
Targeting Genome Integrity in Mycobacterium Tuberculosis: From Nucleotide Synthesis to DNA Replication and Repair


Department of Pharmaceutical Sciences, University of Piemonte Orientale, Via Bovio 6, 28100 Novara, Italy
*
Authors to whom correspondence should be addressed.
Molecules 2020, 25(5), 1205; https://doi.org/10.3390/molecules25051205
Submission received: 10 February 2020
/
Revised: 4 March 2020
/
Accepted: 5 March 2020
/
Published: 7 March 2020
(This article belongs to the Special Issue Recent Advances in Antitubercular Drug Discovery)
Abstract
:Mycobacterium tuberculosis (MTB) is the causative agent of tuberculosis (TB), an ancient disease which still today causes 1.4 million deaths worldwide per year. Long-term, multi-agent anti-tubercular regimens can lead to the anticipated non-compliance of the patient and increased drug toxicity, which in turn can contribute to the emergence of drug-resistant MTB strains that are not susceptible to first- and second-line available drugs. Hence, there is an urgent need for innovative antitubercular drugs and vaccines. A number of biochemical processes are required to maintain the correct homeostasis of DNA metabolism in all organisms. Here we focused on reviewing our current knowledge and understanding of biochemical and structural aspects of relevance for drug discovery, for some such processes in MTB, and particularly DNA synthesis, synthesis of its nucleotide precursors, and processes that guarantee DNA integrity and genome stability. Overall, the area of drug discovery in DNA metabolism appears very much alive, rich of investigations and promising with respect to new antitubercular drug candidates. However, the complexity of molecular events that occur in DNA metabolic processes requires an accurate characterization of mechanistic details in order to avoid major flaws, and therefore the failure, of drug discovery approaches targeting genome integrity.
1. Introduction
According to the most recent Global Tuberculosis Report (2019) edited by the World Health Organization (WHO), 10 million new Tuberculosis (TB) cases worldwide were estimated in 2018; moreover, there were 1.2 million TB deaths amongst HIV-negative patients, and an additional 250,000 deaths amongst HIV-positive patients [1]. The global control and management of TB is facing the worsening scenarios of the spreading of multi-drug resistant (MDR) MTB strains, mostly referred to as rifampicin-resistant TB, as well as extensively-drug resistant (XDR) MTB strains that are resistant to first- and second-line anti-tubercular drugs [2]. In addition to drug resistance events, the adherence to long-term antitubercular therapy, the hepatotoxicity of most currently used drugs and reduced drug tolerance [3,4] all strongly support the development of new therapies for both drug-sensitive and drug-resistant TB with novel mechanisms of action. The genotypic information derived from MTB genome sequencing [5] guides the current anti-TB pipeline for which new drugs (bedaquiline and delamanid) have recently been approved, and several candidates entered Phase II and Phase III clinical trials [6]. However, for a definitive solution to the clinical management of drug-resistant tuberculosis, other innovative drugs are urgently needed.
The criteria for developing new anti-TB drug candidates are well established [7,8,9,10]. In addition to a low toxicity level and reduced adverse reactions, there are a number of other factors that any promising anti-TB molecule candidate should fulfil: (i) to be more potent than those currently available, with the aim of shortening the duration of treatment; (ii) to be active on new targets in order to be effective on MDR-TB and XDR-TB; (iii) to be compatible with antiretroviral therapy, as many patients are co-infected with HIV; and (iv) to show no antagonism to other TB drugs or drug candidates so that a multi-component regimen can be applied. Considering the unique feature of the MTB cell envelope—being mainly built on the mycolylarabinogalactan-peptidoglycan complex, which connects the peptidoglycan to the mycobacterial outer membrane—a number of antitubercular drugs target the synthetic pathway of key components of the cell envelope. Among these, the frontline agents isoniazide and ethambutol constitute the major TB drug arsenal active on cell wall biogenesis, together with the second line agent d-cycloserine. More recently, other drugs or drug candidates have been developed that act on new targets, including PBTZ169, targeting ecaprenylphospho-β-d-ribofuranose 2-oxidase [11], and BM212 and analogs inhibiting mycobacterial membrane protein Large 3 [12] and benzofuran TAM16, active on polyketide synthase Pks13 [13]. Alternative validated target pathways include DNA transcription, which is inhibited by rifampicin, protein synthesis, which is blocked by oxazolidinones [14], ATP synthesis by Q203 (acting on cytochrome bc1 complex) [15] and Bedaquiline, which inhibits F0F1ATP synthase [16].
Despite its central role in cell development and function, a limited number of approved TB drugs target DNA metabolism, which includes all the reactions involved in DNA replication and repair. Indeed, these have been limited exclusively to the fluoroquinolones that interfere with DNA gyrase and DNA topoisomerase activity, and are frequently used as second-line drugs for the treatment of MDR-TB. However, fluoroquinolones’ capability in reducing the duration of therapy in murine models of TB is strengthening their use as first-line drugs [17]. Several reviews have been published recently that focus on the current regimens and emerging drugs acting on the key cellular process of DNA metabolism in pathogenic bacteria; a number of excellent works investigate the DNA replication and repair pathways as sources of molecular targets [18,19,20,21], as well as the biosynthesis of nucleic acids precursors—the nucleotides [22]. The present review gives a concise overview of our current knowledge and understanding of key processes involved in genome maintenance in MTB, particularly on purines and pyrimidine biosynthesis, DNA replication and DNA repair, with the aim to highlight their potential impact for the development of novel drugs against TB. Particular emphasis will be placed on recent results concerning the phenotypic screenings and biochemical characterization of macromolecular complexes acting in DNA metabolic pathways, as well as the innovative strategies that have been proposed to interfere with the formation of such complexes as potential sources of new targets for TB drug development.
2. Targeting Purine and Pyrimidine Ribonucleotide Synthesis
2.1. Purine Biosynthesis
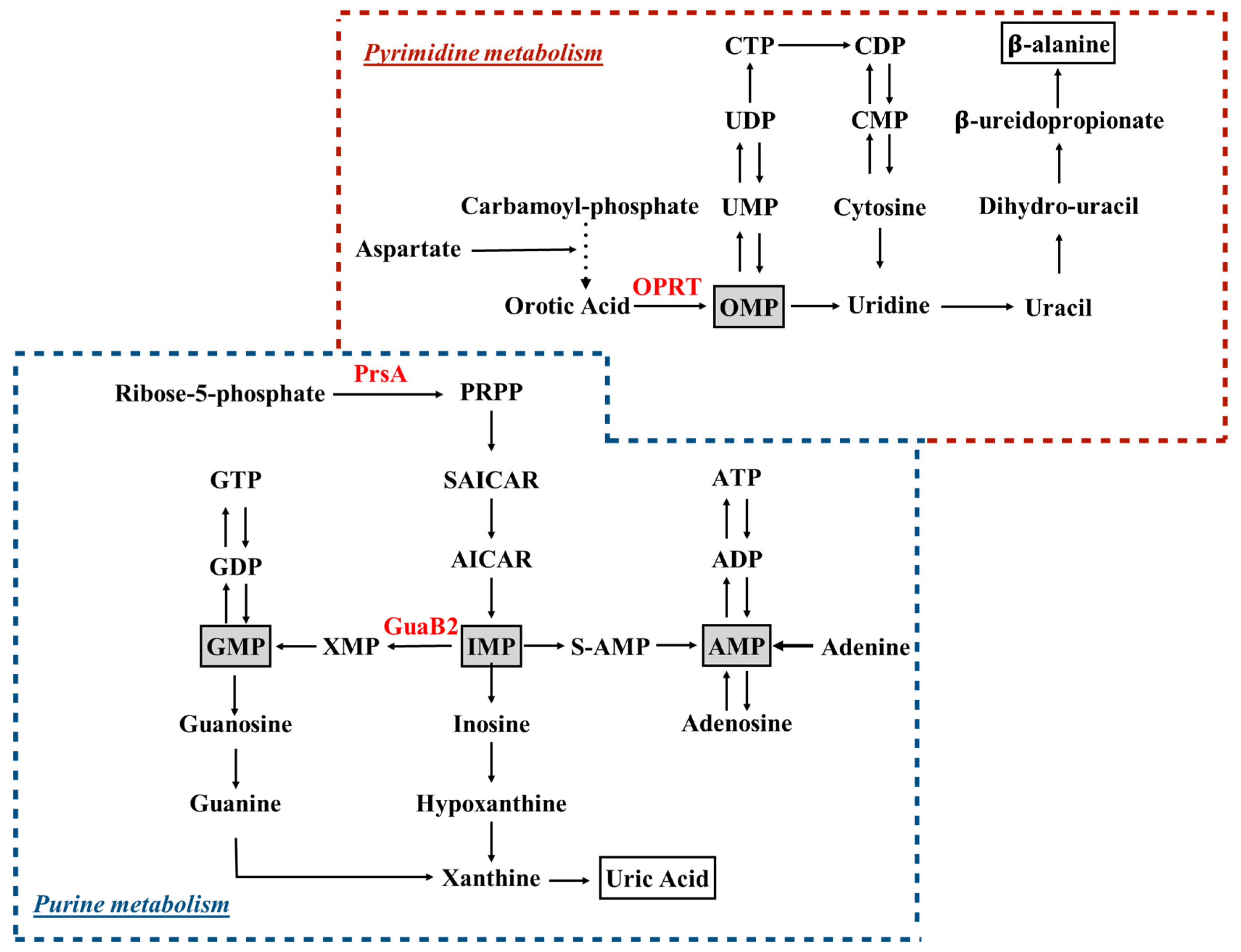
Purine nucleotides are of key importance in living organisms, since they provide the building blocks for DNA and RNA. Two main pathways exist in most organisms, including mycobacteria, to which from here onward we specifically refer for the production of these compounds: the de novo biosynthetic pathway (Figure 1), in which nucleotides are synthesized from 5-phosphoribosylpyrophosphate (PRPP) in a multi enzymatic process, and the salvage pathways, in which nucleotides are retrieved after the catabolism of nucleic acids and coenzymes.
