
Structure and Aggregation Mechanisms in Amyloids


Chemistry Department and Coimbra Chemistry Centre, Faculty of Sciences and Technology, University of Coimbra, 3004-535 Coimbra, Portugal
*
Author to whom correspondence should be addressed.
Molecules 2020, 25(5), 1195; https://doi.org/10.3390/molecules25051195
Submission received: 15 January 2020
/
Revised: 17 February 2020
/
Accepted: 19 February 2020
/
Published: 6 March 2020
(This article belongs to the Special Issue Amyloids in Neurodegenerative Diseases)
Abstract
:The aggregation of a polypeptide chain into amyloid fibrils and their accumulation and deposition into insoluble plaques and intracellular inclusions is the hallmark of several misfolding diseases known as amyloidoses. Alzheimer′s, Parkinson′s and Huntington’s diseases are some of the approximately 50 amyloid diseases described to date. The identification and characterization of the molecular species critical for amyloid formation and disease development have been the focus of intense scrutiny. Methods such as X-ray and electron diffraction, solid-state nuclear magnetic resonance spectroscopy (ssNMR) and cryo-electron microscopy (cryo-EM) have been extensively used and they have contributed to shed a new light onto the structure of amyloid, revealing a multiplicity of polymorphic structures that generally fit the cross-β amyloid motif. The development of rational therapeutic approaches against these debilitating and increasingly frequent misfolding diseases requires a thorough understanding of the molecular mechanisms underlying the amyloid cascade. Here, we review the current knowledge on amyloid fibril formation for several proteins and peptides from a kinetic and thermodynamic point of view, the structure of the molecular species involved in the amyloidogenic process, and the origin of their cytotoxicity.
1. Background
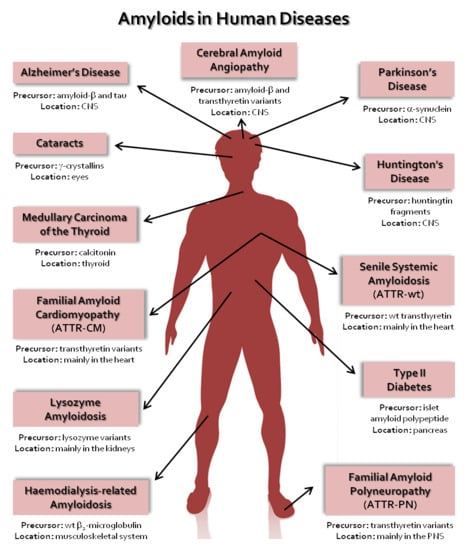
The conversion of normally soluble proteins into insoluble and highly stable amyloid deposits has become the focus of attention by researchers from diverse scientific fields, from physics to chemistry, from biology to medicine. This interest results from the recognition that many of the diseases related with amyloid formation are amongst the most common and debilitating disorders of the modern era [1,2]. Many of these disorders are strongly associated with ageing, such as Alzheimer’s disease (AD) and wild-type TTR amyloidosis (ATTRwt), formerly known as senile systemic amyloidosis (SSA). Alzheimer’s disease affects approximately 50 million people worldwide and it is estimated that this number will exceed 150 million in 2050 [3]. Wild-type TTR amyloidosis affects more than 25% of people over 80 years old, and the disease is becoming more common due to the fast growth of the average age of world population and the increased awareness of medical doctors and positive diagnosis of the disease [4,5]. Presently, there are approximately 50 known amyloid disorders generally referred as protein conformational diseases, misfolding diseases or amyloidosis, with a multitude of distinct symptoms, which are associated with the misfolding and aggregation of several peptides or normally soluble and functional proteins. Table 1 lists some of the amyloid diseases and associated proteins that have been identified to date [1,2]. In the case of local amyloidosis, the amyloid deposits are observed in the organ/tissue where the precursor protein is synthesized; whereas in systemic amyloidosis, the deposition of aggregates and fibrils occurs at locations different than the sites where the precursor protein is expressed [6,7].
As the name suggests, protein misfolding is associated with the formation of an altered protein fold relative to the native structure of a protein. Misfolding can occur as a consequence of several events [8,9,10,11,12,13]: (1) mutations in the gene sequence leading to the production of a protein unable to adopt the native fold; (2) errors in the processes of transcription or translation leading to the production of modified proteins unable to properly fold; (3) failure of the chaperone machinery; (4) mistakes on post-translational modifications or intracellular trafficking of proteins; (5) structural modifications produced by environmental changes; (6) seeding and cross-seeding by pre-formed aggregates or surfaces. Additionally, protein misfolding generally leads to protein aggregation, with specific interactions occurring between intermediate molecular species that tend to form large ordered aggregates, which may evolve into amyloid fibrils which might deposit in the form of insoluble extracellular plaques or intracellular inclusions.
