
Out of Sight, Out of Mind: The Effect of the Equilibration Protocol on the Structural Ensembles of Charged Glycolipid Bilayers

 ,
,  ,
,  ,
,  and
and
Abstract
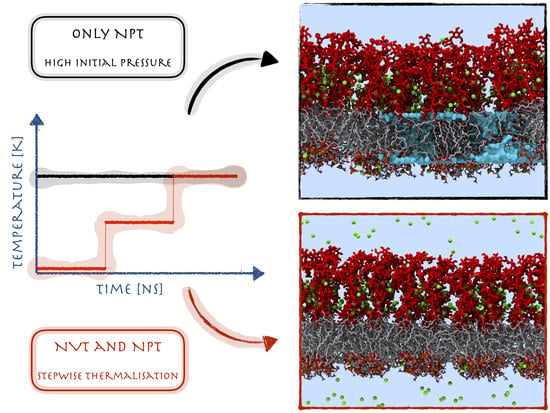
:
1. Introduction
2. Results and Discussion
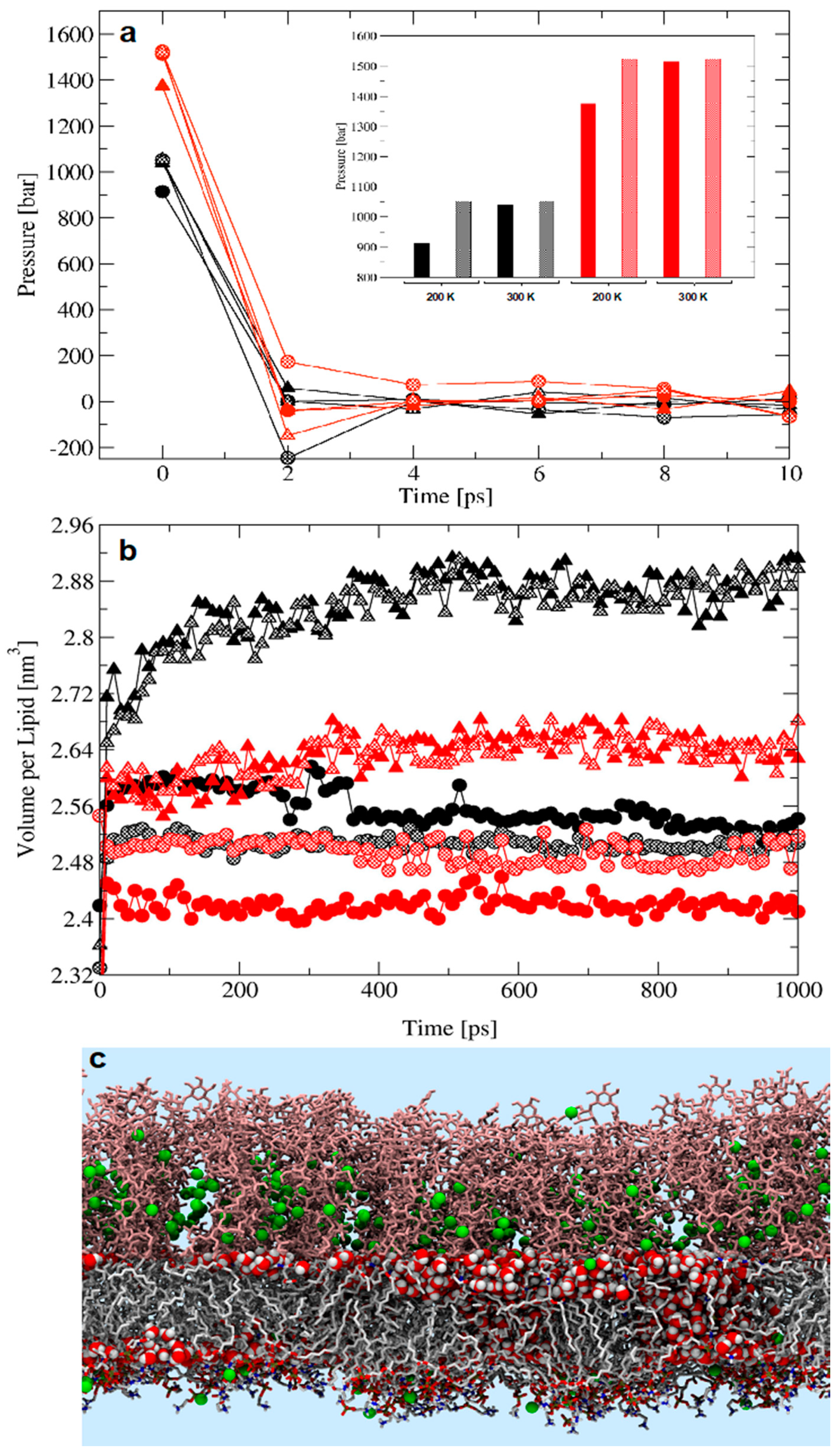
2.1. The Matter with Standard Equilibration Protocols for Glycolipid Membranes
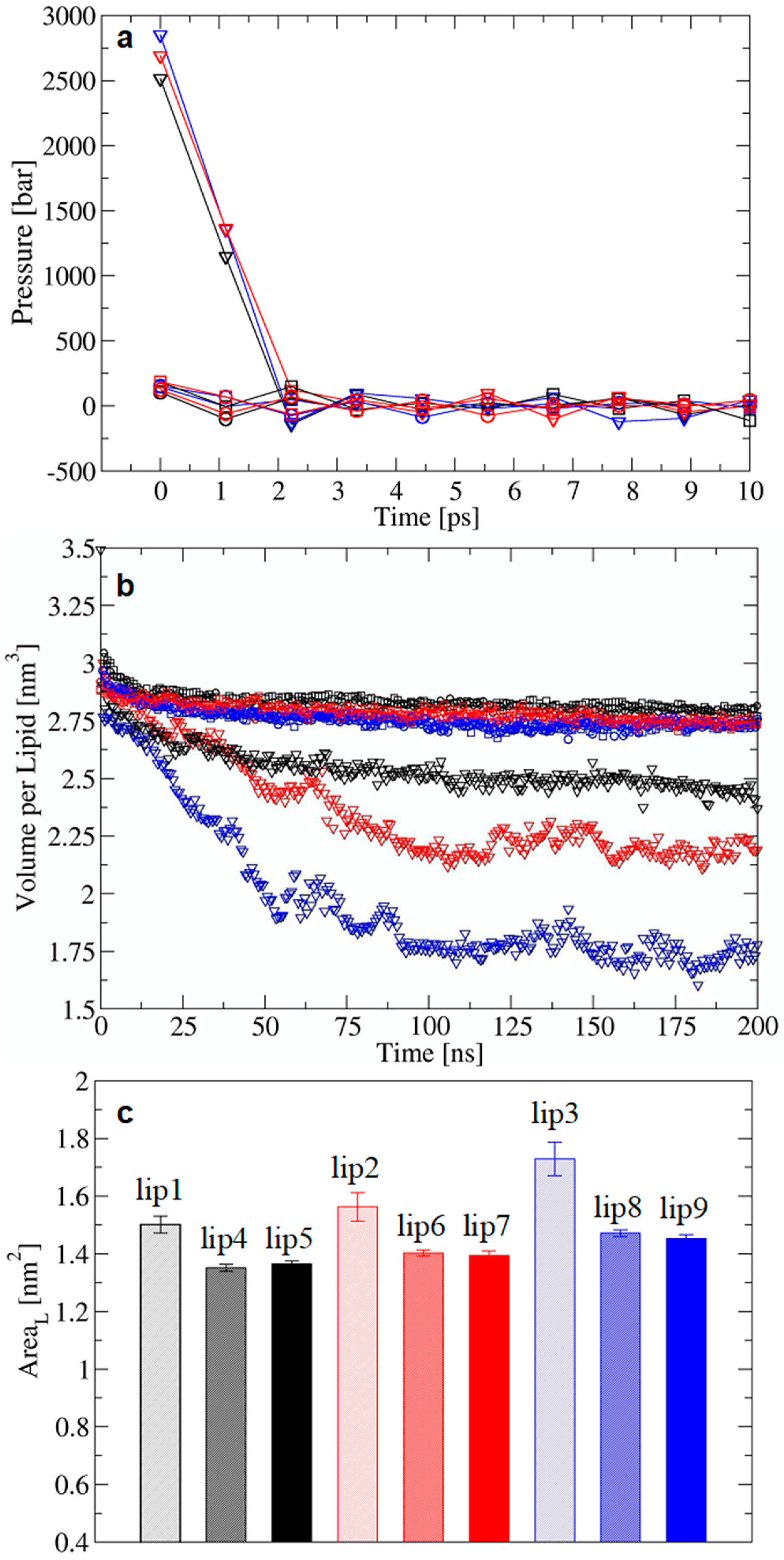
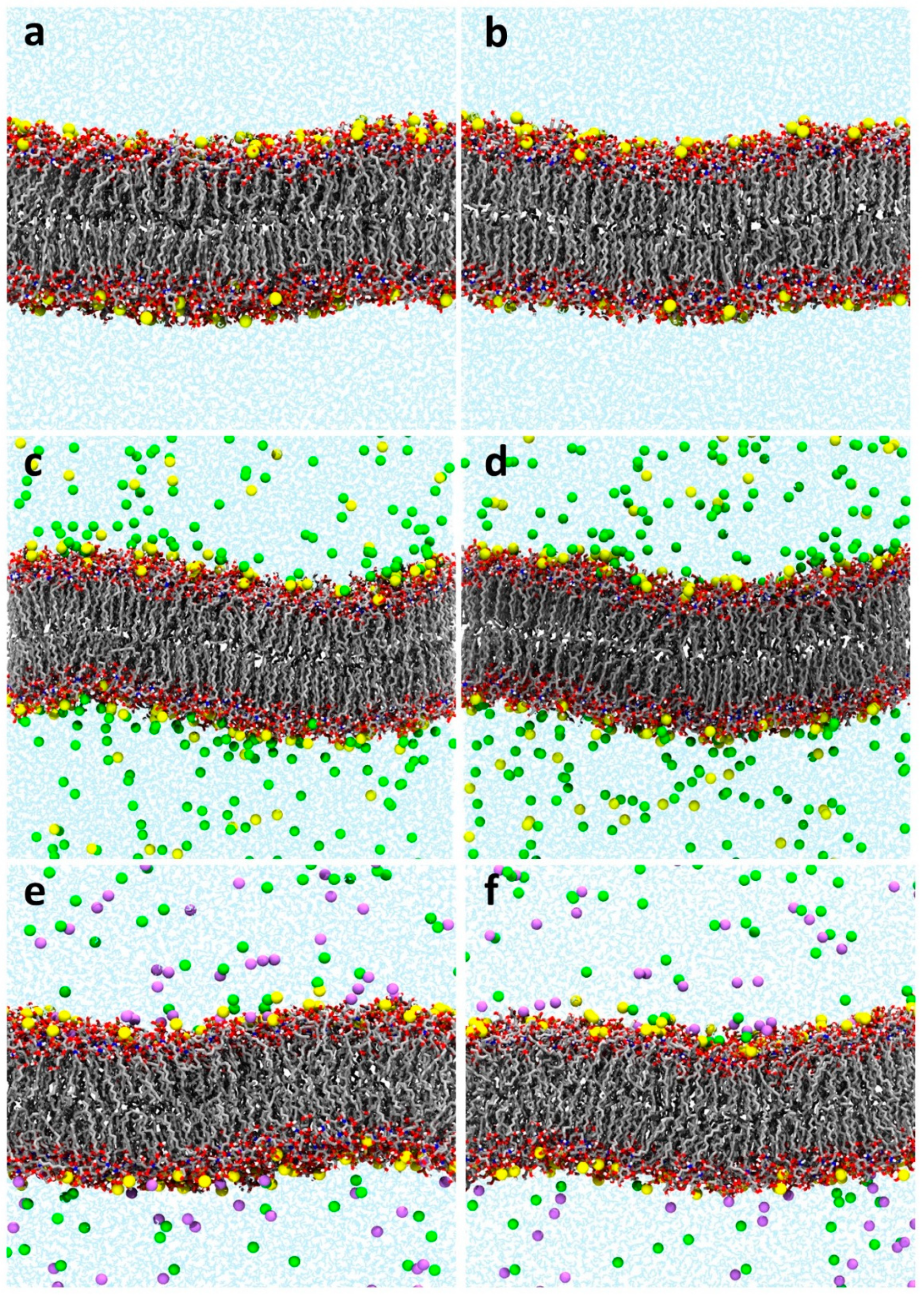
2.2. Validation of the Modified Equilibration Protocol for Glycolipid Membranes
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002, 9, 646–652. [Google Scholar] [CrossRef]
- Schappals, M.; Mecklenfeld, A.; Kröger, L.; Botan, V.; Köster, A.; Stephan, S.; García, E.J.; Rutkai, G.; Raabe, G.; Klein, P.; et al. Round robin study: Molecular simulation of thermodynamic properties from models with internal degrees of freedom. J. Chem. Theory Comput. 2017, 13, 4270–4280. [Google Scholar] [CrossRef]
- Shirts, M.R.; Mobley, D.L.; Chodera, J.D.; Pande, V.S. Accurate and efficient corrections for missing dispersion interactions in molecular simulations. J. Phys. Chem. B 2007, 111, 13052–13063. [Google Scholar] [CrossRef]
- Mobley, D.L.; Bayly, C.I.; Cooper, M.D.; Shirts, M.R.; Dill, K.A. Small molecule hydration free energies in explicit solvent: An extensive test of fixed-charge atomistic simulations. J. Chem. Theory Comput. 2009, 5, 350–358. [Google Scholar] [CrossRef]
- Basconi, J.E.; Shirts, M.R. Effects of temperature control algorithms on transport properties and kinetics in molecular dynamics simulations. J. Chem. Theory Comput. 2013, 9, 2887–2899. [Google Scholar] [CrossRef]