In mycobacteria, the first half of the de novo purine biosynthetic pathway involves the generation of an aminoimidazole moiety attached to a ribose; the series of reactions starts from PRPP synthetized by phosphoribosylpyrophosphate synthetase [23,24,25], whose enzymatic activity has been shown to strongly depend on inorganic phosphate, another key molecule for MTB survival [26,27]. In the second half of the pathway, the carbon atoms at positions 4 and 5 of the imidazole moiety are modified, allowing the final cyclization reaction that generates inosine 5′-monophosphate (IMP) [28]. The conversion of IMP to guanosine 5′-monophosphate (GMP) begins with the NAD+-dependent oxidation of IMP to xanthosine 5′-monophosphate by IMP dehydrogenase (IMPDH, GuaB), which was deeply explored as a drug target. MTB encodes three apparent homologues of IMPDH on its chromosome (guaB1, guaB2 and guaB3), but only guaB2 (Rv3411c) was found to encode a functional IMPDH enzyme (GuaB2) [29,30]. GuaB2 has a key role in guanine nucleotide metabolism, since it is a rate-limiting enzyme of the cascade, and its inhibition results in a depletion of cellular guanine nucleotides [31]. Usha and co-workers identified diphenyl urea-based derivatives as inhibitors of MTB GuaB2 with potent antimycobacterial activity, with the most potent compounds, DPU-2 and DPU-3, behaving as noncompetitive and uncompetitive inhibitors with respect to IMP, respectively [29]. The peculiar mechanism of inhibition sustains the potential species-specificity of such molecules avoiding cross-reactivity with the human enzyme, which shares 41% of sequence identity with the mycobacterial enzyme [32]. Further inhibition studies identified a novel series of triazole-linked mycophenolic adenine inhibitors as NAD cofactor mimics; the activity of these inhibitors was tested against both human IMPDH isoforms and against MTB GuaB2, and showed no selectivity for the bacterial enzyme [33]. Taking into account the difficulties of translating the products of target-based drug discovery into hits that show antibacterial activity, a number of target-agnostic approaches were developed based on whole-cell screenings, which select an initial set of hit compounds based on their capability to inhibit bacterial growth [34]. The drug discovery potential of such methods, namely phenotypic screening, is further reinforced by the whole genome sequencing of resistant mutants in order to identify SNPs, which suggests the potential target(s) of tested compounds are subsequently genetically and pharmacologically validated [35]. This empiric approach guided the research pipeline of two independent groups, which identified a new class of lead compound that targets MTB GuaB2 with promising antitubercular activity and limited mammalian cell toxicity [36,37]. In particular, Park et al. characterized a number of indazole sulfonamides that showed micromolar potency against MTB, behaving as uncompetitive inhibitors of the enzyme that were only effective against replicating MTB cells [36]. Amongst them, the most powerful, N-(1H-indazol-6-yl)-3,5-dimethyl-1H-pyrazole-4-sulfonamide, showed a Ki of 0.22 μM and an MIC of 2 μM. On the other hand, a parallel phenotypic screening of a library with thousands of compounds allowed the identification of the VCC234718 molecule (5-(4-cyclohexanecarbonylpiperazine-1-sulfonyl)isoquinoline, belonging to the class of sulfonamides), which inhibits MTB GuaB2 with a Ki of 0.1 μM and a uncompetitive mechanism with respect to the IMP and NAD+ cofactors; this was entirely characterized through structural and biochemical investigations. Moreover, VCC234718 displayed a MIC90 value of 2 μM [37]. Interestingly, both compounds, which were discovered independently by the two research teams, target the same enzyme, belong to the same chemical class and act with the same inhibitory mechanism by binding the enzyme active site in a comparable manner, and being recognized by the same molecular determinants as clearly showed by the X-ray structures. Moreover, the structural analysis of the enzyme-inhibitor complexes also explained the remarkable selectivity shown by the compounds toward the MTB GuaB2 versus the human enzyme (with a selectivity index higher than 30)—a very relevant aspect when targeting an enzyme that is also present in humans [36,37]. Finally, the structural studies clearly demonstrated why the mutation that features the Guab2 variant (Y487C) conferring MTB resistance to VCC234718 results in a still-active enzyme, which is, however, insensitive to the inhibitor action [36,37]. This observation is of great value to already start the structure-based rational design of VCC234718 derivatives, which could overcome a possible emergence of resistance.
The validation of GuaB2 as a new TB drug target stimulated further studies aiming at the discovery of hit compounds with bactericidal activity [38,39]; indeed, on the basis of previous published results [40,41,42,43,44], an expansion of the structure–activity relationship of benzoxazole-based derivatives revealed potent inhibitors of MTB GuaB2 displaying a Ki of 14 nM, though with a moderate antibacterial activity [39].
2.2. Pyrimidine Biosynthesis
The de novo synthesis of pyrimidine nucleotides (Figure 1) involves the coordination of a multi-enzymatic cascade in which six catalyzed reactions culminate in the formation of uridine monophosphate (UMP), the precursor of all pyrimidine nucleotides [28]. In MTB, the genes encoding five of the enzymes acting in the pyrimidine synthesis map on the pyr operon, and some of these genes are essential for the growth of MTB in vitro [45]. However, biochemical insights on pyrimidine biosynthesis in MTB derive from investigations performed on other bacteria. The fifth step in the pathway, which is catalyzed by orotate phosphoribosyltransfrase (OPRT) [46,47]—a type I PRTase that converts orotate to orotidine 5′-monophosphate (OMP)—has been investigated as a potential drug target, and submicromolar pyrimidin-2(1H)-one-based inhibitors of the MTB OPRT enzyme have been identified, but as yet have not been tested on MTB to evaluate antibacterial activity [48]. As reported in the introduction, both purine and pyrimidine biosynthesis strongly rely on PRPP availability, and therefore the PRPP synthetase is another attractive target for the development of antibacterial agents. In particular, MTB PRPP synthetase (PrsA, encoded by the prsA gene) has been validated as a robust target, and extensively biochemically and structurally characterized [49,50,51]. Therefore, at least three enzymes—namely PRPP synthase (PrsA), IMP dehydrogenase (GuaB2) and orotate phosphoribosyltransferase (PyrE), whose crystal structures has been deposited and are available for further structure-based drug design [23,36,37,47]—are now considered as strong targets, and are being investigated for the development of novel drugs against TB.
Although the current review does not focus and therefore does not detail the synthesis and maintenance of deoxynucleotide triphosphates (dNTPs), such processes are of course of major relevance for a correct DNA homeostasis, and have been targeted in drug discovery events against MTB with, amongst others, investigations on thymidylate synthase [52,53] and dUTPase superfamily enzymes [54].