2. Protein Aggregation
In addition to their native structure that intimately correlates with function, proteins also coexist in other states, including disordered, partially unfolded, or multiple aggregation assemblies. Figure 1 summarizes the most relevant protein states, from a mechanistic and biological point of view.
In a functional living system, the multiple conformational states adopted by proteins involve an extremely complex series of thermodynamic equilibria and kinetic barriers, which ultimately are defined by the protein amino acid sequence. Although their intrinsic amino acid sequences and biological environments in which proteins function have co-evolved to preserve proteins in their native and soluble states (non-amyloidogenic pathway), in some conditions, proteins can interconvert into non-functional and cytotoxic protein aggregates (amyloidogenic pathway) (Figure 1) [13,14].
Protein aggregation has been shown to involve natively unfolded or intrinsically disordered systems, such as in amyloid-β (Aβ) peptide in Alzheimer’s disease, or even folded or globular proteins, such as in transthyretin (TTR) associated amyloidoses, where the misfolding process occurs through the formation of partially unfolded states (Table 1 and Figure 1). In most cases, the oligomeric species formed during the amyloidogenic pathway are assemblies of monomeric units. Such aggregates can adopt highly disordered structures, well-defined fibrils with cross-β structure, or also native-like conformations, when originated from unfolded, partially unfolded, or folded monomeric states, respectively (Figure 1). All these types of aggregates have connections with amyloid disorders as they accumulate in well-characterized pathological states and represent a significant manifestation of the multiplicity of mechanisms, structures and morphologies observed during protein aggregation and disease progression (Table 1) [15].
Although the term “amyloid” is often associated with amyloid diseases, the amyloid state can also be present in functional biological processes and contribute to normal cell and tissue physiology (Figure 1). Functional amyloids can be found in bacteria, unicellular eukaryotes, fungi, plants, insects and vertebrates, playing roles as diverse as surface protection and modification, mediation of pathogen-host interactions, pigment biosynthesis, homeostasis control, hormone storage and release, signal transduction, among others [16,17,18,19].
3. Amyloid Fibrils
In 1854, Rudolph Virchow found a macroscopic tissue abnormality that exhibited a positive iodine staining reaction, suggesting the presence of starch or amylin like material (from amylum in Latin and amylon in Greek). He then suggested that the abnormal deposits could be cellulose in origin and coined the name “amyloid”. Friedrich and Kekule, in 1859, showed that the deposits were actually protein aggregates, but the designation remained and is still used today [20]. Thus, abnormal accumulations of insoluble highly ordered and fibrillar protein structures in tissues and organs, such as in “misfolding diseases” (Table 1), are known as amyloid fibrils (Figure 1). No sequence, structural or functional correlations are apparent between many of the proteins that display the ability to form amyloid fibrils. The number of amino acids is diverse ranging, for example, from 37 to 44 in Aβ peptide to 691 in lactoferrin (Table 1). Despite these dissimilarities, amyloid fibrils from different protein precursors share many common structural features, namely, the formation of long, unbranched filaments with a cross-β structural motif [21,22,23].
3.1. The Tinctorial Properties of Amyloid Fibrils
From the early days in amyloid research, molecular probes have been used in order to both characterize the mechanisms of amyloid fibril formation, and detect the presence of amyloid in tissues and samples, taking advantage of the distinctive structural features and tinctorial properties of the amyloid material. These molecular probes display changes in spectroscopic properties upon binding to amyloid fibrils. Usually, they are aromatic heterocyclic compounds, some of which toxic [24]. Congo red (CR) and thioflavin-T (ThT), shown in Figure 2, are the most commonly used dyes to identify amyloid material and study amyloid aggregation [25,26,27].
Since the beginning of the 1920s, Congo red has been used for detection of amyloid fibrils. Upon binding to amyloid fibrils, CR displays green birefringence under polarized light, which is associated with a characteristic red shift in the absorbance maximum (from 490 to 512 nm), and presence of a characteristic shoulder peak at approximately 540 nm [28]. CR is the most commonly applied dye in the identification of amyloid in ex vivo tissue slices, using mainly polarization microscopy, but also light microscopy [29] and fluorescence microscopy [30]. However, compared to ThT, CR is less sensitive in the detection of amyloid fibrils [31]. In addition, CR interferes with the aggregation mechanisms and is not appropriate for in situ detection, since it is known to inhibit or to enhance amyloid fibril formation depending on the protein [32,33,34,35,36,37].
More recently, thioflavin-T became a standard dye for amyloid detection showing fluorescence enhancement upon interaction with amyloid deposits in tissue sections [38]. ThT fluorescence emission increases upon binding to amyloid fibrils, with excitation at 450 nm and emission maxima at 482 nm, as opposed to excitation at 385 nm and emission maxima at 445 nm for the free dye [31].