- Braun, E.; Moosavi, S.M.; Smit, B. Anomalous effects of velocity rescaling algorithms: The flying ice cube effect revisited. J. Chem. Theory Comput. 2018, 14, 5262–5272. [Google Scholar] [CrossRef]
- Soares, T.A.; Vanni, S.; Milano, G.; Cascella, M. Toward chemically resolved computer simulations of dynamics and remodeling of biological membranes. J. Phys. Chem. Lett. 2017, 8, 3586–3594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalid, S.; Piggot, T.J.; Samsudin, F. Atomistic and coarse grain simulations of the cell envelope of gram-negative bacteria: What have we learned? Acc. Chem. Res. 2019, 52, 180–188. [Google Scholar] [CrossRef] [Green Version]
- Enkavi, G.; Javanainen, M.; Kulig, W.; Róg, T.; Vattulainen, I. Multiscale simulations of biological membranes: The challenge to understand biological phenomena in a living substance. Chem. Rev. 2019, 119, 5607–5774. [Google Scholar] [CrossRef] [Green Version]
- Marrink, S.J.; Corradi, V.; Souza, P.C.T.; Ingólfsson, H.I.; Tieleman, D.P.; Sansom, M.S.P. Computational modeling of realistic cell membranes. Chem. Rev. 2019, 119, 6184–6226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rice, A.; Rooney, M.T.; Greenwood, A.I.; Cotten, M.L.; Wereszczynski, J. Lipopolysaccharide simulations are sensitive to phosphate charge and ion parameterization. J. Chem. Theory Comput. 2020, 16, 1806–1815. [Google Scholar] [CrossRef] [PubMed]
- Wu, E.L.; Engström, O.; Jo, S.; Stuhlsatz, D.; Yeom, M.S.; Klauda, J.B.; Widmalm, G.; Im, W. Molecular dynamics and NMR spectroscopy studies of E. Coli lipopolysaccharide structure and dynamics. Biophys. J. 2013, 105, 1444–1455. [Google Scholar] [CrossRef] [Green Version]