3. Targeting MTB DNA Replication
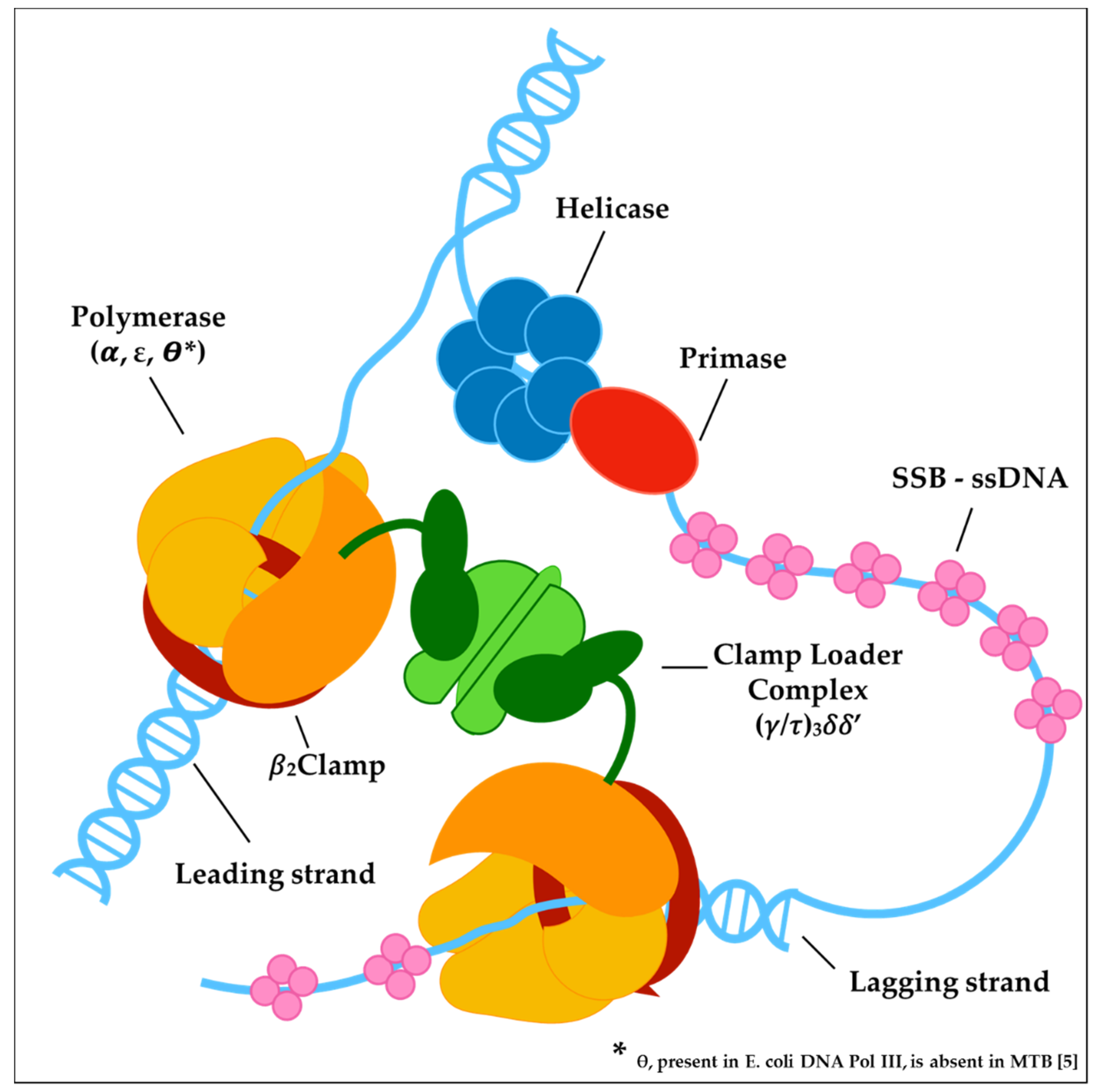
DNA replication in bacteria is performed by a large, multiprotein complex called the replisome, which synthesizes the leading and lagging strands in a highly coordinated manner. The replisome proteins catalyze a huge number of events, such as DNA unwinding, RNA primers synthesis, clamp loading and DNA polymerization. Comparative genomic analyses demonstrated that most of the replisome components are conserved across bacteria [20]; however, this assumption is not valid for MTB, which lacks obvious homologs of several components that perform key functions in model organisms such as Escherichia coli and Bacillus subtilis. As excellently reviewed in the paper of Ditse and co-authors [55], the bacterial replication machinery is based on the concerted action of three catalytic centers: the helicase-primase complex; the core complex; and the clamp loader complex (Figure 2).
3.1. The Helicase-Primase Complex
Although it is common to refer to the Escherichia coli model when considering the overall process of DNA replication in MTB, it is important to note that some notable differences among these species have been described, including the observation that MTB lacks clear homologs of several initiation proteins (DnaC, DnaT, PriB and PriC) [55]. The helicase-primase complex (primosome) mainly refers to the DnaB helicase, the DnaG primase and single-stranded DNA binding proteins (SSB) that constitute the basic replication module in bacterial genomes [20,56]. Despite the absence of a canonical helicase loader (DnaC in Escherichia coli), it was recently demonstrated that MTB possesses an ancestral bacterial replicative operator named DciA (Rv0004), which displayed the attributes of the replicative helicase-operating proteins associated with replication initiation [57,58]. Considering the key role of the helicase-primase complex in the series of events taking place in mycobacterial replication, it should be considered a valid drug target. However, the difficulties encountered in the in vitro reconstruction of such protein complexes have restricted the number of screenings against purified proteins, as well as of the structure-based drug design approach, which at present could finally refer to impressive progress of cryo-EM-based techniques [59]. Most of the drug discovery pipeline targeting the primosome focused on the identification of small-molecule inhibitors of SSB-protein interaction. SSB-protein plays a critical role in protecting unwound single-stranded DNA, and binds target DNA with high affinity and in a sequence-independent manner. In addition to DNA binding, SSB also physically associates with a number of different genome maintenance proteins [60]. Small molecules that disrupt Escherichia coli SSB interaction with the Exonuclease I binding partner have been identified [61,62], and were validated for their antibacterial activity against a diverse panel of bacterial species [63]. The DnaG primase synthesizes primers for lagging strand Okazaki fragments, and a number of inhibition studies aim at targeting bacterial primase. They identified natural products as attractive inhibitors of DnaG from Escherichia coli [64,65]. However, whole-cell activity was only considerable in a mutant strain deficient in the lipopolysaccharide layer, strengthening the role of the bacterial cell wall in the permeation and efflux of antibiotics. The primosome activity is coordinated and facilitated by the replicative DnaB helicase, and despite there being no published results on active molecules blocking the mycobacterial protein, some works reported a number of flavonols that have been shown to inhibit helicase activity in other bacterial species [66,67]. Interestingly, mycobacterial DnaB contains an intein motif whose splicing is blocked under oxidizing conditions, allowing the arrest of replication, which should be advantageous to preserve DNA integrity in the presence of reactive oxygen species [68]. While the drug discovery value of targeting proteins belonging to helicase-primase complex remains to be determined for MTB, these results support the potential of investigating the primosome as a novel anti-mycobacterial target.
3.2. The Core Complex and the Clamp Loader Complex
At present, there are no anti-TB drugs in clinical use which directly target the replisome core complex in MTB; however, by adopting a drug-revisiting approach, Kling et al. validated a natural product called griselimycin (extracted from Streptomyces spp) as a potent inhibitor of the sliding β-clamp. In particular, they improved the griselimycin pharmacological properties by developing one synthetic derivative (cyclohexylgriselimycin) with a higher penetrating capacity in cells of the immune system that harbor the MTB bacilli. Moreover, in combination with other drugs, the griselimycin derivative showed high potency in mice with TB [69]. Cyclohexylgriselimycin binds with dissociation constant in the nanomolar range to the dnaN-encoded sliding β-clamp of MTB, without interfering with the human DNA clamp protein, PCNA, resulting in a very high selectivity index [69].
3.3. DNA Topology Control and Regulation
DNA unwinding by DNA gyrase and topoisomerase could be considered as a parallel molecular event directly linked to the replication of chromosomal DNA. Indeed, DNA topology control is functional to an efficient processive synthesis, as well as the elimination of the stresses resulting from negative supercoiling and the concatenation of double-stranded DNA. The type II topoisomerase, DNA gyrase, is built on two subunits, gyrase A and gyrase B, which together form the catalytically active heterotetrameric enzyme (i.e., GyrA2B2) [70]. The role of the A subunit is the breakage and rejoining of the double DNA strand, while the B subunit possesses the ATPase activity, which provides energy for the DNA supercoiling. MTB lacks topoisomerase IV, and the negative supercoiling of DNA is performed only by gyrase (18). The enzyme is a clinically validated drug target, and the currently available inhibitors can be classified based on their origin, i.e., drugs obtained from natural sources or synthetic drugs. Among the latter group, fluoroquinolones represent the most successful antibacterial agents targeting DNA gyrase; these compounds have been extensively explored to improve the spectrum of activity and potency, and are currently used as second-line anti-TB agents. The mechanism of the action of fluoroquinolones involves the stabilization of the covalent gyrase A subunit-DNA complex, thereby leading to protein-stabilized DNA breaks with a bactericidal outcome [71]. Aminocoumarins such as novobiocin are natural products, and they inhibit the ATPase function of the B subunit [72], which appears less exposed to mutagenic events compared to the A subunit [73]. However, the relatively poor pharmacokinetic profile of this class of natural compounds limited their use in clinics as an anti-TB drug. In the context of DNA topology modulation, type I topoisomerase TopA (Rv3646c), which causes single-stranded nicks in relaxing the DNA, is inhibited by hydroxycamptothecin, a derivative of the anticancer topoisomerase inhibitor camptothecin (18). The functional modification of camptothecin scaffolds led to the discovery of whole-cell activity on both drug-sensitive and XDR MTB strains [74,75], validating MTB TopA as an innovative drug target to be considered in further medicinal chemistry studies. Finally, Okazaki fragments generated during the replication process are ligated by the mycobacterial NAD+-dependent DNA ligase (Rv3014c), which is therefore considered essential, and was deeply investigated as a drug target. However, although several in vitro inhibition studies on recombinant MTB LigA are present in scientific literature, very few molecules have been shown to exhibit whole-cell activity in the micromolar range against MTB [76,77,78,79].
