
Biophysical Screens Identify Fragments That Bind to the Viral DNA-Binding Proteins EBNA1 and LANA

Abstract
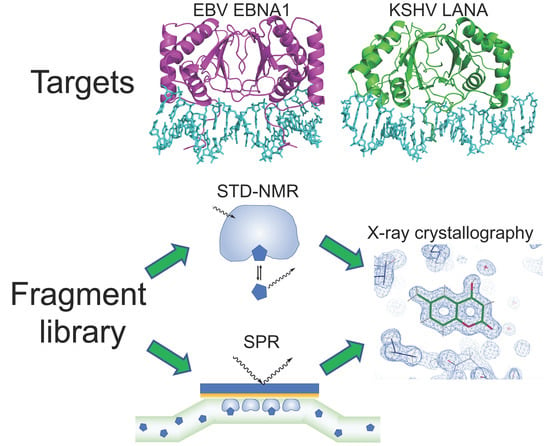
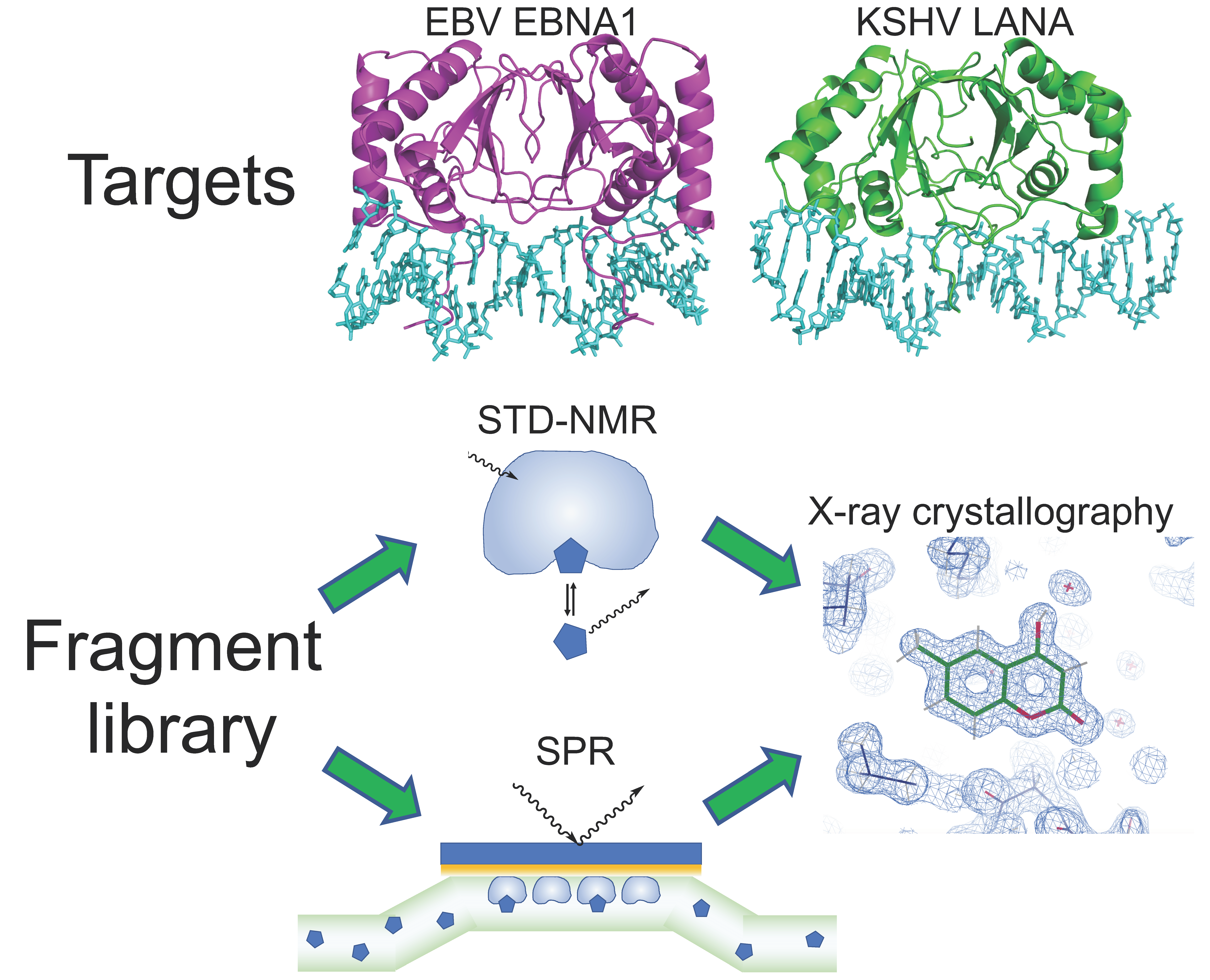
:
1. Introduction
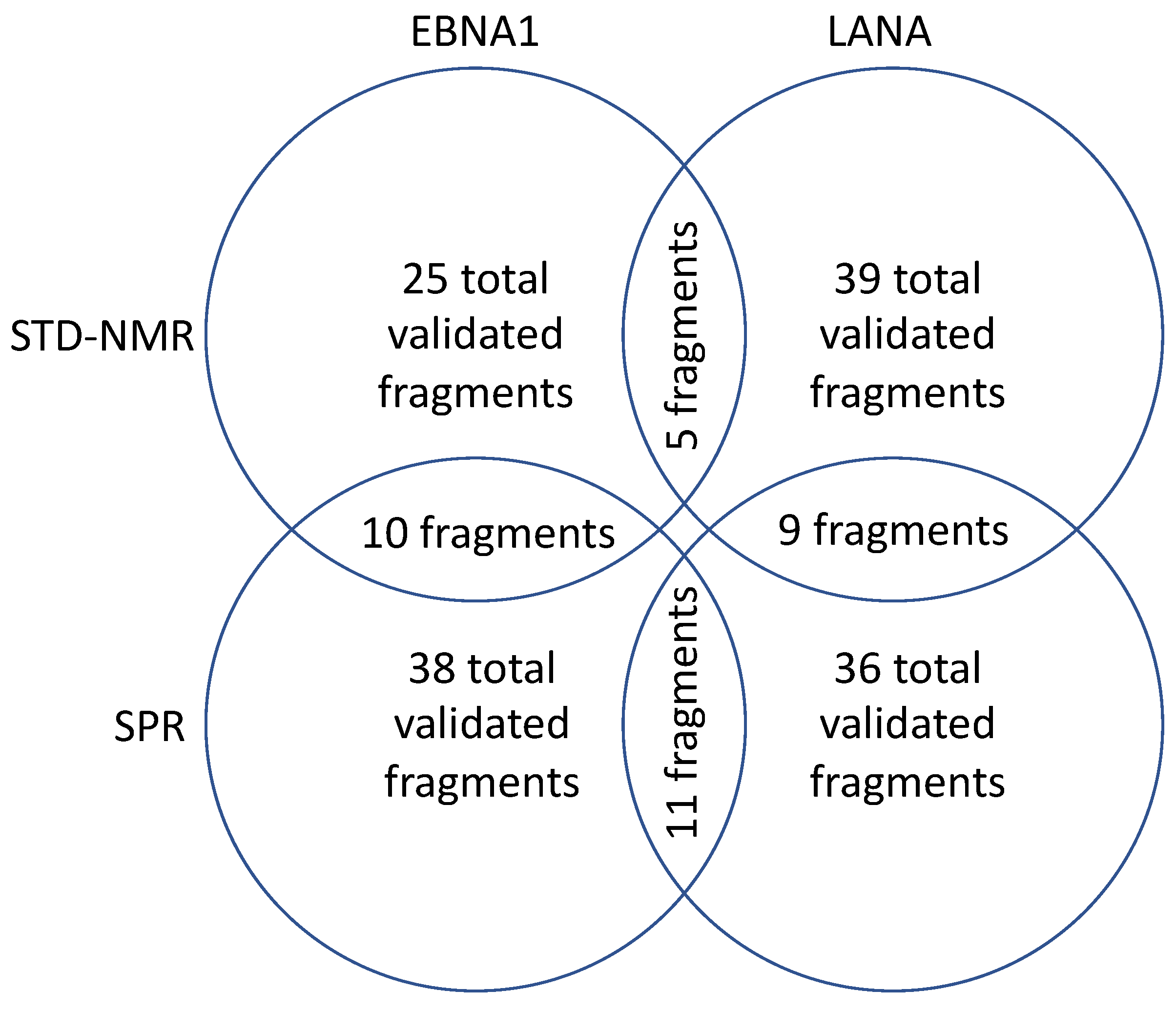
2. Results
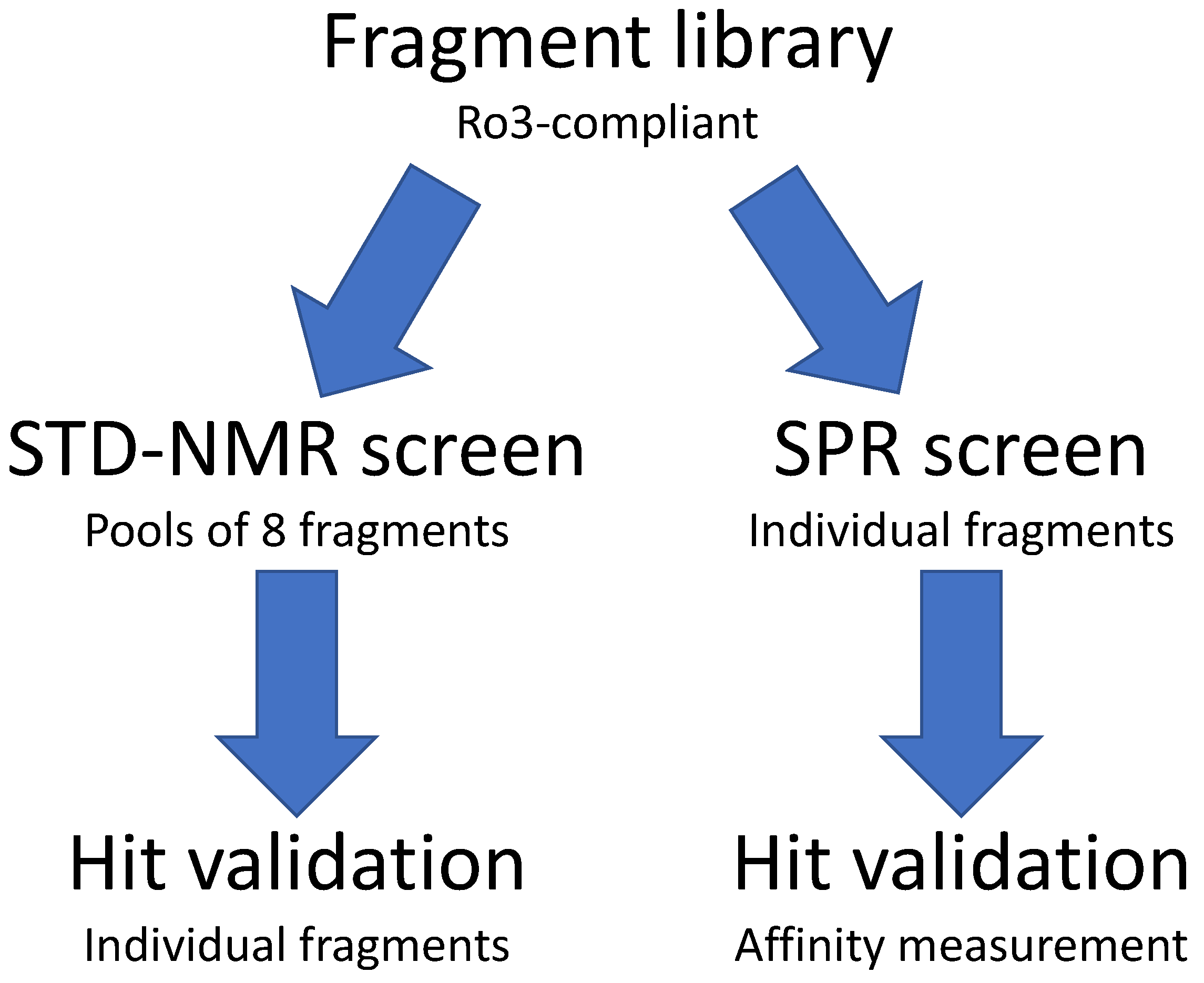
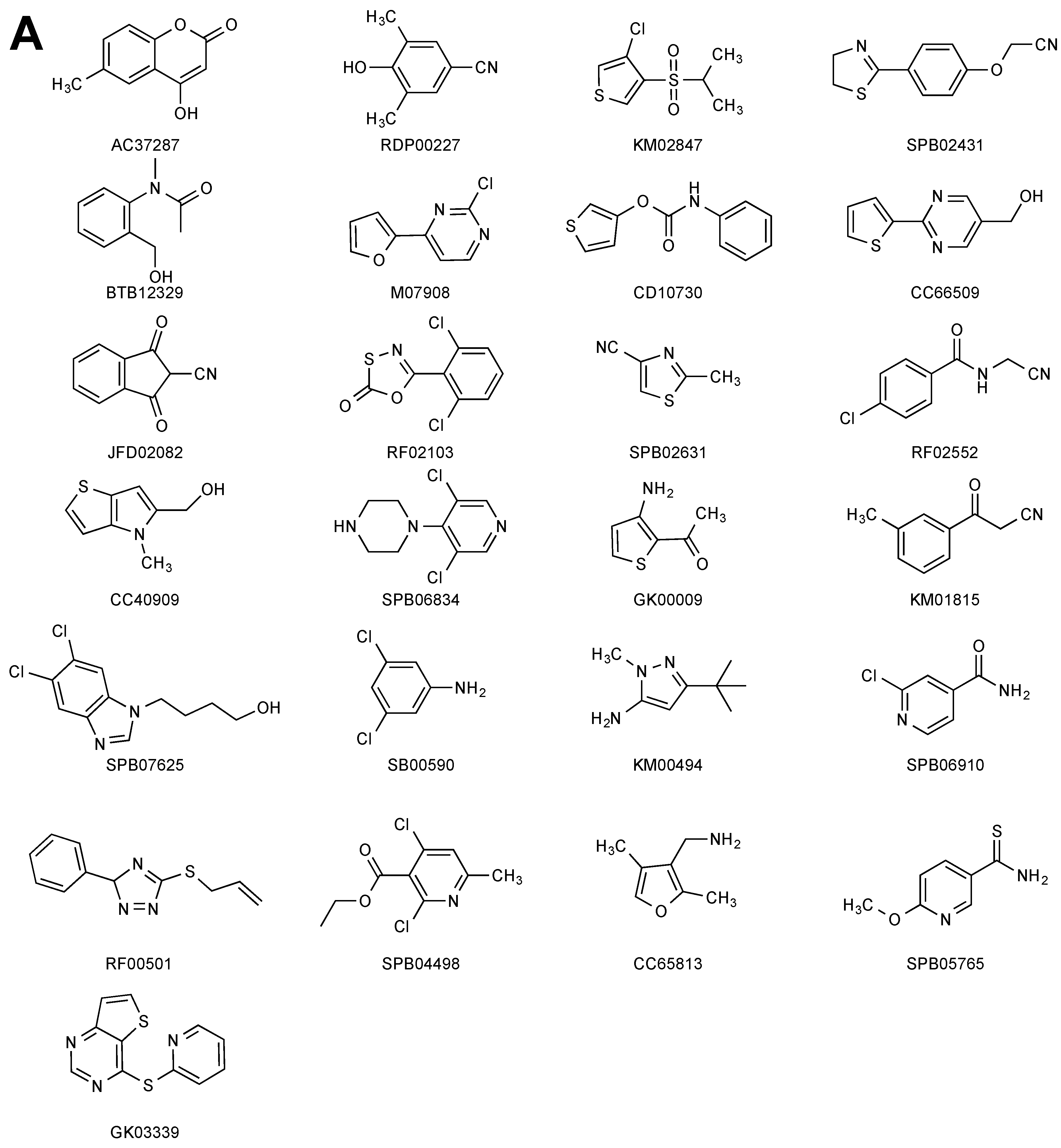
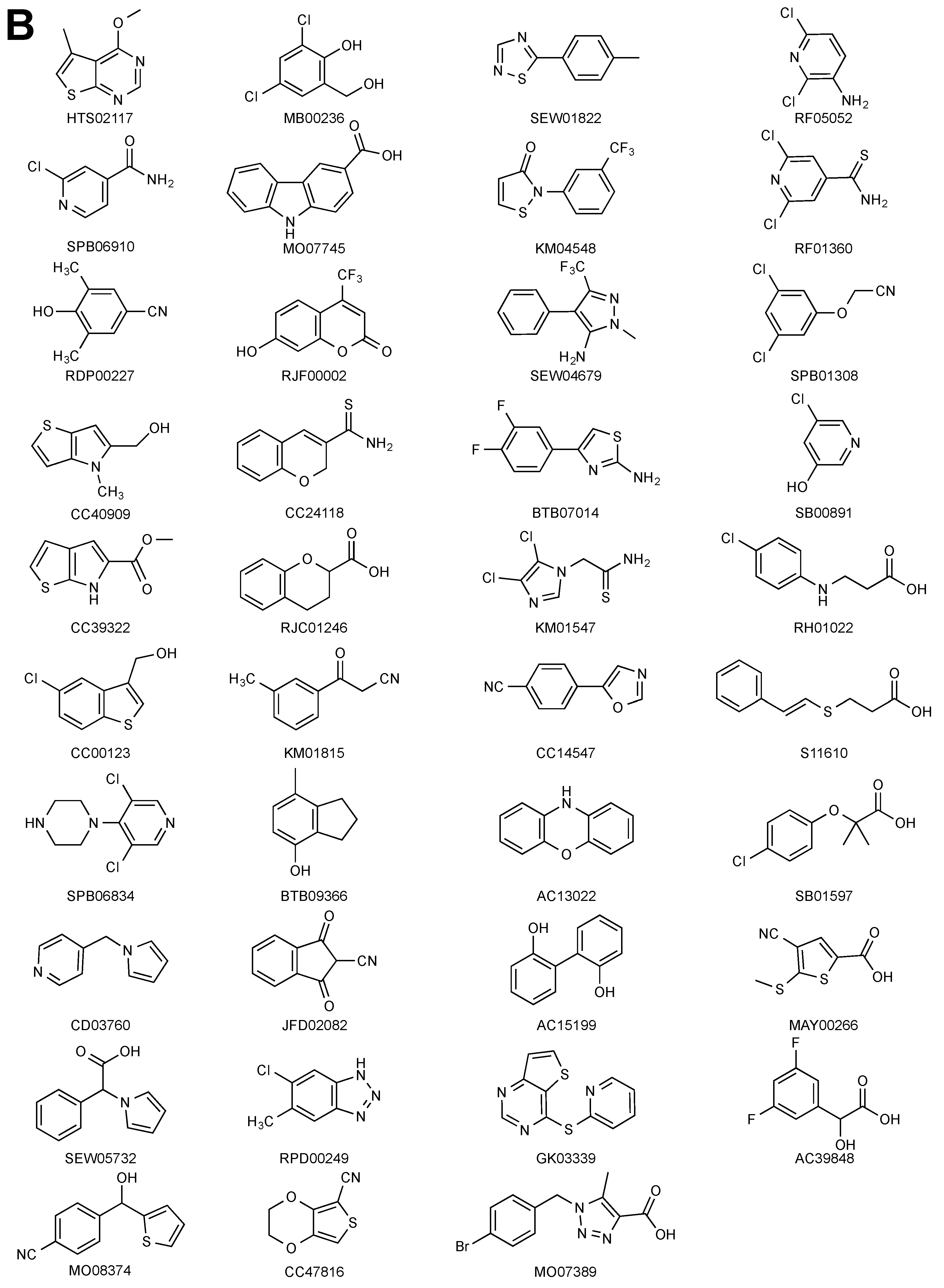
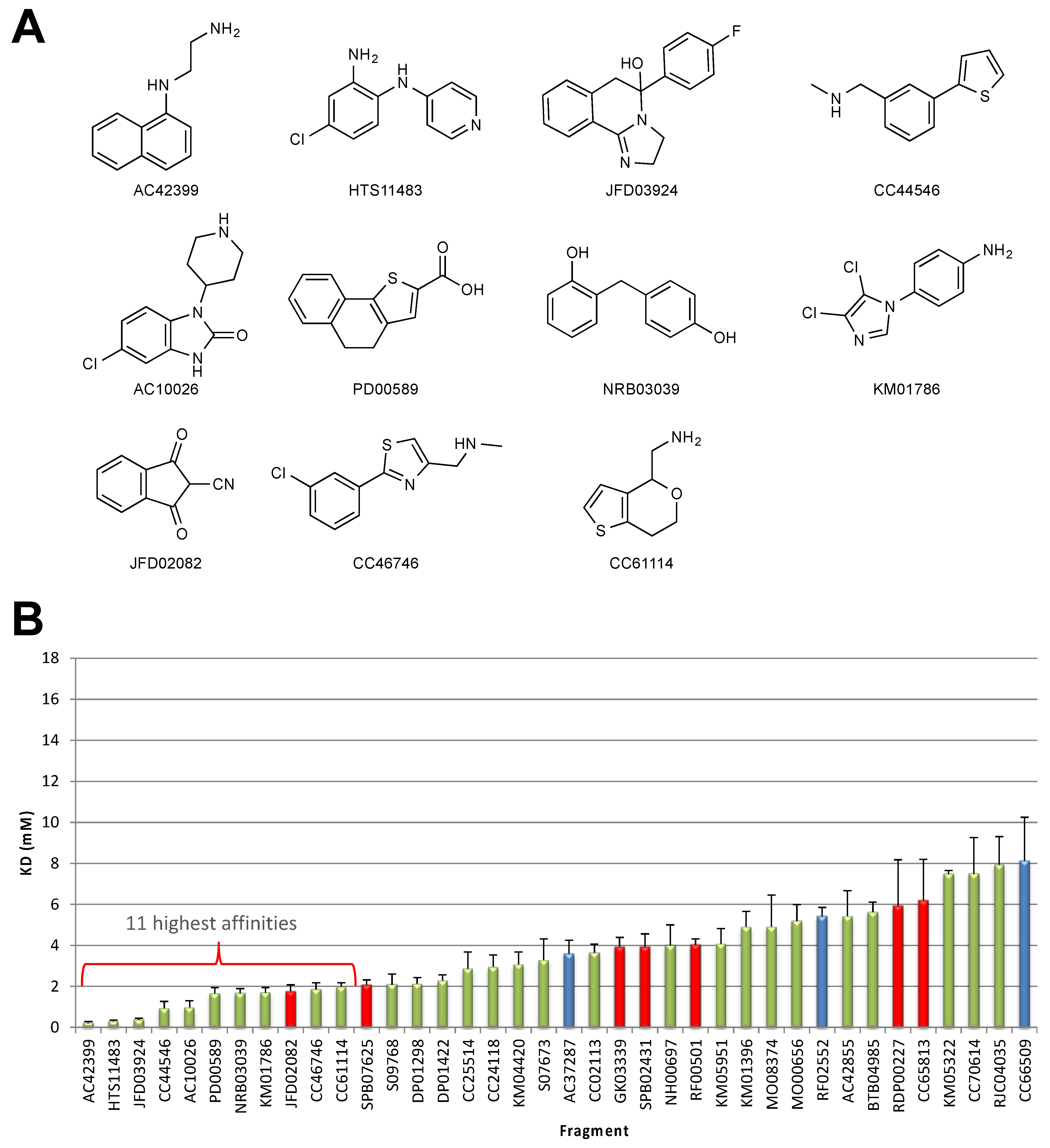
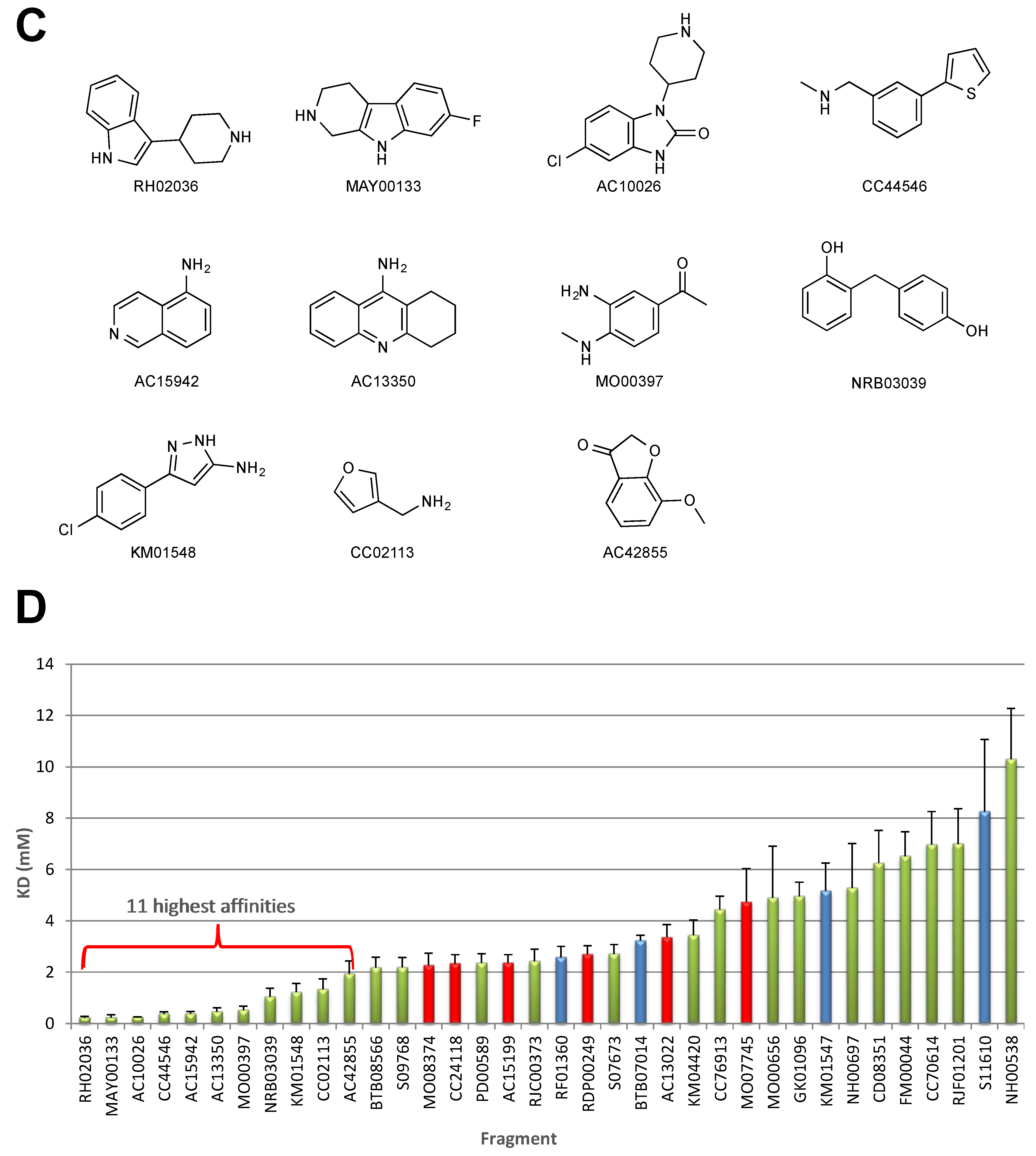
2.1. STD–NMR Screen
2.2. SPR Screen
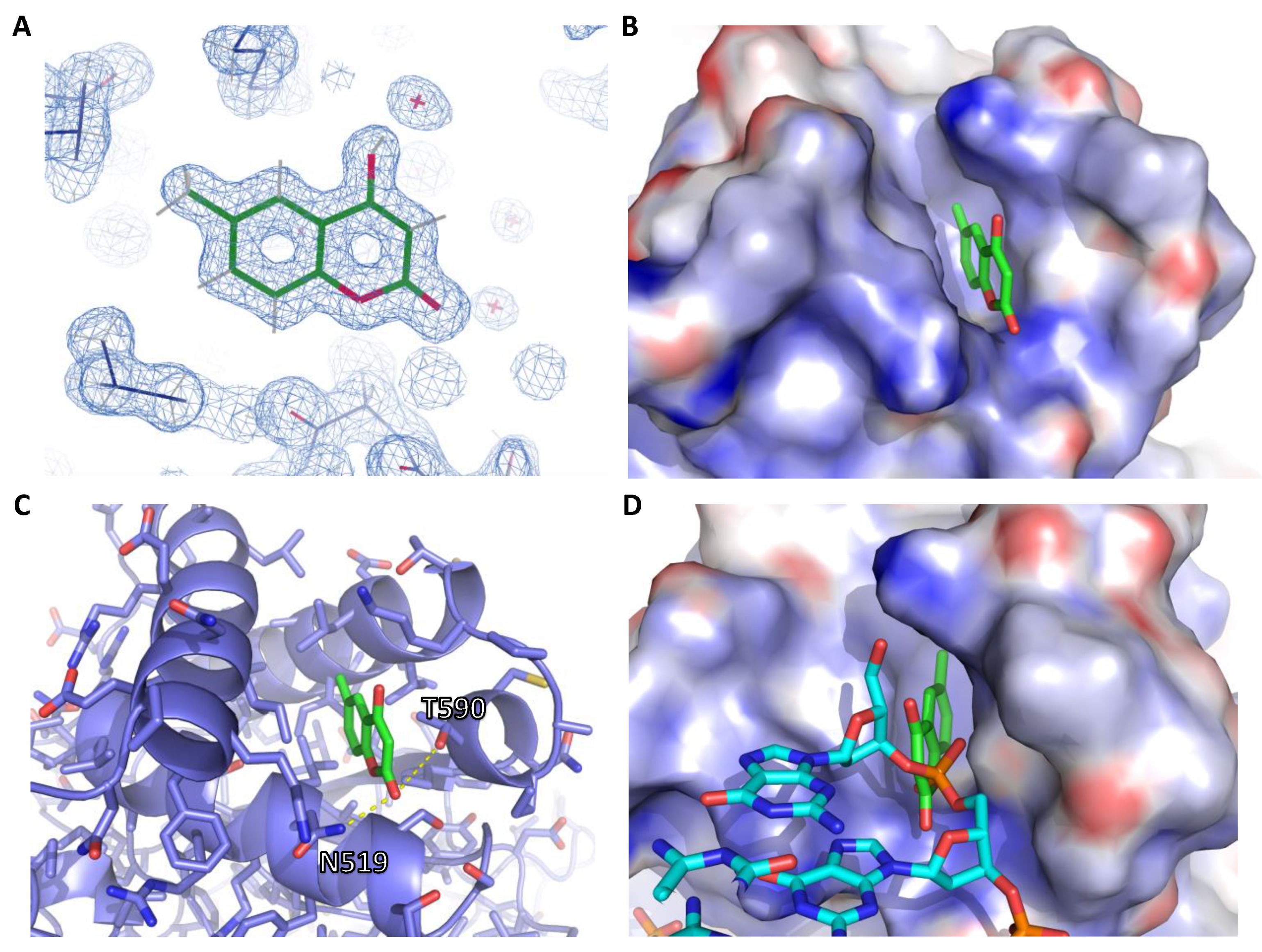
2.3. X-Ray Crystallography
3. Discussion
4. Materials and Methods
4.1. Protein Expression and Purification
4.2. Pooling for NMR Screen
4.3. STD–NMR Screen
4.4. SPR Screen
4.5. X-Ray Crystallography
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DBD | DNA-binding domain |
| DSF | Differential Scanning Fluorimetry |
| EBV | Epstein-Barr Virus |
| EBNA1 | Epstein–Barr Nuclear Antigen 1 |
| FBDD | Fragment-Based Drug Discovery |
| FP | Fluorescence Polarization |
| IE2 | Immediate Early Protein 2 |
| HHV-4 | Human Herpesvirus-4 |
| HHV-6A | Human Herpesvirus-6A |
| HHV-8 | Human Herpesvirus-8 |
| HPV | Human Papillomavirus |