- Kučerka, N.; Papp-Szabo, E.; Nieh, M.P.; Harroun, T.A.; Schooling, S.R.; Pencer, J.; Nicholson, E.A.; Beveridge, T.J.; Katsaras, J. Effect of cations on the structure of bilayers formed by lipopolysaccharides isolated from pseudomonas aeruginosa PAO1. J. Phys. Chem. B 2008, 112, 8057–8062. [Google Scholar] [CrossRef]
- Kirschner, K.N.; Lins, R.D.; Maass, A.; Soares, T.A. A Glycam-based force field for simulations of lipopolysaccharide membranes: Parametrization and validation. J. Chem. Theory Comput. 2012, 8, 4719–4731. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, A.; Pontes, F.J.S.; Lins, R.D.; Soares, T.A. Hydration, ionic valence and cross-linking propensities of cations determine the stability of lipopolysaccharide (LPS) membranes. Chem. Commun. 2014, 50, 231–233. [Google Scholar] [CrossRef]
- Pontes, F.J.S.; Rusu, V.H.; Soares, T.A.; Lins, R.D. The effect of temperature, cations, and number of Acyl chains on the lamellar to non-lamellar transition in lipid-A membranes: A microscopic view. J. Chem. Theory Comput. 2012, 8, 3830–3838. [Google Scholar] [CrossRef]
- Piggot, T.J.; Holdbrook, D.A.; Khalid, S. Electroporation of the E. Coli and S. Aureus membranes: Molecular dynamics simulations of complex bacterial membranes. J. Phys. Chem. B 2011, 115, 13381–13388. [Google Scholar] [CrossRef]
- Li, P.; Merz, K.M. Metal ion modeling using classical mechanics. Chem. Rev. 2017, 117, 1564–1686. [Google Scholar] [CrossRef]
- Wong-ekkabut, J.; Karttunen, M. The good, the bad and the user in soft matter simulations. Biochim. Biophys. Acta Biomembr. 2016, 1858, 2529–2538. [Google Scholar] [CrossRef]
- Baron, R.; de Vries, A.H.; Hünenberger, P.H.; van Gunsteren, W.F. Configurational entropies of lipids in pure and mixed bilayers from atomic-level and coarse-grained molecular dynamics simulations. J. Phys. Chem. B 2006, 110, 15602–15614. [Google Scholar] [CrossRef] [PubMed]