Besides ThT and CR, other compounds have been also used for the detection of amyloid fibrils [39,40,41,42]. The search for novel, more sensitive and blood–brain barrier crossing dyes for detecting the initial stages and more toxic oligomeric species involved in fibril formation in vitro, ex vivo and in vivo is still under way [43,44,45]. Nevertheless, several related structures or derivatives of both CR [46,47,48,49] and ThT [50,51,52,53] have been shown to be quite helpful for both in vitro and ex vivo identification of amyloid fibrils (Figure 2).
The binding mode of the molecular probes to amyloid fibrils is not fully understood. In the case of ThT and CR, a large number of studies examined binding modes and suggested the existence of a binding interface parallel to the long axis of the fibril [54,55].
Features as fibrillar morphology, cross-β structure, and characteristic tinctorial properties are nowadays accepted as benchmarks for defining “amyloid materials”, and any given protein aggregates need to display most of them to be classified as such. In a recent recommendation, the International Society of Amyloidosis (ISA) nomenclature committee suggests the following classification for amyloids based on five main classes, in order to distinguish different natural and synthetic amyloid-like materials: (1) in vivo and ex vivo disease-related fibrils; (2) in vivo and ex vivo functional fibrils; (3) recombinant fibrils of disease-related proteins or functional amyloid proteins; (4) fibrils from synthetic or non-disease-related peptides; and (5) fibrils from hydrogels that produce the cross-β diffraction pattern [56].
3.2. Structure of Amyloid Fibrils at the Subunit Level
Since the cross-β motif was identified [57], many studies have been performed to characterize the structure of amyloid fibrils, using different experimental techniques. The combination of all these data has contributed to reveal the structure of amyloid fibrils on a multi-scale basis and to show how individual protein subunits can form cross-β structures.
The structural feature which all amyloid fibrils share is the cross-β motif, characterized by extended β-sheets with individual β-strands arranged in an orientation perpendicularly to the fibril main axis. Amyloid fibrils are unbranched structures with diameters ranging from 2 to 20 nm and usually present several micrometers in length [21,58,59]. The existence of a repeating cross-β structure was first demonstrated in the late 1960s by X-ray fiber diffraction studies [60,61], and later synchrotron X-ray diffraction studies have shown that amyloid fibrils from different amyloid proteins exhibit the same cross-β diffraction pattern [62]. Characteristic amyloid fibril diffraction patterns show a meridional reflection at 4.7–4.8 Å corresponding to the hydrogen bonding distances found between paired carbonyl and amide groups in adjacent β-strands, and an equatorial reflection at 6–11 Å corresponding to the distance observed between stacked β-sheets [62].
Amyloid fibrils from different origins contain a common cross-β spine formed by a pair of β-sheets with the facing side chains of the two sheets interdigitated in a steric zipper [63]. Steric zippers are thus short peptide segments which provide the structural basis for the hierarchical assembly of an amyloid fibril [63,64]. In this sense, stacks of steric zippers are needed to form the cross-β spine of the amyloid protofilament (Figure 3) [65], which constitutes the basic unit of the mature fibril, whereas the rest of the polypeptide chain assumes either native-like or random coil conformation in a peripheral position to the spine [63,66].
Nevertheless, significant variations may be found among the structural arrangement of steric zippers. Assembly of the strands in a sheet produces four patterns of sheet symmetry (parallel versus antiparallel, and antifacial versus equifacial). These four β-sheet patterns are each capable of self-pairing to form cross-β spines. Thus, in total, ten distinct symmetry classes of steric zippers can be enumerated. These classes are distinguishable by the particular sheet pattern and by the symmetry operation that relates the pair of sheets (Figure 3) [65].
Mature fibrils also reveal a periodic structure due to the twist of multiple protofilaments around each other. The helical twist of the protofilaments can give rise to a discernible overall helicity of the mature amyloid fibril. Although the basic structural arrangement of the cross-β structure is conserved in different fibrils, there are different possibilities of how protofilaments can pack into the three-dimensional fibril structure, giving rise to several distinct amyloid fibril morphologies. It is possible to distinguish amyloid fibril polymorphism based on the degree of fibril twisting, the number of protofilaments per fibril, and the diameter or weight per length of the fibrils [22,67,68]. In addition to amino acid composition, the fibril morphology is determined by environmental factors, such as pH, temperature, mechanic agitation, shear force, ionic strength or presence of other co-solutes and co-solvents [22,69].
Amyloidogenic regions or short segments forming steric zipper spines in amyloid fibrils are possible to identify/predict by submitting a protein sequence to specific algorithms and web services. Table 2 summarizes the computational tools available online and reported in the literature to predict propensities or incidence levels of a given amino acid sequence to form amyloid fibrils.