| HSQC | Heteronuclear Single-Quantum Coherence |
| KSHV | Kaposi’s Sarcoma-associated Herpesvirus |
| LANA | Latency-Associated Nuclear Antigen |
| PPI | Protein–Protein Interaction |
| Ro3 | Rule of Three |
| SAR | Structure Activity Relationship |
| SPR | Surface Plasmon Resonance |
| STD–NMR | Saturation Transfer Difference-Nuclear Magnetic Resonance |
References
- Scott, D.E.; Bayly, A.R.; Abell, C.; Skidmore, J. Small molecules, big targets: Drug discovery faces the protein-protein interaction challenge. Nat. Rev. Drug Discov. 2016, 15, 533–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenti, D.; Hristeva, S.; Tzalis, D.; Ottmann, C. Clinical candidates modulating protein-protein interactions: The fragment-based experience. Eur. J. Med. Chem. 2019, 167, 76–95. [Google Scholar] [CrossRef] [PubMed]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J.; et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005, 435, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Lambert, M.; Jambon, S.; Depauw, S.; David-Cordonnier, M.H. Targeting transcription factors for cancer treatment. Molecules 2018, 23, 1479. [Google Scholar] [CrossRef] [Green Version]
- De Leo, A.; Calderon, A.; Lieberman, P.M. Control of viral latency by episome maintenance proteins. Trends Microbiol. 2019, 28, 150–162. [Google Scholar] [CrossRef]
- Messick, T.E.; Smith, G.R.; Soldan, S.S.; McDonnell, M.E.; Deakyne, J.S.; Malecka, K.A.; Tolvinski, L.; van den Heuvel, A.P.J.; Gu, B.W.; Cassel, J.A.; et al. Structure-based design of small-molecule inhibitors of EBNA1 DNA binding blocks Epstein-Barr virus latent infection and tumor growth. Sci. Transl. Med. 2019, 11, eaau5612. [Google Scholar] [CrossRef]
- Murray, P.G.; Young, L.S. An etiological role for the Epstein-Barr virus in the pathogenesis of classical Hodgkin lymphoma. Blood 2019, 134, 591–596. [Google Scholar] [CrossRef]
- Young, L.S.; Yap, L.F.; Murray, P.G. Epstein-Barr virus: More than 50 years old and still providing surprises. Nat. Rev. Cancer 2016, 16, 789–802. [Google Scholar] [CrossRef]
- Cesarman, E.; Damania, B.; Krown, S.E.; Martin, J.; Bower, M.; Whitby, D. Kaposi sarcoma. Nat. Rev. Dis. Primers 2019, 5, 9. [Google Scholar] [CrossRef]
