide. Nature 2014, 7512, 49–53. [Google Scholar] [CrossRef] [Green Version]
- Matyskiela, M.E.; Clayton, T.; Zheng, X.; Mayne, C.; Tran, E.; Carpenter, A.; Pagarigan, B.; McDonald, J.; Rolfe, M.; Hamann, L.G.; et al. Crystal Structure of the Sall4-Pomalidomide-Cereblon-Ddb1 Complex. Nat. Struct. Mol. Biol. 2020, 27, 319–322. [Google Scholar] [CrossRef]
- Matyskiela, M.E.; Lu, G.; Ito, T.; Pagarigan, B.; Lu, C.C.; Miller, K.; Fang, W.; Wang, N.Y.; Nguyen, D.; Houston, J.; et al. A Novel Cereblon Modulator Recruits Gspt1 to the Crl4(Crbn) Ubiquitin Ligase. Nature 2016, 535, 252–257. [Google Scholar] [CrossRef]
- Petzold, G.; Fischer, E.S.; Thoma, N.H. Structural Basis of Lenalidomide-Induced Ck1alpha Degradation by the Crl4(Crbn) Ubiquitin Ligase. Nature 2016, 532, 127–130. [Google Scholar] [CrossRef]
- Eriksson, T.; Bjorkman, S.; Roth, B.; Fyge, A.; Hoglund, P. Stereospecific Determination, Chiral Inversion in Vitro and Pharmacokinetics in Humans of the Enantiomers of Thalidomide. Chirality 1995, 7, 44–52. [Google Scholar] [CrossRef]
- Blaschke, G.; Kraft, H.P.; Fickentscher, K.; Kohler, F. Chromatographic Separation of Racemic Thalidomide and Teratogenic Activity of Its Enantiomers (Author’s Transl). Arzneimittelforschung 1979, 29, 1640–1642. [Google Scholar]
- Mori, T.; Ito, T.; Liu, S.; Ando, H.; Sakamoto, S.; Yamaguchi, Y.; Tokunaga, E.; Shibata, N.; Handa, H.; Hakoshima, T. Structural Basis of Thalidomide Enantiomer Binding to Cereblon. Sci. Rep. 2018, 8, 1294. [Google Scholar] [CrossRef]
- Beedie, S.L.; Rore, H.M.; Barnett, S.; Chau, C.H.; Luo, W.; Greig, N.H.; Figg, W.D.; Vargesson, N. In Vivo Screening and Discovery of Novel Candidate Thalidomide Analogs in the Zebrafish Embryo and Chicken Embryo Model Systems. Oncotarget 2016, 7, 33237–33245. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Tweedie, D.; Beedie, S.L.; Vargesson, N.; Figg, W.D.; Greig, N.H.; Scerba, M.T. Design, Synthesis and Biological Assessment of N-Adamantyl, Substituted Adamantyl and Noradamantyl Phthalimidines for Nitrite, Tnf-Alpha and Angiogenesis Inhibitory Activities. Bioorg. Med. Chem. 2018, 26, 1547–1559. [Google Scholar] [CrossRef]
- Ambrozak, A.; Steinebach, C.; Gardner, E.R.; Beedie, S.L.; Schnakenburg, G.; Figg, W.D.; Gutschow, M. Synthesis and Antiangiogenic Properties of Tetrafluorophthalimido and Tetrafluorobenzamido Barbituric Acids. Chem. Med. Chem. 2016, 11, 2621–2629. [Google Scholar] [CrossRef]
- Gutschow, M.; Hecker, T.; Thiele, A.; Hauschildt, S.; Eger, K. Aza Analogues of Thalidomide: Synthesis and Evaluation as Inhibitors of Tumor Necrosis Factor-Alpha Production in Vitro. Bioorg. Med. Chem. 2001, 9, 1059–1065. [Google Scholar] [CrossRef]
- Hashimoto, Y. Structural Development of Biological Response Modifiers Based on Retinoids and Thalidomide. Mini Rev. Med. Chem. 2002, 2, 543–551. [Google Scholar] [CrossRef]
- Ng, S.S.; Gutschow, M.; Weiss, M.; Hauschildt, S.; Teubert, U.; Hecker, T.K.; Luzzio, F.A.; Kruger, E.A.; Eger, K.; Figg, W.D. Antiangiogenic Activity of N-Substituted and Tetrafluorinated Thalidomide Analogues. Cancer Res. 2003, 63, 3189–3194. [Google Scholar]
- Muller, G.W.; Corral, L.G.; Shire, M.G.; Wang, H.; Moreira, A.; Kaplan, G.; Stirling, D.I. Structural Modifications of Thalidomide Produce Analogs with Enhanced Tumor Necrosis Factor Inhibitory Activity. J. Med. Chem. 1996, 39, 3238–3240. [Google Scholar] [CrossRef]