- Gu, R.X.; Ingólfsson, H.I.; de Vries, A.H.; Marrink, S.J.; Tieleman, D.P. Ganglioside-lipid and ganglioside-protein interactions revealed by coarse-grained and atomistic molecular dynamics simulations. J. Phys. Chem. B 2017, 121, 3262–3275. [Google Scholar] [CrossRef] [Green Version]
- Hughes, Z.E.; Mark, A.E.; Mancera, R.L. Molecular dynamics simulations of the interactions of DMSO with DPPC and DOPC phospholipid membranes. J. Phys. Chem. B 2012, 116, 11911–11923. [Google Scholar] [CrossRef]
- Pluhackova, K.; Kirsch, S.A.; Han, J.; Sun, L.; Jiang, Z.; Unruh, T.; Böckmann, R.A. A critical comparison of biomembrane force fields: Structure and dynamics of model DMPC, POPC, and POPE bilayers. J. Phys. Chem. B 2016, 120, 3888–3903. [Google Scholar] [CrossRef]
- van den Bogaart, G.; Hermans, N.; Krasnikov, V.; de Vries, A.H.; Poolman, B. On the decrease in lateral mobility of phospholipids by sugars. Biophys. J. 2007, 92, 1598–1605. [Google Scholar] [CrossRef] [Green Version]
- Falck, E.; Róg, T.; Karttunen, M.; Vattulainen, I. Lateral diffusion in lipid membranes through collective flows. J. Am. Chem. Soc. 2008, 130, 44–45. [Google Scholar] [CrossRef]
- Siwko, M.E.; de Vries, A.H.; Mark, A.E.; Kozubek, A.; Marrink, S.J. Disturb or stabilize? A molecular dynamics study of the effects of resorcinolic lipids on phospholipid bilayers. Biophys. J. 2009, 96, 3140–3153. [Google Scholar] [CrossRef] [Green Version]
- Anézo, C.; de Vries, A.H.; Höltje, H.D.; Tieleman, D.P.; Marrink, S.J. Methodological issues in lipid bilayer simulations. J. Phys. Chem. B 2003, 107, 9424–9433. [Google Scholar] [CrossRef] [Green Version]