- Damania, B.; Munz, C. Immunodeficiencies that predispose to pathologies by human oncogenic gamma-herpesviruses. Fems Microbiol. Rev. 2019, 43, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Shannon-Lowe, C.; Rickinson, A. The global landscape of EBV-associated tumors. Front. Oncol. 2019, 9, 713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, K.M.; Young, L.S. Epstein-Barr virus and carcinogenesis: Beyond Burkitt’s lymphoma. Clin. Microbiol. Infect. 2009, 15, 982–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oksenhendler, E.; Boutboul, D.; Galicier, L. Kaposi sarcoma-associated herpesvirus/human herpesvirus 8-associated lymphoproliferative disorders. Blood 2019, 133, 1186–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domsic, J.F.; Chen, H.S.; Lu, F.; Marmorstein, R.; Lieberman, P.M. Molecular basis for oligomeric-DNA binding and episome maintenance by KSHV LANA. PLoS Pathog. 2013, 9, e1003672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bochkarev, A.; Bochkareva, E.; Frappier, L.; Edwards, A.M. The 2.2 A structure of a permanganate-sensitive DNA site bound by the Epstein-Barr virus origin binding protein, EBNA1. J. Mol. Biol. 1998, 284, 1273–1278. [Google Scholar] [CrossRef]