4. Targeting MTB DNA Repair
During its entire life cycle, MTB must face a multitude of DNA-damaging stresses and a continuous exposure to harmful agents, which could compromise bacterial fitness as the result of increased genomic instability. The relative contribution of different DNA repair activities to the maintenance of MTB chromosome stability at different stages of the infection has been exhaustively reviewed [80]. In particular, one of the hallmarks of MTB infection is the bacilli’s ability to survive the hostile environment of the host’s infected macrophages, and the release of chemicals by the host cells that lead to bacterial DNA damage, mainly induced by endogenous DNA-alkylating agents originated by the action of highly reactive oxidative (ROS) and nitrosative (RNI) radicals [81,82]. The reaction of nitrogen monoxide radicals with oxygen produces nitrous anhydride, which nitrosates amines and amides to produce compounds that are converted by a metabolic pathway into potent DNA alkylating agents. Significant progress has been made in the knowledge of bacterial physiology as result of in-depth genomic analyses and gene inactivation studies. Aside from the notable exception of canonical Mismatch Repair (MMR) components, the DNA damage response of MTB includes most of the DNA repair pathways described in other bacterial species: (i) multi-enzymatic systems like Nucleotide Excision Repair (NER) and Base Excision Repair (BER); (ii) recombination repair systems; and (iii) single proteins responsible for the direct reversal of DNA damage [83]. Interestingly, a NucS-dependent DNA repair system that potentially replaces the MutS/MutL-based MMR was recently identified in Mycobacterium smegmatis [84]. DNA repair pathways should be considered an attractive source of drug targets, since they provide essential functions to the bacteria [45], and in many cases, the enzymatic cascade requires proteins that are distinct from the human ones at the biochemical and structural level, potentially ensuring drug selectivity. Oxidative and pro-alkylation stresses are mainly counteracted by the action of the multi-step NER pathway, in which the damage recognition and subsequent endonucleolytic reactions are carried out by the coordinated action of the UvrA, UvrB and UvrC proteins [85]. In MTB, as observed in other bacteria, the NER cascade begins with formation of a macromolecular complex between UvrA and UvrB proteins that were structurally and biochemically characterized [86,87]. It was demonstrated that the two proteins interact in solutions in the absence of ligands, supporting the hypothesis that the scouting of damaged sites inside DNA could involve pre-assembled UvrA2/UvrB2 heterotetramers, and suggesting the possibility of targeting UvrA-UvrB in order to block the entire NER cascade in MTB [88,89]. Moreover, an inhibitor of the endonuclease activity of the UvrABC complex [2-(5-amino-1,3,4-thiadiazol-2-ylbenzo[f]chromen-3-one) (ATBC)] was identified, which was active at a micromolar concentration [90]. However, the mechanistic aspects of ATBC inhibitory activity and the direct target inside the UvrABC system are still unknown. The full complement of genes that encode for homologs involved in the BER pathway have been detected in MTB [5]. Despite proteins belonging to BER pathways in mycobacteria being highly conserved, they have not yet been exploited for the design of new compounds that target this pathway. As an alternative to the multi-enzymatic DNA repair response, MTB also counteracts the deleterious effect of alkylating agents by the expression of inducible genes of Ada response [91,92]. The domains of Ada (i.e., AdaA and AdaB), AlkA and AlkB proteins exist in different combinations in different bacteria. In particular, MTB shows a gene fusion of adaA with alkA (AdaA-AlkA), and an independent adaB gene, also annotated as ogt, which encodes for the OGT protein (Rv1316c). MTB OGT, as the orthologous proteins of other organisms, invariably performs the removal of alkyl adducts on modified guanines through a suicidal mechanism, by catalyzing the stoichiometric transfer of the O6-alkyl group to the strictly conserved cysteine residue in the protein active site, which is hosted in the C-terminal domain [93,94,95]. The protein’s overall structure and mechanistic aspects of the suicidal reaction are highly conserved among prokaryotic OGTs, as well as in the human equivalent enzyme [96,97,98,99,100], which is a validated target for cancer chemotherapy [101]. Small-molecule inhibitors of the human enzyme, which are currently used as adjuvants in antineoplastic therapeutic regimens, have not been tested on MTB; indeed, their potential exploitability as anti-tubercular drugs is limited by their cross-reactivity with the human protein, possibly resulting in a genotoxic effect. While nucleotide synthesis and DNA replication have both been exploited for the development of antibacterial agents and are therefore logical and solid targets for the discovery of novel antitubercular drugs, DNA repair is proposed as a new target for the design of drugs with a completely new mechanism of action, and therefore potentially active against drug-resistant strains. However, this poses significant challenges not only for selectivity of drug action and drug resistance, but also for a potential risk of emergence of hypervirulent bacteria. Indeed, as the DNA repair is a major mechanism limiting the occurrence of spontaneous and/or induced mutations, its inactivation could result in hypermutability with the selection of a hypervirulent strain.
5. Conclusions
A number of biochemical events ensure the genome integrity in all organisms; many of these are highly conserved and essential. Moreover, most of the protein complexes active in the maintenance of DNA homeostasis in bacteria, including MTB, are distinct from those in eukaryotes, and should be considered a valid source of molecular targets for antibiotic design and development. To this end, we summarize in this review current understanding of biochemical and structural features of mycobacterial DNA metabolism pathways, and particularly focused on the three main processes described below: (i) synthesis of nucleotide precursors, describing the drug discovery approaches targeting purine and pyrimidine biosynthesis at level of the key enzymatic steps; (ii) DNA synthesis, analyzing the components of replication machinery (i.e., the helicase-primase complex, the core complex and the clamp loader complex) that have been exploited for the development of antibacterial agents; and (iii) DNA repair and damage reversal, widely unexploited as a drug target, discussing the innovative strategies that have been suggested to interfere with mycobacterial responses to DNA damage. The classes of approved drugs targeting DNA metabolism, along with those that have been characterized in preliminary in vitro studies, described in the manuscript, are listed in Table 1.
Author Contributions
Conceptualization, R.M. and M.R.; resources, M.R.; writing—original draft preparation, R.M., C.M., F.R., M.R., writing—review and editing, R.M., C.M., F.R., M.R; supervision, M.R.; funding acquisition, R.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by FONDAZIONE CARIPLO, grant number 2016-0604.
Acknowledgments
In this section you can acknowledge any support given which is not covered by the author contribution or funding sections. This may include administrative and technical support, or donations in kind (e.g., materials used for experiments).
Conflicts of Interest
The authors declare no conflict of interest.