3.3. The Cross-α Amyloid-Like Fibril
Cross-β motifs and amyloid fibrils can be formed both by β-sheet rich and α-helical proteins. Alpha-helical conformations have also been associated to amyloid fibril formation. Certain peptides and proteins that present well defined α-helical structures undergo conformational changes into β-pleated structures that in turn aggregate into amyloid fibrils [103,104,105,106,107], while other peptides or proteins maintain its α-helical conformation during the aggregation cascade, forming “cross-α” structures. Cross-α structures are also formed by elongated unbranched fibrils, and display the ability to bind thioflavin-T with the characteristic enhanced fluorescence emission. These fibrils result from the stacking of α-helices perpendicular to the fibril axis [108]. The cross-α structure, although rare, was found in the functional amyloid phenol soluble modulin α3 (PSMα3) [108], in the yeast prion Ure2p [109], in de novo-designed amphiphilic peptides [110,111,112], and among proteins of multiple tandem copies of a helix–loop–helix unit [113].
3.4. Hetero-Amyloid Fibrils
The terms “hetero-amyloid” or “hetero-fibrils” relate to amyloid fibrils structures formed by two different β-sheets along a compact hydrophobic interface, featuring an unusual ladder of sequential stacking. The hetero-fibril is stabilized by hydrophobic packing and enriched with interactions along the fibrillar axis, such as hydrogen bonds between neighboring β-strands, analogous to homo-amyloids. This type of amyloid structure was found in the interaction between the M45 protein with other proteins, namely the receptor-interacting protein kinase 1 and 3 (RIPK1 and RIPK3), and the Z-DNA binding protein-1 (ZBP1) [114]; as well as in the RIPK1-RIPK3 necrosome human signaling complex [115].
In addition, it has also been observed that some amyloidogenic proteins may support cross-seeding by amyloid aggregates of different proteins, building a link between different amyloid forms. Heterologous cross-seeding has been reported between amyloid-β peptide (Aβ) and islet amyloid polypeptide [116,117], Aβ and prion protein (PrP) [118], and Aβ and α-synuclein [119]. Furthermore, Aβ fibrils also induce the formation of tau neurofibrillary tangles in vivo [120], and Aβ aggregates promote tau aggregation in vitro [121]. Heterologous cross-seeding was also found when tau K18 interacts with the C-terminus of α-synuclein [122], when functional amyloids curli and Sup35 induce amyloid protein A amyloidosis [123], when PrPs promote Sup35 aggregation [124], and when the in vitro aggregation of CsgA is seeded by CsgB fibrils [125]. Moreover, the observation of heterologous cross-seeding cases suggests that interactions between different amyloidogenic proteins might enhance the onset of certain types of amyloidoses.
3.5. The Major Differences between the Techniques that Inform on Amyloid Structure
Recent progresses in instrumentation have improved quite significantly the ability to characterize the structure of complex biological molecules and macromolecular complexes. The knowledge of the three-dimensional structure of amyloid fibrils from different origins provides important insights into the mechanisms of amyloid formation, and thereby helps in the rational design of novel therapeutic agents. The combination of structural biology data from several approaches and experimental techniques provides new insights on how individual protein subunits form fibrillar structures, at an atomic level of resolution. In the past decades, many structural biology studies have been performed based on X-ray fiber diffraction [126], X-ray crystallography [23], solid state NMR (ssNMR) [127], and cryo-electron microscopy (cryo-EM) [128], to mention just a few. Table 3 reports the main differences between these powerful techniques. However, is important to mention that no method is completely effective by itself, since all of them offer unique advantages and limitations.
4. Toxic Species in Amyloid Diseases
The pathogenic or the most toxic species in amyloidosis can result from extracellular amyloid fibrillar deposits affecting organ integrity [134,135,136,137], as in the case of cardiac amyloid in transthyretin amyloidosis, and/or soluble protein oligomers that can either populate during the process of amyloid fibril formation or be released by mature deposits causing direct cellular dysfunction [137], as in the case of several neurodegenerative amyloid diseases.
Although the β-sheet structure present in amyloid oligomers is a favorable structural component, it is not an important prerequisite for toxicity [138]. The structural determinants by which these misfolded oligomers cause cellular damage may be related with the exposure of hydrophobic groups on the oligomer surface [139,140] and with the small size of the oligomers with high diffusion coefficient [140,141]. Thus, the toxicity of amyloidogenic oligomers is likely to be a result of their intrinsic misfolded nature and aggregation propensity [137,142]. Such structural properties will cause them to engage in a multitude of abnormal interactions with a range of cellular components, such as phospholipid bilayers, soluble peptides and proteins, protein receptors, RNAs, and cellular metabolites [139,143,144,145,146,147] where some or all of them have the potential to cause cell damage and ultimately cell death. In fact, it is known that several amyloid neurodegenerative diseases have an important inflammatory component which may be the result of the natural response against the “unknown” molecular species formed during the amyloidogenic cascade or against by-products of their action.