- Hellert, J.; Weidner-Glunde, M.; Krausze, J.; Lunsdorf, H.; Ritter, C.; Schulz, T.F.; Luhrs, T. The 3D structure of Kaposi sarcoma herpesvirus LANA C-terminal domain bound to DNA. Proc. Natl. Acad. Sci. USA 2015, 112, 6694–6699. [Google Scholar] [CrossRef] [Green Version]
- Reitz, A.B.; Smith, G.R.; Tounge, B.A.; Reynolds, C.H. Hit triage using efficiency indices after screening of compound libraries in drug discovery. Curr. Top. Med. Chem. 2009, 9, 1718–1724. [Google Scholar] [CrossRef]
- Velvadapu, V.; Farmer, B.T.; Reitz, A.B. Chapter 7—Fragment-based drug discovery. In The Practice of Medicinal Chemistry, 4th ed.; Wermuth, C.G., Aldous, D., Raboisson, P., Rognan, D., Eds.; Academic Press: San Diego, CA, USA, 2015; pp. 161–180. [Google Scholar] [CrossRef]
- Erlanson, D.A.; McDowell, R.S.; O’Brien, T. Fragment-based drug discovery. J. Med. Chem. 2004, 47, 3463–3482. [Google Scholar] [CrossRef]
- Jhoti, H.; Williams, G.; Rees, D.C.; Murray, C.W. The ‘rule of three’ for fragment-based drug discovery: Where are we now? Nat. Rev. Drug Discov. 2013, 12, 644–645. [Google Scholar] [CrossRef] [Green Version]
- Carr, R.A.; Congreve, M.; Murray, C.W.; Rees, D.C. Fragment-based lead discovery: Leads by design. Drug. Discov. Today 2005, 10, 987–992. [Google Scholar] [CrossRef]