- Muller, G.W.; Shire, M.G.; Wong, L.M.; Corral, L.G.; Patterson, R.T.; Chen, Y.; Stirling, D.I. Thalidomide Analogs and Pde4 Inhibition. Bioorg Med. Chem. Lett. 1998, 8, 2669–2674. [Google Scholar] [CrossRef]
- Beedie, S.L.; Peer, C.J.; Pisle, S.; Gardner, E.R.; Mahony, C.; Barnett, S.; Ambrozak, A.; Gutschow, M.; Chau, C.H.; Vargesson, N.; et al. Anticancer Properties of a Novel Class of Tetrafluorinated Thalidomide Analogues. Mol. Cancer Ther. 2015, 14, 2228–2237. [Google Scholar] [CrossRef] [Green Version]
- Bauer, K.S.; Cude, K.J.; Dixon, S.C.; Kruger, E.A.; Figg, W.D. Carboxyamido-Triazole Inhibits Angiogenesis by Blocking the Calcium-Mediated Nitric-Oxide Synthase-Vascular Endothelial Growth Factor Pathway. J. Pharmacol Exp. Ther. 2000, 292, 31–37. [Google Scholar]
- Iqbal, F.; Gratch, Y.S.; Szaraz, P.; Librach, C.L. The Aortic Ring Co-Culture Assay: A Convenient Tool to Assess the Angiogenic Potential of Mesenchymal Stromal Cells in Vitro. J. Vis. Exp. 2017, 127, e56083. [Google Scholar] [CrossRef]
- Murai, T.; Kawashita, N.; Tian, Y.S.; Takagi, T. In Silico Analysis of Enantioselective Binding of Immunomodulatory Imide Drugs to Cereblon. Springerplus 2016, 5, 1122. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, M.D.; Boichenko, I.; Coles, M.; Lupas, A.N.; Hernandez Alvarez, B. Structural Dynamics of the Cereblon Ligand Binding Domain. PLoS ONE 2015, 10, e0128342. [Google Scholar] [CrossRef]
- Fu, D.Y.; Meiler, J. Rosettaligandensemble: A Small-Molecule Ensemble-Driven Docking Approach. Acs Omega 2018, 3, 3655–3664. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.Y.; Li, M.; Wang, J.; Pan, Y. Hybriddock: A Hybrid Protein-Ligand Docking Protocol Integrating Protein- and Ligand-Based Approaches. J. Chem. Inf. Model. 2016, 56, 1078–1087. [Google Scholar] [CrossRef]
- Lexa, K.W.; Carlson, H.A. Protein Flexibility in Docking and Surface Mapping. Q Rev. Biophys. 2012, 45, 301–343. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the Role of the Crystal Environment in Determining Protein Side-Chain Conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef]
- Lopez-Girona, A.; Mendy, D.; Ito, T.; Miller, K.; Gandhi, A.K.; Kang, J.; Karasawa, S.; Carmel, G.; Jackson, P.; Abbasian, M.; et al. Cereblon Is a Direct Protein Target for Immunomodulatory and Antiproliferative Activities of Lenalidomide and Pomalidomide. Leukemia 2012, 26, 2326–2335. [Google Scholar] [CrossRef]
- Matyskiela, M.E.; Zhang, W.; Man, H.W.; Muller, G.; Khambatta, G.; Baculi, F.; Hickman, M.; LeBrun, L.; Pagarigan, B.; Carmel, G.; et al. A Cereblon Modulator (Cc-220) with Improved Degradation of Ikaros and Aiolos. J. Med. Chem. 2018, 61, 535–542. [Google Scholar] [CrossRef]
- Price, D.K.; Ando, Y.; Kruger, E.A.; Weiss, M.; Figg, W.D. 5′-Oh-Thalidomide, a Metabolite of Thalidomide, Inhibits Angiogenesis. Drug Monit. 2002, 24, 104–110. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kasserra, C.; Reyes, J.; Schafer, P.; Kosek, J.; Capone, L.; Parton, A.; Kim-Kang, H.; Surapaneni, S.; Kumar, G. Absorption, Metabolism and Excretion of [14c]Pomalidomide in Humans Following Oral Administration. Cancer Chemother. Pharm. 2013, 71, 489–501. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, H.; Smith, R.L.; Williams, R.T. The Metabolism of Thalidomide: The Spontaneous Hydrolysis of Thalidomide in Solution. Br. J. Pharm. Chemother. 1965, 25, 324–337. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Zhou, S.; Palmisano, M. Clinical Pharmacokinetics and Pharmacodynamics of Lenalidomide. Clin. Pharm. 2017, 56, 139–152. [Google Scholar] [CrossRef] [Green Version]