5. Kinetics and Thermodynamics of Amyloid Fibril Formation
The aggregation mechanisms in amyloid systems are prone to multiple pathways, depending on the ensemble of co-existing amyloidogenic conformations and environmental factors. Thus, several aggregation mechanisms and multiple pathways have been described depending on protein sequence, conformational states adopted by the amyloidogenic monomer and experimental conditions (for instance, temperature, pH, protein concentration, and solvent effects). The aggregation processes take place over a wide time range, spanning several orders of magnitude, with conformational changes occurring in the milliseconds and formation of particles observable with the naked eye in days, weeks or months. Elucidation of the mechanisms of amyloid formation and characterization of the most relevant molecular species involved are crucial to devise new rational therapeutic strategies against amyloid diseases. A brief overview of the most common amyloid formation mechanisms is presented in Figure 4, and a summary of the events and conditions that may trigger protein aggregation is shown in Table 4.
The aggregation kinetics of amyloidogenic proteins is highly dependent on protein concentration, and may reflect a nucleation-dependent or a nucleation-independent process (Figure 4) [149,150,151,152].
5.1. Aggregation Via a Nucleation-Dependent Mechanism
The nucleation-dependent mechanism of amyloid formation (Figure 4), also known as nucleation–elongation polymerization, displays a typical sigmoidal shape curve as a function of time and consists of three consecutive steps: (1) initial lag or nucleation phase; (2) elongation, growth, polymerization, or fibrillation phase; (3) equilibrium, stationary, or saturation phase [211,212].
The nucleation phase corresponds to the assembly of transient, critical nuclei that next will act as seeding intermediates where additional monomeric subunits can latch on, driving the assembly of oligomers with cross-β structure. At this stage, the rate constants for monomer addition and dissociation are similar, making the global process of nucleation slow and the rate limiting step in fibril formation. The nucleation phase can be shortened or eliminated by the addition of pre-formed aggregates or fibrillar species, a process known as seeding [213,214,215]. In the elongation phase, monomers, nucleus and oligomers continue to interact, assembling into prefibrillar structures that rapidly grow to form ordered fibrillar structures known as protofibrils. Because this phase gives rise to more stable protofibrils, this is a faster and thermodynamically favorable process. Lastly, the saturation phase, where monomer concentration is low and approximately constant, involves the assembly of protofibrils into mature amyloid fibrils with different morphological structures and different levels of polymorphism.
The Finke–Watzky aggregation model (Scheme 1) is one of the numerous models proposed for nucleation–elongation polymerization and has been applied to more than 40 different aggregating proteins [216,217]. As shown in Scheme 1, the Finke–Watzky model consists of two simple steps: (1) nucleation and (2) growth. Due to its simplicity, this model does have some limitations, including: (1) a vast number of aggregation steps is condensed into two elementary steps; (2) the rate constants, knucleation and kgrowth, are average rate constants and independent of the size of aggregating species; (3) a higher kinetic order in [M] may be kinetically hidden in the nucleation step in particular; (4) all growing polydisperse aggregates are hidden behind the descriptor “An”; (5) the descriptor “An” can also hide processes such as fragmentation. However, the simplicity and quality of the fits obtained in many practical examples suggest that the Finke–Watzky two-step mechanism encompasses the main characteristics and it is a good general kinetic model for nucleation-growth aggregation.
5.1.1. Primary Nucleation Mechanisms
In the amyloidogenesis cascade, primary nucleation is a critical step in a variety of oligomerization mechanisms and includes the initial formation of amyloidogenic nuclei without contributions from pre-formed oligomers. Two types of primary nucleation mechanisms have been described: homogeneous and heterogeneous. Homogeneous primary nucleation comprises monomers aggregation in bulk solution, while heterogeneous primary nucleation involves association of monomeric subunits on the surface of a different object, such as the wall of a reaction container [191,192,193,194], other proteins [202], phospholipid bilayers [203,204,205,206,207,208,209,210], or the air–water interface [175,176,177,178,179,180].
From a structural point of view, the simplest manifestation of a primary nucleation mechanism is the nucleated polymerization (NP) mechanism. In this case, amyloidogenic monomers aggregate and originate the nucleus, which further grows into amyloid protofilaments and protofibrils through an elongation process involving mostly monomer addition [219,220,221]. This is the preferential mechanism at relatively low protein concentrations favoring the presence of monomeric species in solution.
However, in several instances, it has been observed the presence of multiple conformational heterogeneous oligomers and transient intermediate species during fibril formation which NP mechanisms cannot explain. In these cases, a nucleated conformational conversion (NCC) mechanism has been proposed. NCC comprises structurally organized oligomers as intermediates which are able to subsequently conformationally transition into cross-β dominated fibrillar species. The formation of these conformationally dynamic oligomers may be favored at higher protein concentrations and they undergo a rate-limiting conformational change to form protofibrils and then amyloid fibrils [222]. This type of nucleation was observed, in particular, in the yeast prion protein (Sup35) [223], among variants of the amyloid-β peptide [224,225,226], SH3 domain [227,228,229], Ure2p yeast prion [230], polyglutamine (polyGln) peptides [231], and lysozyme [232,233].