- Poger, D.; Mark, A.E. Lipid bilayers: The effect of force field on ordering and dynamics. J. Chem. Theory Comput. 2012, 8, 4807–4817. [Google Scholar] [CrossRef]
- Poger, D.; Mark, A.E. On the validation of molecular dynamics simulations of saturated and cis -monounsaturated phosphatidylcholine lipid bilayers: A comparison with experiment. J. Chem. Theory Comput. 2010, 6, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Jarerattanachat, V.; Karttunen, M.; Wong-ekkabut, J. Molecular dynamics study of oxidized lipid bilayers in NaCl solution. J. Phys. Chem. B 2013, 117, 8490–8501. [Google Scholar] [CrossRef]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; van Gunsteren, W.F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. 2011, 40, 843–856. [Google Scholar] [CrossRef]
- Lins, R.D.; Hünenberger, P.H. A new GROMOS force field for hexopyranose-based carbohydrates. J. Comput. Chem. 2005, 26, 1400–1412. [Google Scholar] [CrossRef]
- Pol-Fachin, L.; Rusu, V.H.; Verli, H.; Lins, R.D. GROMOS 53A6 GLYC, an improved GROMOS force field for hexopyranose-based carbohydrates. J. Chem. Theory Comput. 2012, 8, 4681–4690. [Google Scholar] [CrossRef] [PubMed]
- Santos, D.E.S.; Pol-Fachin, L.; Lins, R.D.; Soares, T.A. Polymyxin binding to the bacterial outer membrane reveals cation displacement and increasing membrane curvature in susceptible but not in resistant lipopolysaccharide chemotypes. J. Chem. Inf. Modeling 2017, 57, 2181–2193. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Adcock, S.A.; McCammon, J.A. Molecular dynamics: Survey of methods for simulating the activity of proteins. Chem. Rev. 2006, 106, 1589–1615. [Google Scholar] [CrossRef] [Green Version]