- Gianti, E.; Messick, T.E.; Lieberman, P.M.; Zauhar, R.J. Computational analysis of EBNA1 “druggability” suggests novel insights for Epstein-Barr virus inhibitor design. J. Comput. Aided Mol. Des. 2016, 30, 285–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiebel, J.; Radeva, N.; Koster, H.; Metz, A.; Krotzky, T.; Kuhnert, M.; Diederich, W.E.; Heine, A.; Neumann, L.; Atmanene, C.; et al. One question, multiple answers: Biochemical and biophysical screening methods retrieve deviating fragment hit lists. ChemMedChem 2015, 10, 1511–1521. [Google Scholar] [CrossRef] [PubMed]
- Schiebel, J.; Radeva, N.; Krimmer, S.G.; Wang, X.; Stieler, M.; Ehrmann, F.R.; Fu, K.; Metz, A.; Huschmann, F.U.; Weiss, M.S.; et al. Six biophysical screening methods miss a large proportion of crystallographically discovered fragment hits: A case study. ACS Chem. Biol. 2016, 11, 1693–1701. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, P.; Jakob, V.; Oberhausen, K.; Stein, S.C.; Cucarro, I.; Schulz, T.F.; Empting, M. Fragment-based Discovery of a Qualified Hit Targeting the Latency-Associated Nuclear Antigen of the Oncogenic Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8. J. Med. Chem. 2019, 62, 3924–3939. [Google Scholar] [CrossRef] [PubMed]