5.1.2. Secondary Nucleation Mechanisms
Although conceptually appealing and observed in several instances, a simple homogeneous primary nucleation mechanism is not always observed [149,223,234]. Several studies have pointed out that simple homogeneous nucleation could not fit certain experimental aggregation kinetics data [235,236]. Simple homogeneous primary nucleation does not take into account other nucleation mechanisms and events, such as fibril-catalyzed secondary nucleation (a monomer-dependent process) and fibril fragmentation (a monomer-independent process) (Figure 4), both contributing to the formation of new aggregation nuclei [237,238,239,240,241]. In fibril-catalyzed secondary nucleation nucleus formation occurs on the surface of an already existing oligomer (Figure 4). No foreign surface is involved in this type of nucleation as in the case of heterogeneous primary nucleation. This nucleation mechanism appears to be highly dependent on the structural compatibility of the amyloid precursor protein [241].
Secondary nucleation has been inferred for several proteins, including amyloid-β peptides [235,242], tau protein [243], α-synuclein [244,245], islet amyloid polypeptide (IAPP) [246], insulin [247], and bovine carbonic anhydrase [248].
Amyloid fibril formation may also be seeded by the presence of pre-formed aggregates. In this case, the primary nucleation event is negligible, leading directly to the growth phase, the absence of secondary mechanisms, and the polymerization process is expected to follow a single exponential function [249]. This is a consequence of the slower rate of primary nucleation when compared with the rate of addition of monomers onto an existing fibril (growth). This seeding process has been proposed to be an important factor in the propagation of the pathogenesis in most, if not all, amyloidoses [250,251,252,253,254,255,256].
5.2. Aggregation Via a Nucleation-Independent Mechanism
The nucleation-independent mechanism of protein aggregation (Figure 4 and Scheme 2) is an isodesmic or linear polymerization mechanism and may be exemplified by the simplest possible model for the formation of spherical oligomers or linear multimers [152]. This model is characterized by an infinite number of steps with identical rate constants (k) independent of the size of the aggregate (Scheme 2), resulting in an exponential polymerization curve with the absence of a lag phase. Once aggregation starts, the process undergoes downhill-polymerization. In this case, aggregation proceeds through a sequence of multiple energetically favorable steps, where the successive addition of amyloidogenic monomers to the growing aggregate is energetically favorable without the need of a multimeric nucleus.
Generally, seeding does not increase the aggregation rate in a downhill-polymerization process. However, this model disregards other aggregation processes that can change the number, size and shape of oligomeric species. For these reasons, this model sometimes predicts incorrect length distributions of amyloid fibrils at equilibrium. Nevertheless, this kinetic model has been used to investigate the effect of mutations on the rate of amyloid fibril formation [259,260,261].
This aggregation mechanism was observed in transthyretin [262,263], among variants of the amyloid-β peptide [219,264,265], and also in the four-repeat domain of tau (Tau4RD) [266], β2-microglobulin [152], human and bovine serum albumins [267,268], HypF-N [269], FF domain [270], human muscle acylphosphatase [271], apolipoprotein C-II [272,273], and several SH3 domains [228,259,274,275,276].
5.3. The Energy Landscape View of Protein Aggregation
The protein folding energy landscape available to each polypeptide chain includes a wide range of different conformational states and a multitude of pathways en route to the folded state. The energy landscape in the case of short polypeptide sequences tends to be a smooth funnel-shaped surface where the polypeptide chain folds quickly towards a single folded state [277]. On the other hand, larger proteins have rougher energy landscapes, with local minima and a population of intermediate states that eventually interconvert to the low energy folded state (Figure 5) [278,279].
At the top of the funnel, as depicted in Figure 5, the unfolded state of the polypeptide chain has high Gibbs free energy and high conformational entropy. Upon polypeptide chain folding, the number of conformational states and thus the conformational entropy decreases. Concurrently, the hydrophobic collapse and the increase in the number of intramolecular contacts leads to a decrease in free energy toward the native state occupying the global free energy minimum, yielding the necessary conformational stability of the folded state. However, changes in amino acid sequence, and/or chemical or biological environment, including changes in pH, temperature, ionic strength, pressure, agitation, shear forces, interaction with surfaces and many other factors, may tip the energetic balance towards a different free energy minimum. This is highlighted in Figure 5, where in parallel with the common folding funnel for a protein it is also depicted an aggregation funnel [280,281,282]. Regarding the protein aggregation process, the funnel-shaped free energy surface is potentially rougher and more complex, since the energy landscape encodes not only the relative stability of unfolded states, partially unfolded states, and folded states, but also the relative stability of amorphous aggregates, β-sheet-rich amyloid fibrils, and native-like aggregates (Figure 1 and Figure 5).