- Allen, M.P. Introduction to molecular dynamics simulation. In Computational Soft Matter: From Synthetic Polymers to Proteins; Lecture Notes; Gustav-Stresemann-Institut: Bonn, Germany, 2004; Volume 23, pp. 1–28. [Google Scholar]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Lima, M.; Nader, M.; Santos, D.; Soares, T. Compatibility of GROMOS-derived atomic parameters for lipopolysaccharide membranes with the SPC/E water model and alternative long-range electrostatic treatments using single nonbonded cutoff and atom-based charge schemes. J. Braz. Chem. Soc. 2019, 30, 2219–2230. [Google Scholar] [CrossRef]
- Harvey, S.C.; Tan, R.K.-Z.; Cheatham, T.E. The flying ice cube: Velocity rescaling in molecular dynamics leads to violation of energy equipartition. J. Comput. Chem. 1998, 19, 726–740. [Google Scholar] [CrossRef]
- Snyder, S.; Kim, D.; McIntosh, T.J. Lipopolysaccharide bilayer structure: Effect of chemotype, core mutations, divalent cations, and temperature. Biochemistry 1999, 38, 10758–10767. [Google Scholar] [CrossRef]
- Brandenburg, K.; Funari, S.S.; Koch, M.H.; Seydel, U. Investigation into the Acyl chain packing of endotoxins and phospholipids under near physiological conditions by WAXS and FTIR spectroscopy. J. Struct. Biol. 1999, 128, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekhar, I.; Kastenholz, M.; Lins, R.D.; Oostenbrink, C.; Schuler, L.D.; Tieleman, D.P.; van Gunsteren, W.F. A consistent potential energy parameter set for lipids: Dipalmitoylphosphatidylcholine as a benchmark of the GROMOS96 45A3 force field. Eur. Biophys. J. 2003, 32, 67–77. [Google Scholar] [CrossRef]
- Oostenbrink, C.; Soares, T.A.; van der Vegt, N.F.A.; van Gunsteren, W.F. Validation of the 53A6 GROMOS Force Field. Eur. Biophys. J. 2005, 34, 273–284. [Google Scholar] [CrossRef] [Green Version]