- Kruger, E.A.; Figg, W.D. Tnp-470: An Angiogenesis Inhibitor in Clinical Development for Cancer. Expert Opin. Investig. Drugs 2000, 9, 1383–1396. [Google Scholar] [CrossRef]
- Halgren, T.A. Mmff Vi. Mmff94s Option for Energy Minimization Studies. J. Comput. Chem. 1999, 20, 720–729. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. Opls3: A Force Field Providing Broad Coverage of Drug-Like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The Vsgb 2.0 Model: A Next Generation Energy Model for High Resolution Protein Structure Modeling. Proteins 2011, 79, 2794–2812. [Google Scholar] [CrossRef] [Green Version]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.L.; Smondyrev, A.M.; Rao, S.N. Phase: A Novel Approach to Pharmacophore Modeling and 3d Database Searching. Chem. Biol. Drug Des. 2006, 67, 370–372. [Google Scholar] [CrossRef]
- Lepper, E.R.; Ng, S.S.; Gutschow, M.; Weiss, M.; Hauschildt, S.; Hecker, T.K.; Luzzio, F.A.; Eger, K.; Figg, W.D. Comparative Molecular Field Analysis and Comparative Molecular Similarity Indices Analysis of Thalidomide Analogues as Angiogenesis Inhibitors. J. Med. Chem. 2004, 47, 2219–2227. [Google Scholar] [CrossRef]
- Godin, A.M.; Araujo, D.P.; Menezes, R.R.; Brito, A.M.; Melo, I.S.; Coura, G.M.; Soares, D.G.; Bastos, L.F.; Amaral, F.A.; Ribeiro, L.S.; et al. Activities of 2-Phthalimidethanol and 2-Phthalimidethyl Nitrate, Phthalimide Analogs Devoid of the Glutarimide Moiety, in Experimental Models of Inflammatory Pain and Edema. Pharm. Biochem. Behav. 2014, 122, 291–298. [Google Scholar] [CrossRef]
- Matijevic-Sosa, J.; Cvetnic, Z. Antimicrobial Activity of N-Phthaloylamino Acid Hydroxamates. Acta Pharm. 2005, 55, 387–399. [Google Scholar]
- Yamamoto, T.; Shibata, N.; Takashima, M.; Nakamura, S.; Toru, T.; Matsunaga, N.; Hara, H. Enzymatic Resolution and Evaluation of Enantiomers of Cis-5’-Hydroxythalidomide. Org. Biomol. Chem. 2008, 6, 1540–1543. [Google Scholar] [CrossRef]
- Avila, C.M.; Romeiro, N.C.; da Silva, G.M.; Sant’Anna, C.M.; Barreiro, E.J.; Fraga, C.A. Development of New Comfa and Comsia 3d-Qsar Models for Anti-Inflammatory Phthalimide-Containing Tnfalpha Modulators. Bioorg. Med. Chem. 2006, 14, 6874–6885. [Google Scholar] [CrossRef]
- Dean, P.M. Molecular Similarity in Drug Design; Blackie Academic & Professional: New York, NY, USA, 1995. [Google Scholar]
- Kubinyi, H. Qsar and 3d Qsar in Drug Design.1. Methodology. Drug Discov. 1997, 2, 457–467. [Google Scholar] [CrossRef]
- Boichenko, I.; Bar, K.; Deiss, S.; Heim, C.; Albrecht, R.; Lupas, A.N.; Hernandez Alvarez, B.; Hartmann, M.D. Chemical Ligand Space of Cereblon. ACS Omega. 2018, 3, 11163–11171. [Google Scholar] [CrossRef]
- Brunhofer, G.; Granig, W.H.; Studenik, C.R.; Erker, T. A Journey from Benzanilides to Dithiobenzanilides: Synthesis of Selective Spasmolytic Compounds. Bioorg. Med. Chem. 2011, 19, 994–1001. [Google Scholar] [CrossRef]
- Fischer, A.; Schmidt, C.; Lachenicht, S.; Grittner, D.; Winkler, M.; Wrobel, T.; Rood, A.; Lemoine, H.; Frank, W.; Braun, M. Synthesis of Benzofuran, Benzothiophene, and Benzothiazole-Based Thioamides and Their Evaluation as K(Atp) Channel Openers. Chem. Med. Chem. 2010, 5, 1749–1759. [Google Scholar] [CrossRef]