Protein aggregation involves several processes that are interconnected, such as folding, unfolding and partial folding/unfolding, conformational changes, formation of intermolecular interactions, and fibril nucleation, elongation and stationary phases (Figure 1 and Figure 4) [237,238,283,284,285]. There is well documented evidence that protein aggregation states may be formed not only by amyloidogenic intermediates but also by denatured and native states, with polypeptide chains establishing critical contacts with neighboring molecules through intermolecular interactions. Aggregated states are in general thermodynamically and kinetically favorable and it is a fine balance of forces that tip the processes towards a native soluble state or any type of aggregated state. It seems that most polypeptide chains under the right “stress” conditions tend to form extended β-sheet structures and thus amyloid aggregates [14,284,286,287]. The energy landscape of protein systems forming large aggregates is described by numerous peaks corresponding to different conformational states, which is the case of amyloids due to the heterogeneity of fibrillar morphology. Even under the same experimental conditions, a large number of polymorphic fibrils with distinct morphologies might be formed at the same time, emphasizing the complexity and multiplicity of the aggregation pathways [67,288]. The energy minimum of mature fibrils is deeper and sharper than the native state of a given protein (Figure 5), as suggested by the high stability of the fibrillar state [286,289,290].
Under specified conditions, a given polypeptide chain has its own folding and aggregation surface funnel. Each point on this surface expresses a specific and unique conformation of the protein. The profile of the energy landscape is affected by enthalpic contributions due to interactions between amino acids residues, and enthalpic and entropic contributions due to the interaction with the aqueous environment, as well as entropic contributions due to changes in conformational freedom of the polypeptide chain. The driving forces towards the low free energy state for both protein folding and aggregation are mainly hydrophobic in nature, with additional contributions from electrostatic and polar interactions, as well as hydrogen bonds [14,280,281,287]. In the case of amyloid fibrils, the cross-β motif conformation is stabilized essentially by polar interactions due to intermolecular hydrogen bonds, and intermediate aggregated species are formed by intermolecular hydrophobic and electrostatic interactions [284,291].
In the late 1990s [292] amyloid-like aggregates or fibrils were found to be formed in vitro under specific experimental conditions by proteins entirely unrelated to well-established amyloid diseases [293]. As we previously mentioned, under the right conditions, many if not most polypeptide chains may form amyloid. The term “amylome” was thus coined to describe all the proteins that can form amyloid-like fibrils [294]. There are authors that consider that the fibrillar amyloid state represents a standard state that every polypeptide chain can adopt under appropriate conditions, and that this state is the thermodynamic ground state. In this sense, the amyloid fibrillar conformation would be the universal global free-energy minimum of any polypeptide chain [295].
6. Conclusions
With the progresses being made in medical research and drug discovery, there is an optimistic view that protein misfolding diseases will become successfully diagnosed, prevented and treated. At the moment, the research into the “amyloid problem” is reaching a turning point, since the structures and properties of different amyloidogenic species involved in the amyloid cascade are finally being identified and, in some cases, structurally characterized. The improvement in resolution of molecular structures, with the contribution from X-ray, ssNMR and cryo-electron microscopy, has allowed a better understanding of how amyloid polymorphism may relate with different manifestations of amyloid diseases, and how different amyloid structures influence cellular function and may be influenced by tissue environment. This seems to be the perfect time to finally correlate protein aggregation mechanisms, structure of amyloid species, and how they impact the onset, severity and progression of different misfolding diseases. In turn, this is also the opportunity to set up therapeutic solutions based on rational approaches, and thus provide new hope to those many affected by amyloid diseases.
Author Contributions
Writing—original draft preparation, Z.L.A.; writing—review and editing, Z.L.A. and R.M.M.B.; supervision, R.M.M.B. All authors have read and agreed to the published version of the manuscript.
Funding
The authors acknowledge funding from the Fundação para a Ciência e a Tecnologia (FCT), Portuguese Agency for Scientific Research, through project UID/QUI/00313/2019 (to Coimbra Chemistry Centre, University of Coimbra) and programs COMPETE and CENTRO-2020. Z.L.A. also thanks FCT for doctoral fellowship SFRH/BD/137991/2018.