- Malecka, K.A.; Dheekollu, J.; Deakyne, J.S.; Wiedmer, A.; Ramirez, U.D.; Lieberman, P.M.; Messick, T.E. Structural basis for cooperative binding of EBNA1 to the Epstein-Barr virus dyad symmetry minimal origin of replication. J. Virol. 2019, 93, e00487-19. [Google Scholar] [CrossRef] [PubMed]
- Studier, F.W. Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 2005, 41, 207–234. [Google Scholar] [CrossRef]
- Arroyo, X.; Goldflam, M.; Feliz, M.; Belda, I.; Giralt, E. Computer-aided design of fragment mixtures for NMR-based screening. PLoS ONE 2013, 8, e58571. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the compounds are not available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EBNA1:AC37287 PDB Code 6VH6 | |
|---|---|
| Spacegroup | P212121 |
| Wavelength | 0.97919 |
| a, b, c (Å) α, β, γ (°) | 59.52, 68.34, 69.69 90.00, 90.00, 90.00 |
| Resolution (Å) | 50.0-1.30 (1.32-1.30) |
| I/σ | 26.4 (1.03) |
| Completeness | 99.1% (91.9%) |
| Redundancy | 6.4 (3.1) |
| Reflections | 69927 (3190) |
| Rwork/Rfree | 0.1500/0.1608 |
| Wilson B factor (Å2) | 14.6 |
| Bond lengths (Å) | 0.009 |
| Bond angles (°) | 1.004 |
| Ramachandran favored | 99% |