References
- World Health Organization. Global Tuberculosis Report 2019; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Dheda, K.; Gumbo, T.; Gandhi, N.R.; Murray, M.; Theron, G.; Udwadia, Z.; Migliori, G.B.; Warren, R. Global control of tuberculosis: From extensively drug-resistant to untreatable tuberculosis. Lancet Respir. Med. 2014, 4, 321–338. [Google Scholar] [CrossRef] [Green Version]
- Nahid, P.; Dorman, S.E.; Alipanah, N.; Barry, P.M.; Brozek, J.L.; Cattamanchi, A.; Chaisson, L.H.; Chaisson, R.E.; Daley, C.L.; Grzemska, M.; et al. Official American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America Clinical Practice Guidelines: Treatment of Drug-Susceptible Tuberculosis. Clin. Infect. Dis. 2016, 63, 147–195. [Google Scholar] [CrossRef] [PubMed]
- Sotgiu, G.; Centis, R.; D’ambrosio, L.; Migliori, G.B. Tuberculosis treatment and drug regimens. Cold Spring Harb. Perspect. Med. 2015, 5, a017822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, S.T.; Brosch, R.; Parkhill, J.; Garnier, T.; Churcher, C.; Harris, D.; Gordon, S.V.; Eiglmeier, K.; Gas, S.; Barry III, C.E.; et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Wallis, R.S.; Maeurer, M.; Mwaba, P.; Chakaya, J.; Rustomjee, R.; Migliori, G.B.; Marais, B.; Schito, M.; Churchyard, G.; Swaminathan, S.; et al. Tuberculosis—Advances in development of new drugs, treatment regimens, host-directed therapies, and biomarkers. Lancet Infect. Dis. 2016, 16, 34–46. [Google Scholar] [CrossRef] [Green Version]
- Cole, S.T.; Riccardi, G. New tuberculosis drugs on the horizon. Curr. Opin. Microbiol. 2011, 14, 570–576. [Google Scholar] [CrossRef] [PubMed]
- Koul, A.; Arnoult, E.; Lounis, N.; Guillemont, J.; Andries, K. The challenge of new drug discovery for tuberculosis. Nature 2011, 469, 483–490. [Google Scholar] [CrossRef]
- Global Alliance for TB Drug Development. Tuberculosis. Scientific blueprint for tuberculosis drug development. Tuberculosis (Edinb) 2001, 81, 1–52. [Google Scholar] [CrossRef]
- Zumla, A.; Nahid, P.; Cole, S.T. Advances in the development of new tuberculosis drugs and treatment regimens. Nat. Rev. Drug Discov. 2013, 12, 388–404. [Google Scholar] [CrossRef]
- Makarov, V.; Lechartier, B.; Zhang, M.; Neres, J.; van der Sar, A.M.; Raadsen, S.A.; Hartkoorn, R.C.; Ryabova, O.B.; Vocat, A.; Decosterd, L.A.; et al. Towards a new combination therapy for tuberculosis with next generation benzothiazinones. EMBO Mol. Med. 2014, 6, 372–383. [Google Scholar] [CrossRef]
- Xu, Z.; Meshcheryakov, V.A.; Poce, G.; Chng, S.S. MmpL3 is the flippase for mycolic acids in mycobacteria. Proc. Natl. Acad. Sci. USA 2017, 114, 7993–7998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, A.; Parai, M.K.; Shetty, N.; Wallis, D.; Woolhiser, L.; Hastings, C.; Dutta, N.K.; Galaviz, S.; Dhakal, R.C.; Shrestha, R.; et al. Development of a Novel Lead that Targets M. tuberculosis Polyketide Synthase 13. Cell 2017, 170, 249–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jadhavar, P.S.; Vaja, M.D.; Dhameliya, T.M.; Chakraborti, A.K. Oxazolidinones as Anti-tubercular Agents: Discovery, Development and Future Perspectives. Curr. Med. Chem. 2015, 22, 4379–4397. [Google Scholar] [CrossRef] [PubMed]
- Pethe, K.; Bifani, P.; Jang, J.; Kang, S.; Park, S.; Ahn, S.; Jiricek, J.; Jung, J.; Jeon, H.K.; Cechetto, J.; et al. Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat. Med. 2013, 19, 1157–1160. [Google Scholar] [CrossRef] [PubMed]
- Pym, A.S.; Diacon, A.H.; Tang, S.J.; Conradie, F.; Danilovits, M.; Chuchottaworn, C.; Vasilyeva, I.; Andries, K.; Bakare, N.; De Marez, T.; et al. Bedaquiline in the treatment of multidrug- and extensively drug-resistant tuberculosis. Eur. Respir. J. 2016, 47, 564–574. [Google Scholar] [CrossRef] [Green Version]
- Nuermberger, E.L.; Yoshimatsu, T.; Tyagi, S.; O’Brien, R.J.; Vernon, A.N.; Chaisson, R.E.; Bishai, W.R.; Grosset, J.H. Moxifloxacin-containing regimen greatly reduces time to culture conversion in murine tuberculosis. Am. J. Respir. Crit. Care Med. 2004, 169, 421–426. [Google Scholar] [CrossRef]
- Reiche, M.A.; Warner, D.F.; Mizrahi, V. Targeting DNA Replication and Repair for the Development of Novel Therapeutics against Tuberculosis. Front. Mol. Biosci. 2017, 4, 75. [Google Scholar] [CrossRef] [Green Version]
- Sanyal, G.; Doig, P. Bacterial DNA replication enzymes as targets for antibacterial drug discovery. Expert Opin. Drug Discov. 2012, 7, 327–339. [Google Scholar] [CrossRef]
- Robinson, A.; Causer, R.J.; Dixon, N.E. Architecture and conservation of the bacterial DNA replication machinery, an underexploited drug target. Curr. Drug Targets. 2012, 13, 352–372. [Google Scholar] [CrossRef] [Green Version]
- Van Eijk, E.; Wittekoek, B.; Kuijper, E.J.; Smits, W.K. DNA replication proteins as potential targets for antimicrobials in drug-resistant bacterial pathogens. J. Antimicrob. Chemother. 2017, 72, 1275–1284. [Google Scholar] [CrossRef]
- Płocinska, R.; Korycka-Machala, M.; Plocinski, P.; Dziadek, J. Mycobacterial DNA Replication as a Target for Antituberculosis. Drug Discov. Curr. Top. Med. Chem. 2017, 17, 2129–2142. [Google Scholar] [CrossRef] [PubMed]
- Donini, S.; Garavaglia, S.; Ferraris, D.M.; Miggiano, R.; Mori, S.; Shibayama, K.; Rizzi, M. Biochemical and structural investigations on phosphoribosylpyrophosphate synthetase from Mycobacterium smegmatis. PLoS ONE 2017, 12, e0175815. [Google Scholar] [CrossRef] [PubMed]
- Alderwick, L.J.; Lloyd, G.S.; Lloyd, A.J.; Lovering, A.L.; Eggeling, L.; Besra, G.S. Biochemical characterization of the Mycobacterium tuberculosis phosphoribosyl-1-pyrophosphate synthetase. Glycobiology 2011, 21, 410–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breda, A.; Martinelli, L.K.; Bizarro, C.V.; Rosado, L.A.; Borges, C.B.; Santos, D.S.; Basso, L.A. Wild-type phosphoribosylpyrophosphate synthase (PRS) from Mycobacterium tuberculosis: A bacterial class II PRS? PLoS ONE 2012, 7, e39245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Weerd, R.; Boot, M.; Maaskant, J.; Sparrius, M.; Verboom, T.; van Leeuwen, L.M.; Burggraaf, M.J.; Paauw, N.J.; Dainese, E.; Manganelli, R.; et al. Inorganic Phosphate Limitation Modulates Capsular Polysaccharide Composition in Mycobacteria. J. Biol. Chem. 2016, 291, 11787–11799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferraris, D.M.; Spallek, R.; Oehlmann, W.; Singh, M.; Rizzi, M. Crystal structure of the Mycobacterium tuberculosis phosphate binding protein PstS3. Proteins 2014, 82, 2268–2274. [Google Scholar] [CrossRef] [Green Version]
- Warner, D.F.; Evans, J.C.; Mizrahi, V. Nucleotide Metabolism and DNA Replication. In Molecular Genetics of Mycobacteria, 2nd ed.; Hatfull, G.F., Jacobs, W.R., Jr., Eds.; American Society for Microbiology: Washington, WA, USA, 2014. [Google Scholar]
- Usha, V.; Gurcha, S.S.; Lovering, A.L.; Lloyd, A.J.; Papaemmanouil, A.; Reynolds, R.C.; Besra, G.S. Identification of novel diphenyl urea inhibitors of Mt-GuaB2 active against Mycobacterium tuberculosis. Microbiology 2011, 157, 290–299. [Google Scholar] [CrossRef] [Green Version]
- Griffin, J.E.; Gawronski, J.D.; Dejesus, M.A.; Ioerger, T.R.; Akerley, B.J.; Sassetti, C.M. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 2011, 7, e1002251. [Google Scholar] [CrossRef] [Green Version]
- Hedstrom, L. IMP dehydrogenase: Structure, mechanism, and inhibition. Chem. Rev. 2009, 109, 2903–2928. [Google Scholar] [CrossRef] [Green Version]
- Shu, Q.; Nair, V. Inosine monophosphate dehydrogenase (IMPDH) as a target in drug discovery. Med. Res. Rev. 2008, 28, 219–232. [Google Scholar] [CrossRef]