- Faro, T.M.C.; Thim, G.P.; Skaf, M.S. A Lennard-Jones plus Coulomb potential for Al3+ ions in aqueous solutions. J. Chem. Phys. 2010, 132, 114509. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; Hermans, J. Intermolecular Forces; Pullman, B., Ed.; D. Reidel: Dordrecht, The Netherlands, 1981. [Google Scholar]
- Soares, T.A.; Straatsma, T.P. Assessment of the convergence of molecular dynamics simulations of lipopolysaccharide membranes. Mol. Simul. 2008, 34, 295–307. [Google Scholar] [CrossRef]
- Martyna, G.J.; Klein, M.L.; Tuckerman, M. Nosé-Hoover chains: The canonical ensemble via continuous dynamics. J. Chem. Phys. 1992, 97, 2635–2643. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Tironi, I.G.; Sperb, R.; Smith, P.E.; Van Gunsteren, W.F. A generalized reaction field method for molecular dynamics simulations. J. Chem. Phys. 1995, 102, 5451–5459. [Google Scholar] [CrossRef]
- Essex, J.W. The application of the reaction-field method to the calculation of dielectric constants. Mol. Simul. 1998, 20, 159–178. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N log( N ) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Santos, D.E.S.; Pontes, F.; Lins, R.D.; Coutinho, K.; Soares, T.A. SuAVE: A tool for analyzing curvature-dependent properties in chemical interfaces. J. Chem. Inf. Modeling 2019. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Systems | NVT Step | NPT Step | τp | τT | LRE |
|---|---|---|---|---|---|
| lps1 | 300 K | 200 K | 1.0 | 0.4 | PME |
| lps2 | 300 K | 200 K | 0.1 | 0.4 | PME |
| lps3 | 300 K | 200 K | 1.0 | 0.4 | RF |
| lps4 | 300 K | 200 K | 0.1 | 0.4 | RF |
| lps5 | 300 K | 300 K | 1.0 | 0.4 | PME |