| Ramachandran allowed | 1% |
| Ramachandran outliers | 0% |
| Clash score | 4.0 |
| Average B, all atoms (Å2) | 23.0 |
| Number of protein atoms | 4572 |
| Number of solvent atoms | 454 |
| Number of ligand atoms | 21 |
| Total number of atoms | 5047 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Messick, T.E.; Tolvinski, L.; Zartler, E.R.; Moberg, A.; Frostell, Å.; Smith, G.R.; Reitz, A.B.; Lieberman, P.M. Biophysical Screens Identify Fragments That Bind to the Viral DNA-Binding Proteins EBNA1 and LANA. Molecules 2020, 25, 1760. https://doi.org/10.3390/molecules25071760
Messick TE, Tolvinski L, Zartler ER, Moberg A, Frostell Å, Smith GR, Reitz AB, Lieberman PM. Biophysical Screens Identify Fragments That Bind to the Viral DNA-Binding Proteins EBNA1 and LANA. Molecules. 2020; 25(7):1760. https://doi.org/10.3390/molecules25071760
Chicago/Turabian StyleMessick, Troy E., Lois Tolvinski, Edward R. Zartler, Anna Moberg, Åsa Frostell, Garry R. Smith, Allen B. Reitz, and Paul M. Lieberman. 2020. "Biophysical Screens Identify Fragments That Bind to the Viral DNA-Binding Proteins EBNA1 and LANA" Molecules 25, no. 7: 1760. https://doi.org/10.3390/molecules25071760