- Chen, L.; Wilson, D.J.; Xu, Y.; Aldrich, C.C.; Felczak, K.; Sham, Y.Y.; Pankiewicz, K.W. Triazole-linked inhibitors of inosine monophosphate dehydrogenase from human and Mycobacterium tuberculosis. J. Med. Chem. 2010, 53, 4768–4778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payne, D.J.; Gwynn, M.N.; Holmes, D.J.; Pompliano, D.L. Drugs for bad bugs: Confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 2007, 6, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Ioerger, T.R.; O’Malley, T.; Liao, R.; Guinn, K.M.; Hickey, M.J.; Mohaideen, N.; Murphy, K.C.; Boshoff, H.I.; Mizrahi, V.; Rubin, E.J.; et al. Identification of new drug targets and resistance mechanisms in Mycobacterium tuberculosis. PLoS ONE 2013, 8, e75245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.; Pacitto, A.; Bayliss, T.; Cleghorn, L.A.T.; Wang, Z.; Hartman, T.; Arora, K.; Ioerger, T.R.; Sacchettini, J.; Rizzi, M.; et al. Essential but Not Vulnerable: Indazole Sulfonamides Targeting Inosine Monophosphate Dehydrogenase as Potential Leads against Mycobacterium tuberculosis. ACS Infect. Dis. 2017, 3, 18–33. [Google Scholar] [CrossRef]
- Singh, V.; Donini, S.; Pacitto, A.; Sala, C.; Hartkoorn, R.C.; Dhar, N.; Keri, G.; Ascher, D.B.; Mondésert, G.; Vocat, A.; et al. The Inosine Monophosphate Dehydrogenase, GuaB2, Is a Vulnerable New Bactericidal Drug Target for Tuberculosis. ACS Infect. Dis. 2017, 3, 5–17. [Google Scholar] [CrossRef]
- Sahu, N.U.; Singh, V.; Ferraris, D.M.; Rizzi, M.; Kharkar, P.S. Hit discovery of Mycobacterium tuberculosis inosine 5′-monophosphate dehydrogenase, GuaB2, inhibitors. Bioorg. Med. Chem. Lett. 2018, 28, 1714–1718. [Google Scholar] [CrossRef]
- Chacko, S.; Boshoff, H.I.M.; Singh, V.; Ferraris, D.M.; Gollapalli, D.R.; Zhang, M.; Lawson, A.P.; Pepi, M.J.; Joachimiak, A.; Rizzi, M.; et al. Expanding Benzoxazole-Based Inosine 5′-Monophosphate Dehydrogenase (IMPDH) Inhibitor Structure-Activity As Potential Antituberculosis Agents. J. Med. Chem. 2018, 61, 4739–4756. [Google Scholar] [CrossRef]
- Makowska-Grzyska, M.; Kim, Y.; Gorla, S.K.; Wei, Y.; Mandapati, K.; Zhang, M.; Maltseva, N.; Modi, G.; Boshoff, H.I.; Gu, M.; et al. Mycobacterium tuberculosis IMPDH in Complexes with Substrates, Products and Antitubercular Compounds. PLoS ONE 2015, 10, e0138976. [Google Scholar] [CrossRef] [Green Version]
- Umejiego, N.N.; Gollapalli, D.; Sharling, L.; Volftsun, A.; Lu, J.; Benjamin, N.N.; Stroupe, A.H.; Riera, T.V.; Striepen, B.; Hedstrom, L. Targeting a prokaryotic protein in a eukaryotic pathogen: Identification of lead compounds against cryptosporidiosis. Chem. Biol. 2008, 15, 70–77. [Google Scholar] [CrossRef] [Green Version]
- Maurya, S.K.; Gollapalli, D.R.; Kirubakaran, S.; Zhang, M.; Johnson, C.R.; Benjamin, N.N.; Hedstrom, L.; Cuny, G.D. Triazole inhibitors of Cryptosporidium parvum inosine 5′-monophosphate dehydrogenase. J. Med. Chem. 2009, 52, 4623–4630. [Google Scholar] [CrossRef] [Green Version]
- Kirubakaran, S.; Gorla, S.K.; Sharling, L.; Zhang, M.; Liu, X.; Ray, S.S.; MacPherson, I.S.; Striepen, B.; Hedstrom, L.; Cuny, G.D. Structure-activity relationship study of selective benzimidazole-based inhibitors of Cryptosporidium parvum IMPDH. Bioorg. Med. Chem. Lett. 2012, 22, 1985–1988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharling, L.; Liu, X.; Gollapalli, D.R.; Maurya, S.K.; Hedstrom, L.; Striepen, B. A screening pipeline for antiparasitic agents targeting cryptosporidium inosine monophosphate dehydrogenase. PLoS Negl. Trop. Dis. 2010, 4, e794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sassetti, C.M.; Boyd, D.H.; Rubin, E.J. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 2003, 48, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Breda, A.; Rosado, L.A.; Lorenzini, D.M.; Basso, L.A.; Santos, D.S. Molecular, kinetic and thermodynamic characterization of Mycobacterium tuberculosis orotate phosphoribosyltransferase. Mol. Biosyst. 2012, 8, 572–586. [Google Scholar] [CrossRef] [PubMed]
- Donini, S.; Ferraris, D.M.; Miggiano, R.; Massarotti, A.; Rizzi, M. Structural investigations on orotate phosphoribosyltransferase from Mycobacterium tuberculosis, a key enzyme of the de novo pyrimidine biosynthesis. Sci. Rep. 2017, 7, 1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breda, A.; Machado, P.; Rosado, L.A.; Souto, A.A.; Santos, D.S.; Basso, L.A. Pyrimidin-2(1H)-ones based inhibitors of Mycobacterium tuberculosis orotate phosphoribosyltransferase. Eur. J. Med. Chem. 2012, 54, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Kolly, G.S.; Boldrin, F.; Sala, C.; Dhar, N.; Hartkoorn, R.C.; Ventura, M.; Serafini, A.; McKinney, J.D.; Manganelli, R.; Cole, S.T. Assessing the essentiality of the decaprenyl-phospho-d-arabinofuranose pathway in Mycobacterium tuberculosis using conditional mutants. Mol. Microbiol. 2014, 92, 194–211. [Google Scholar] [CrossRef] [Green Version]
- Wolucka, B.A. Biosynthesis of D-arabinose in mycobacteria - a novel bacterial pathway with implications for antimycobacterial therapy. FEBS J. 2008, 275, 2691–2711. [Google Scholar] [CrossRef]
- Lucarelli, A.P.; Buroni, S.; Pasca, M.R.; Rizzi, M.; Cavagnino, A.; Valentini, G.; Riccardi, G.; Chiarelli, L.R. Mycobacterium tuberculosis phosphoribosylpyrophosphate synthetase: Biochemical features of a crucial enzyme for mycobacterial cell wall biosynthesis. PLoS ONE 2010, 5, e15494. [Google Scholar] [CrossRef] [Green Version]
- Fivian-Hughes, A.S.; Houghton, J.; Davis, E.O. Mycobacterium tuberculosis thymidylate synthase gene thyX is essential and potentially bifunctional, while thyA deletion confers resistance to p-aminosalicylic acid. Microbiology 2012, 158, 308–318. [Google Scholar] [CrossRef]
- Luciani, R.; Saxena, P.; Surade, S.; Santucci, M.; Venturelli, A.; Borsari, C.; Marverti, G.; Ponterini, G.; Ferrari, S.; Blundell, T.L.; et al. Virtual Screening and X-ray Crystallography Identify Non-Substrate Analog Inhibitors of Flavin-Dependent Thymidylate Synthase. J. Med. Chem. 2016, 59, 9269–9275. [Google Scholar] [CrossRef] [PubMed]
- Hirmondo, R.; Lopata, A.; Suranyi, E.V.; Vertessy, B.G.; Toth, J. Differential control of dNTP biosynthesis and genome integrity maintenance by the dUTPase superfamily enzymes. Sci. Rep. 2017, 7, 6043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ditse, Z.; Lamers, M.H.; Warner, D.F. DNA Replication in Mycobacterium tuberculosis. In Tuberculosis and the Tubercle Bacillus, 2nd ed.; Jacobs, W.R., Jr., McShane, H., Mizrahi, V., Orme, I.M., Eds.; American Society for Microbiology: Washington, WA, USA, 2018. [Google Scholar]
- McHenry, C.S. DNA replicases from a bacterial perspective. Annu. Rev. Biochem. 2011, 80, 403–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brézellec, P.; Vallet-Gely, I.; Possoz, C.; Quevillon-Cheruel, S.; Ferat, J.L. DciA is an ancestral replicative helicase operator essential for bacterial replication initiation. Nat. Commun. 2016, 7, 13271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, K.M.; Huang, D.L.; Hooppaw, A.J.; Logsdon, M.M.; Richardson, K.; Lee, H.J.; Kimmey, J.M.; Aldridge, B.B.; Stallings, C.L. Rv0004 is a new essential member of the mycobacterial DNA replication machinery. PLoS Genet. 2017, 13, e1007115. [Google Scholar] [CrossRef] [PubMed]
- Frank, J. Time-resolved cryo-electron microscopy: Recent progress. J. Struct. Biol. 2017, 200, 303–306. [Google Scholar] [CrossRef]
- Shereda, R.D.; Kozlov, A.G.; Lohman, T.M.; Cox, M.M.; Keck, J.L. SSB as an organizer/mobilizer of genome maintenance complexes. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 289–318. [Google Scholar] [CrossRef] [Green Version]