| lps6 | 300 K | 300 K | 0.1 | 0.4 | PME |
| lps7 | 300 K | 300 K | 1.0 | 0.4 | RF |
| lps8 | 300 K | 300 K | 0.1 | 0.4 | RF |
| lps9 | - | 100 K→200 K→300 K | 0.1 | 0.4 | PME |
| lps10 | 100 K | 100 K→200 K→300 K | 0.1 | 0.4 | PME |
| lps11 * | - | 100 K→200 K→300 K | 5.0 | 0.5 | PME |
| lps12 * | 100 K | 100 K→200 K→300 K | 5.0 | 0.5 | PME |
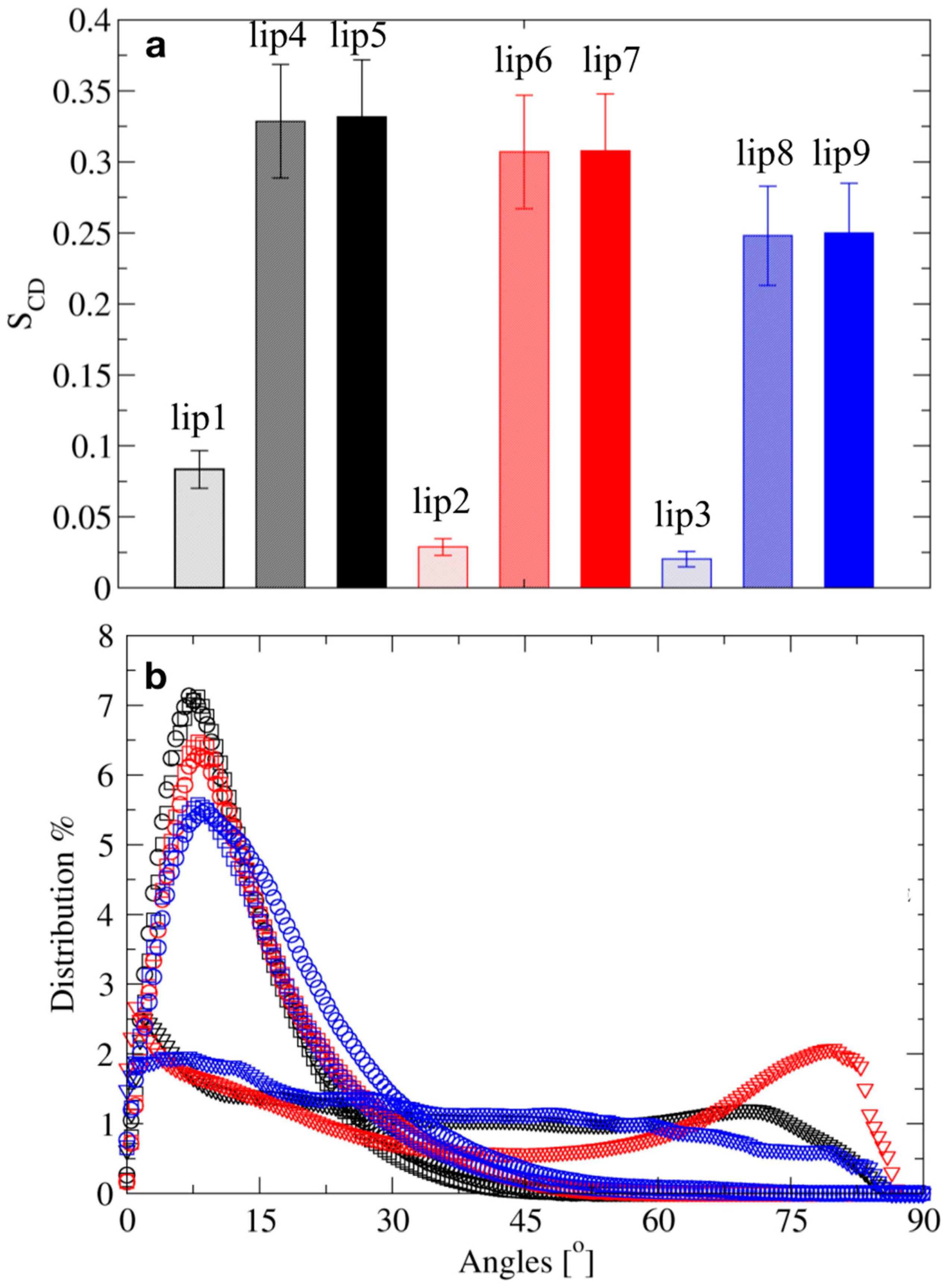
| Systems | Protocol | LRE | Ions | ||
|---|---|---|---|---|---|
| Al3+ | Na+ | Cl− | |||
| lip1 | NpT | RF | 108 | 0 | 0 |
| lip2 | NpT | RF | 219 | 0 | 333 |
| lip3 | NpT | RF | 108 | 111 | 111 |
| lip4 | NVT/NpT | RF | 108 | 0 | 0 |
| lip5 | NVT/NpT | PME | 108 | 0 | 0 |
| lip6 | NVT/NpT | RF | 219 | 0 | 333 |
| lip7 | NVT/NpT | PME | 219 | 0 | 333 |
| lip8 | NVT/NpT | RF | 108 | 111 | 111 |
| lip9 | NVT/NpT | PME | 108 | 111 | 111 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Messias, A.; Santos, D.E.S.; Pontes, F.J.S.; Lima, F.S.; Soares, T.A. Out of Sight, Out of Mind: The Effect of the Equilibration Protocol on the Structural Ensembles of Charged Glycolipid Bilayers. Molecules 2020, 25, 5120. https://doi.org/10.3390/molecules25215120
Messias A, Santos DES, Pontes FJS, Lima FS, Soares TA. Out of Sight, Out of Mind: The Effect of the Equilibration Protocol on the Structural Ensembles of Charged Glycolipid Bilayers. Molecules. 2020; 25(21):5120. https://doi.org/10.3390/molecules25215120
Chicago/Turabian StyleMessias, Andresa, Denys E. S. Santos, Frederico J. S. Pontes, Filipe S. Lima, and Thereza A. Soares. 2020. "Out of Sight, Out of Mind: The Effect of the Equilibration Protocol on the Structural Ensembles of Charged Glycolipid Bilayers" Molecules 25, no. 21: 5120. https://doi.org/10.3390/molecules25215120