- Wanka, L.; Iqbal, K.; Schreiner, P.R. The Lipophilic Bullet Hits the Targets: Medicinal Chemistry of Adamantane Derivatives. Chem. Rev. 2013, 113, 3516–3604. [Google Scholar] [CrossRef] [Green Version]
- Cady, S.D.; Wang, J.; Wu, Y.; DeGrado, W.F.; Hong, M. Specific Binding of Adamantane Drugs and Direction of Their Polar Amines in the Pore of the Influenza M2 Transmembrane Domain in Lipid Bilayers and Dodecylphosphocholine Micelles Determined by Nmr Spectroscopy. J. Am. Chem. Soc. 2011, 133, 4274–4284. [Google Scholar] [CrossRef] [Green Version]
- Kaefer, V.; Semedo, J.G.; Silva Kahl, V.F.; Von Borowsky, R.G.; Gianesini, J.; Ledur Kist, T.B.; Pereira, P.; Picada, J.N. DNA Damage in Brain Cells and Behavioral Deficits in Mice after Treatment with High Doses of Amantadine. J. Appl. Toxicol. 2010, 30, 745–753. [Google Scholar] [CrossRef]
- Shimizu, K.; Costa Gomes, M.F.; Padua, A.A.; Rebelo, L.P.; Canongia Lopes, J.N. On the Role of the Dipole and Quadrupole Moments of Aromatic Compounds in the Solvation by Ionic Liquids. J. Phys. Chem. B. 2009, 113, 9894–9900. [Google Scholar] [CrossRef]
- Steinebach, C.; Ambrozak, A.; Dosa, S.; Beedie, S.L.; Strope, J.D.; Schnakenburg, G.; Figg, W.D.; Gutschow, M. Synthesis, Structural Characterization, and Antiangiogenic Activity of Polyfluorinated Benzamides. Chem. Med. Chem. 2018, 13, 2080–2089. [Google Scholar] [CrossRef]
- Beedie, S.L.; Huang, P.A.; Harris, E.M.; Strope, J.D.; Mahony, C.; Chau, C.H.; Vargesson, N.; Figg, W.D. Role of Cereblon in Angiogenesis and in Mediating the Antiangiogenic Activity of Immunomodulatory Drugs. Faseb J. 2020, 34, 11395–11404. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Tanatani, A.; Nagasawa, K.; Miyachi, H. Thalidomide as a Multitarget Drug and Its Application as a Template for Drug Design. Drugs Future 2004, 29, 383–391. [Google Scholar] [CrossRef]
- Sahn, J.J.; Su, J.Y.; Martin, S.F. Facile and Unified Approach to Skeletally Diverse, Privileged Scaffolds. Org. Lett. 2011, 13, 2590–2593. [Google Scholar] [CrossRef] [Green Version]
- Sharma, U.; Kumar, P.; Kumar, N.; Singh, B. Recent Advances in the Chemistry of Phthalimide Analogues and Their Therapeutic Potential. Mini. Rev. Med. Chem. 2010, 10, 678–704. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peach, M.L.; Beedie, S.L.; Chau, C.H.; Collins, M.K.; Markolovic, S.; Luo, W.; Tweedie, D.; Steinebach, C.; Greig, N.H.; Gütschow, M.; et al. Antiangiogenic Activity and in Silico Cereblon Binding Analysis of Novel Thalidomide Analogs. Molecules 2020, 25, 5683. https://doi.org/10.3390/molecules25235683
Peach ML, Beedie SL, Chau CH, Collins MK, Markolovic S, Luo W, Tweedie D, Steinebach C, Greig NH, Gütschow M, et al. Antiangiogenic Activity and in Silico Cereblon Binding Analysis of Novel Thalidomide Analogs. Molecules. 2020; 25(23):5683. https://doi.org/10.3390/molecules25235683
Chicago/Turabian StylePeach, Megan L., Shaunna L. Beedie, Cindy H. Chau, Matthew K. Collins, Suzana Markolovic, Weiming Luo, David Tweedie, Christian Steinebach, Nigel H. Greig, Michael Gütschow, and et al. 2020. "Antiangiogenic Activity and in Silico Cereblon Binding Analysis of Novel Thalidomide Analogs" Molecules 25, no. 23: 5683. https://doi.org/10.3390/molecules25235683