- Lu, D.; Bernstein, D.A.; Satyshur, K.A.; Keck, J.L. Small-molecule tools for dissecting the roles of SSB/protein interactions in genome maintenance. Proc. Natl. Acad. Sci. USA 2010, 107, 633–638. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.C.; Smith, K.C. Mechanism of sbcB-suppression of the recBC-deficiency in postreplication repair in UV-irradiated Escherichia coli K-12. Mol. Gen. Genet. 1985, 201, 186–191. [Google Scholar] [CrossRef]
- Marceau, A.H.; Bernstein, D.A.; Walsh, B.W.; Shapiro, W.; Simmons, L.A.; Keck, J.L. Protein interactions in genome maintenance as novel antibacterial targets. PLoS ONE 2013, 8, e58765. [Google Scholar] [CrossRef] [Green Version]
- Hegde, V.R.; Pu, H.; Patel, M.; Black, T.; Soriano, A.; Zhao, W.; Gullo, V.P.; Chan, T.-M. Two new bacterial DNA primase inhibitors from the plant Polygonum cuspidatum. Bioorg. Med. Chem. Lett. 2004, 14, 2275–2277. [Google Scholar] [CrossRef] [PubMed]
- Chu, M.; Mierzwa, R.; Xu, L.; He, L.; Terracciano, J.; Patel, M.; Gullo, V.; Black, T.; Zhao, W.; Chan, T.-M.; et al. Isolation and structure elucidation of Sch 642305, a novel bacterial DNA primase inhibitor produced by Penicillium verrucosum. J. Nat. Prod. 2003, 66, 1527–1530. [Google Scholar] [CrossRef] [PubMed]
- Griep, M.A.; Blood, S.; Larson, M.A.; Koepsell, S.A.; Hinrichs, S.H. Myricetin inhibits Escherichia coli DnaB helicase but not primase. Bioorg. Med. Chem. 2007, 15, 7203–7208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.H.; Huang, C.Y. Characterization of flavonol inhibition of DnaB helicase: Real-time monitoring, structural modeling, and proposed mechanism. J. Biomed. Biotechnol. 2012, 2012, 735368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, D.S.; Lennon, C.W.; Li, Z.; Miller, M.R.; Banavali, N.K.; Li, H.; Belfort, M. Mycobacterial DnaB helicase intein as oxidative stress sensor. Nat. Commun. 2018, 9, 4363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kling, A.; Lukat, P.; Almeida, D.V.; Bauer, A.; Fontaine, E.; Sordello, S.; Zaburannyi, N.; Herrmann, J.; Wenzel, S.C.; König, C.; et al. Targeting DnaN for tuberculosis therapy using novel griselimycins. Science 2015, 348, 1106–1112. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.; Sankhe, K.; Suvarna, V.; Sherje, A.; Patel, K.; Dravyakar, B. DNA gyrase inhibitors: Progress and synthesis of potent compounds as antibacterial agents. Biomed. Pharmacother. 2018, 103, 923–938. [Google Scholar] [CrossRef]
- Kohanski, M.A.; Dwyer, D.J.; Collins, J.J. How antibiotics kill bacteria: From targets to networks. Nat. Rev. Microbiol. 2010, 8, 423–435. [Google Scholar] [CrossRef] [Green Version]
- Lewis, R.J.; Singh, O.M.; Smith, C.V.; Skarzynski, T.; Maxwell, A.; Wonacott, A.J.; Wigley, D.B. The nature of inhibition of DNA gyrase by the coumarins and the cyclothialidines revealed by X-ray crystallography. EMBO J. 1996, 15, 1412–1420. [Google Scholar] [CrossRef]
- Chopra, S.; Matsuyama, K.; Tran, T.; Malerich, J.P.; Wan, B.; Franzblau, S.G.; Lun, S.; Guo, H.; Maiga, M.C.; Bishai, W.R.; et al. Evaluation of gyrase B as a drug target in Mycobacterium tuberculosis. J. Antimicrob. Chemother. 2012, 67, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Godbole, A.A.; Ahmed, W.; Bhat, R.S.; Bradley, E.K.; Ekins, S.; Nagaraja, V. Inhibition of Mycobacterium tuberculosis topoisomerase I by m-AMSA, a eukaryotic type II topoisomerase poison. Biochem. Biophys. Res. Commun. 2014, 446, 916–920. [Google Scholar] [CrossRef] [PubMed]
- Godbole, A.A.; Ahmed, W.; Bhat, R.S.; Bradley, E.K.; Ekins, S.; Nagaraja, V. Targeting Mycobacterium tuberculosis topoisomerase I by small-molecule inhibitors. Antimicrob. Agents Chemother. 2015, 59, 1549–1557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, C.; Martins, A.; Bongiorno, P.; Glickman, M.; Shuman, S. Biochemical and genetic analysis of the four DNA ligases of mycobacteria. J. Biol. Chem. 2004, 279, 20594–20606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, S.K.; Dube, D.; Tewari, N.; Dwivedi, N.; Tripathi, R.P.; Ramachandran, R. Mycobacterium tuberculosis NAD+-dependent DNA ligase is selectively inhibited by glycosylamines compared with human DNA ligase I. Nucleic Acids Res. 2005, 33, 7090–7101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, S.K.; Dube, D.; Kukshal, V.; Jha, A.K.; Hajela, K.; Ramachandran, R. NAD+-dependent DNA ligase (Rv3014c) from Mycobacterium tuberculosis: Novel structure-function relationship and identification of a specific inhibitor. Proteins 2007, 69, 97–111. [Google Scholar] [CrossRef]
- Srivastava, S.K.; Tripathi, R.P.; Ramachandran, R. NAD+-dependent DNA Ligase (Rv3014c) from Mycobacterium tuberculosis. Crystal structure of the adenylation domain and identification of novel inhibitors. J. Biol. Chem. 2005, 280, 30273–30281. [Google Scholar]
- Gorna, A.E.; Bowater, R.P.; Dziadek, J. DNA repair systems and the pathogenesis of Mycobacterium tuberculosis: Varying activities at different stages of infection. Clin. Sci. (Lond.) 2010, 119, 187–202. [Google Scholar] [CrossRef] [Green Version]
- Durbach, S.I.; Springer, B.; Machowski, E.E.; North, R.J.; Papavinasasundaram, K.G.; Colston, M.J.; Böttger, E.C.; Mizrahi, V. DNA alkylation damage as a sensor of nitrosative stress in Mycobacterium tuberculosis. Infect. Immun. 2003, 71, 997–1000. [Google Scholar] [CrossRef] [Green Version]
- Drabløs, F.; Feyzi, E.; Aas, P.A.; Vaagbø, C.B.; Kavli, B.; Bratlie, M.S.; Peña-Diaz, J.; Otterlei, M.; Slupphaug, G.; Krokan, H.E. Alkylation damage in DNA and RNA--repair mechanisms and medical significance. DNA Repair 2004, 3, 1389–1407. [Google Scholar] [CrossRef]
- Mizrahi, V.; Andersen, S.J. DNA repair in Mycobacterium tuberculosis. What have we learnt from the genome sequence? Mol. Microbiol. 1998, 29, 1331–1339. [Google Scholar] [CrossRef]
- Castañeda-García, A.; Prieto, A.I.; Rodríguez-Beltrán, J.; Alonso, N.; Cantillon, D.; Costas, C.; Pérez-Lago, L.; Zegeye, E.D.; Herranz, M.; Plociński, P.; et al. A non-canonical mismatch repair pathway in prokaryotes. Nat. Commun. 2017, 8, 14246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truglio, J.J.; Croteau, D.L.; Van Houten, B.; Kisker, C. Prokaryotic nucleotide excision repair: The UvrABC system. Chem. Rev. 2006, 106, 233–252. [Google Scholar] [CrossRef] [PubMed]
- Rossi, F.; Khanduja, J.S.; Bortoluzzi, A.; Houghton, J.; Sander, P.; Güthlein, C.; Davis, E.O.; Springer, B.; Böttger, E.C.; Relini, A.; et al. The biological and structural characterization of Mycobacterium tuberculosis UvrA provides novel insights into its mechanism of action. Nucleic Acids Res. 2011, 39, 7316–7328. [Google Scholar] [CrossRef] [PubMed]
- Thakur, M.; Kumar, M.B.; Muniyappa, K. Mycobacterium tuberculosis UvrB Is a Robust DNA-Stimulated ATPase That Also Possesses Structure-Specific ATP-Dependent DNA Helicase Activity. Biochemistry 2016, 55, 5865–5883. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, S.; Rizzi, M.; Rossi, F.; Miggiano, R. Mycobacterium tuberculosis UvrB forms dimers in solution and interacts with UvrA in the absence of ligands. Proteins 2018, 86, 98–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferraris, D.M.; Miggiano, R.; Rossi, F.; Rizzi, M. Mycobacterium tuberculosis Molecular Determinants of Infection, Survival Strategies, and Vulnerable Targets. Pathogens 2018, 7, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazloum, N.; Stegman, M.A.; Croteau, D.L.; Houten, B.V.; Kwon, N.S.; Ling, Y.; Dickinson, C.; Venugopal, A.; Towheed, M.A.; Nathan, C. Identification of a chemical that inhibits the mycobacterial UvrABC complex in nucleotide excision repair. Biochemistry 2011, 50, 1329–1335. [Google Scholar] [CrossRef]
- Yang, M.; Aamodt, R.M.; Dalhus, B.; Balasingham, S.; Helle, I.; Andersen, P.; Tønjum, T.; Alseth, I.; Rognes, T.; Bjørås, M. The ada operon of Mycobacterium tuberculosis encodes two DNA methyltransferases for inducible repair of DNA alkylation damage. DNA Repair 2011, 10, 595–602. [Google Scholar] [CrossRef]
- Shrivastav, N.; Li, D.; Essigmann, J.M. Chemical biology of mutagenesis and DNA repair: Cellular responses to DNA alkylation. Carcinogenesis 2010, 31, 59–70. [Google Scholar] [CrossRef] [Green Version]
- Miggiano, R.; Casazza, V.; Garavaglia, S.; Ciaramella, M.; Perugino, G.; Rizzi, M.; Rossi, F. Biochemical and structural studies of the Mycobacterium tuberculosis O6-methylguanine methyltransferase and mutated variants. J. Bacteriol. 2013, 195, 2728–2736. [Google Scholar] [CrossRef] [Green Version]
- Miggiano, R.; Perugino, G.; Ciaramella, M.; Serpe, M.; Rejman, D.; Páv, O.; Pohl, R.; Garavaglia, S.; Lahiri, S.; Rizzi, M.; et al. Crystal structure of Mycobacterium tuberculosis O6-methylguanine-DNA methyltransferase protein clusters assembled on to damaged DNA. Biochem. J. 2016, 473, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Miggiano, R.; Valenti, A.; Rossi, F.; Rizzi, M.; Perugino, G.; Ciaramella, M. Every OGT Is Illuminated … by Fluorescent and Synchrotron Lights. Int. J. Mol. Sci. 2017, 18, 2613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perugino, G.; Miggiano, R.; Serpe, M.; Vettone, A.; Valenti, A.; Lahiri, S.; Rossi, F.; Rossi, M.; Rizzi, M.; Ciaramella, M. Structure-function relationships governing activity and stability of a DNA alkylation damage repair thermostable protein. Nucleic Acids Res. 2015, 43, 8801–8816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrone, C.; Miggiano, R.; Serpe, M.; Massarotti, A.; Valenti, A.; del Monaco, G.; Rossi, M.; Rossi, F.; Rizzi, M.; Perugino, G.; et al. Interdomain interactions rearrangements control the reaction steps of a thermostable DNA alkyltransferase. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, H.; Inoue, T.; Nishioka, M.; Fujiwara, S.; Takagi, M.; Imanakac, T.; Kai, Y. Hyperthermostable protein structure maintained by intra and inter-helix ion-pairs in archaeal O6-methylguanine-DNA methyltransferase. J. Mol. Biol. 1999, 292, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.; Pelton, J.G.; Wemmer, D.E. Structural studies of MJ1529, an O6-methylguanine-DNA methyltransferase. Magn. Reson. Chem. 2006, 44, S71–S82. [Google Scholar] [CrossRef] [PubMed]
- Wibley, J.E.; Pegg, A.E.; Moody, P.C. Crystal structure of the human O6-alkylguanine-DNA alkyltransferase. Nucleic Acids Res. 2000, 28, 393–401. [Google Scholar] [CrossRef] [Green Version]
- Pegg, A.E. Multifaceted roles of alkyltransferase and related proteins in DNA repair, DNA damage, resistance to chemotherapy, and research tools. Chem. Res. Toxicol. 2011, 24, 618–639. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Schematic overview of purine and pyrimidine metabolism. Ribose-5-phosphate and carbamoyl-phosphate are the starting points of purine and pyrimidine biosynthetic pathways, respectively. Key intermediates, across de novo biosynthesis and salvage pathways, are highlighted in gray boxes. End-products of purine and pyrimidine catabolism (i.e., uric acid and β-alanine) are in white boxes. Enzymes discussed in the manuscript are depicted in red. PRPP, 5-phosphorybosyl-1-pyrophosphate; OPRT, orotate phosphoribosyltransfrase, PrsA, PRPP synthetase; S-AMP, adenylosuccinate; SAICAR: succinylaminoimidazole carboxamide ribotide.
Figure 1.
Schematic overview of purine and pyrimidine metabolism. Ribose-5-phosphate and carbamoyl-phosphate are the starting points of purine and pyrimidine biosynthetic pathways, respectively. Key intermediates, across de novo biosynthesis and salvage pathways, are highlighted in gray boxes. End-products of purine and pyrimidine catabolism (i.e., uric acid and β-alanine) are in white boxes. Enzymes discussed in the manuscript are depicted in red. PRPP, 5-phosphorybosyl-1-pyrophosphate; OPRT, orotate phosphoribosyltransfrase, PrsA, PRPP synthetase; S-AMP, adenylosuccinate; SAICAR: succinylaminoimidazole carboxamide ribotide.

Figure 2.
Macromolecular complexes assembled on the DNA at the replication fork. Helicase-primase complex constitutes the so-called primosome that binds the lagging strand DNA, unwinding duplex DNA while it synthesizes RNA primers for the lagging strand polymerase. DNA synthesis on both strands is catalyzed by a holoenzyme complex formed by the polymerase and a processivity β-clamp. The clamp is loaded onto the DNA by the clamp loader complex. The leading and lagging strand holoenzymes interact to form a dimer. Single-stranded DNA resulting from helicase activity is coated with single-stranded DNA-binding protein (SSB).
Figure 2.
Macromolecular complexes assembled on the DNA at the replication fork. Helicase-primase complex constitutes the so-called primosome that binds the lagging strand DNA, unwinding duplex DNA while it synthesizes RNA primers for the lagging strand polymerase. DNA synthesis on both strands is catalyzed by a holoenzyme complex formed by the polymerase and a processivity β-clamp. The clamp is loaded onto the DNA by the clamp loader complex. The leading and lagging strand holoenzymes interact to form a dimer. Single-stranded DNA resulting from helicase activity is coated with single-stranded DNA-binding protein (SSB).


{kind=link}
{kind=link}
Table 1.
Essential proteins involved in DNA metabolism targeted by anti-bacterial compounds.
| Target Protein | In Vitro Essentiality [30,45] | Inhibitor Molecules/Classes |
|---|---|---|
| GuaB2 | Essential | diphenyl urea derivatives: DPU-2, DPU-3 [29] |
| triazole-linked mycophenolic adenine [33] | ||
| indazole sulfonamides [36] | ||
| VCC234718 5-(4-cyclohexanecarbonylpiperazine-1-sulfonyl)isoquinoline [37] | ||
| 5-amidophthalide derivative [38] | ||
| Benzoxazole derivatives [39] | ||
| OPRT | Essential | Hydroxy-2-oxo-1,2-dihydropyridine-4-carboxylic acid and its derivative 3-Benzylidene-2,6-dioxo-1,2,3,6-tetrahydropyridine-4-carboxylic acid [48] |
| SSB (E.coli) | Essential | Small-molecule inhibitors 1 [61] |
| DnaG (E.coli) | Essential | Phenolic monosaccharides 1 [64] |
| Bicyclic macrolide 1 [65] | ||
| DnaB (E.coli, K. Pneumoniae) | Essential | Flavonols 1 [66,67] |
| DnaN/β | Essential | Griselimycins [69] |
| GyrA | Essential | Quinolones [71] |
| GyrB | Essential | Novobiocin and coumarin derivatives [72,73] |
| TopA | Essential | m-AMSA [74] |
| Norclomipramine and Imipramin [75] | ||
| Hydroxycamptothecin derivatives [18] | ||
| LigA | Essential | Pyridochromanone [76] |
| Bis-xylofuranosylated diamines [77,79] | ||
| N-substituted tetracyclic indoles [78] | ||
| UvrABC complex | Essential | 2-(5-amino-1,3,4-thiadiazol-2-ylbenzo[f]chromen-3-one) (ATBC) [90] |
1 The compounds refer to inhibition studies performed on homologs of essential MTB proteins belonging to other bacterial species, indicated in brackets.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Miggiano, R.; Morrone, C.; Rossi, F.; Rizzi, M. Targeting Genome Integrity in Mycobacterium Tuberculosis: From Nucleotide Synthesis to DNA Replication and Repair. Molecules 2020, 25, 1205. https://doi.org/10.3390/molecules25051205
AMA Style
Miggiano R, Morrone C, Rossi F, Rizzi M. Targeting Genome Integrity in Mycobacterium Tuberculosis: From Nucleotide Synthesis to DNA Replication and Repair. Molecules. 2020; 25(5):1205. https://doi.org/10.3390/molecules25051205
Chicago/Turabian StyleMiggiano, Riccardo, Castrese Morrone, Franca Rossi, and Menico Rizzi. 2020. "Targeting Genome Integrity in Mycobacterium Tuberculosis: From Nucleotide Synthesis to DNA Replication and Repair" Molecules 25, no. 5: 1205. https://doi.org/10.3390/molecules25051205