Acknowledgments
The authors are grateful to Daniela C. Vaz for her careful and meticulous reading of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest. The funding agencies had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Chiti, F.; Dobson, C.M. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef]
- Iadanza, M.G.; Jackson, M.P.; Hewitt, E.W.; Ranson, N.A.; Radford, S.E. A new era for understanding amyloid structures and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 755–773. [Google Scholar] [CrossRef] [PubMed]
- Patterson, C. World Alzheimer Report 2018. The State of the Art of Dementia Research: New Frontiers; Alzheimer’s Disease International (ADI): London, UK, 2018; pp. 6–7. [Google Scholar]
- Westermark, P.; Sletten, K.; Johansson, B.; Cornwell, G.G. Fibril in senile systemic amyloidosis is derived from normal transthyretin. Proc. Natl. Acad. Sci. USA 1990, 87, 2843–2845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banypersad, S.M.; Moon, J.C.; Whelan, C.; Hawkins, P.N.; Wechalekar, A.D. Updates in cardiac amyloidosis: A review. J. Am. Heart Assoc. 2012, 1, e000364. [Google Scholar] [CrossRef] [Green Version]
- Sipe, J.D. Amyloidosis. Annu. Rev. Biochem. 1992, 61, 947–975. [Google Scholar] [CrossRef] [PubMed]
- Blancas-Mejía, L.M.; Ramirez-Alvarado, M. Systemic Amyloidoses. Annu. Rev. Biochem. 2013, 82, 745–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintas, A.; Saraiva, M.J.M.; Brito, R.M.M. The amyloidogenic potential of transthyretin variants correlates with their tendency to aggregate in solution. Febs Lett. 1997, 418, 297–300. [Google Scholar] [CrossRef] [Green Version]
- Jesus, C.S.H.; Almeida, Z.L.; Vaz, D.C.; Faria, T.Q.; Brito, R.M.M. A New Folding Kinetic Mechanism for Human Transthyretin and the Influence of the Amyloidogenic V30M Mutation. Int. J. Mol. Sci. 2016, 17, 1428. [Google Scholar] [CrossRef] [Green Version]
- Takano, K.; Funahashi, J.; Yutani, K. The stability and folding process of amyloidogenic mutant human lysozymes. Eur. J. Biochem. 2001, 268, 155–159. [Google Scholar] [CrossRef]
- Isaacson, R.L.; Weeds, A.G.; Fersht, A.R. Equilibria and kinetics of folding of gelsolin domain 2 and mutants involved in familial amyloidosis-Finnish type. Proc. Natl. Acad. Sci. USA 1999, 96, 11247–11252. [Google Scholar] [CrossRef] [Green Version]
- Grant, M.A.; Lazo, N.D.; Lomakin, A.; Condron, M.M.; Arai, H.; Yamin, G.; Rigby, A.C.; Teplow, D.B. Familial Alzheimer’s disease mutations alter the stability of the amyloid beta-protein monomer folding nucleus. Proc. Natl. Acad. Sci. USA 2007, 104, 16522–16527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobson, C.M. Protein misfolding, evolution and disease. Trends Biochem. Sci. 1999, 24, 329–332. [Google Scholar] [CrossRef]
- Dobson, C.M. Protein folding and misfolding. Nature 2003, 426, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Functional Amyloid, and Human Disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fowler, D.M.; Koulov, A.V.; Balch, W.E.; Kelly, J.W. Functional amyloid—From bacteria to humans. Trends Biochem. Sci. 2007, 32, 217–224. [Google Scholar] [CrossRef]
- Pham, C.L.L.; Kwan, A.H.; Sunde, M. Functional amyloid: Widespread in Nature, diverse in purpose. Essays Biochem. 2014, 56, 207–219. [Google Scholar]
- Otzen, D.; Riek, R. Functional Amyloids. Cold Spring Harb. Perspect. Biol. 2019, a033860. [Google Scholar] [CrossRef]
- Avni, A.; Swasthi, H.M.; Majumdar, A.; Mukhopadhyay, S. Intrinsically disordered proteins in the formation of functional amyloids from bacteria to humans. In Progress in Molecular Biology and Translational Science; Academic Press, Elsevier: Cambridge, MA, USA, 2019; Volume 166, pp. 109–143. [Google Scholar]
- Sipe, J.D.; Cohen, A.S. Review: History of the Amyloid Fibril. J. Struct. Biol. 2000, 130, 88–98. [Google Scholar] [CrossRef] [Green Version]
- Makin, O.S.; Serpell, L.C. Structures for amyloid fibrils. FEBS J. 2005, 272, 5950–5961. [Google Scholar] [CrossRef]
- Close, W.; Neumann, M.; Schmidt, A.; Hora, M.; Annamalai, K.; Schmidt, M.; Reif, B.; Schmidt, V.; Grigorieff, N.; Fändrich, M. Physical basis of amyloid fibril polymorphism. Nat. Commun. 2018, 9, 699. [Google Scholar] [CrossRef]
- Nelson, R.; Sawaya, M.R.; Balbirnie, M.; Madsen, A.Ø.; Riekel, C.; Grothe, R.; Eisenberg, D. Structure of the cross-β spine of amyloid-like fibrils. Nature 2005, 435, 773–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jalandoni-Buan, A.C.; Decena-Soliven, A.L.A.; Cao, E.P.; Barraquio, V.L.; Barraquio, W.L. Characterization and Identification of Congo Red Decolorizing Bacteria from Monocultures and Consortia. Philipp. J. Sci. 2010, 139, 71–78. [Google Scholar]
- Nilsson, M.R. Techniques to study amyloid fibril formation in vitro. Methods 2004, 34, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Klunk, W.E.; Jacob, R.F.; Mason, R.P. Quantifying Amyloid β-Peptide (Aβ) Aggregation Using the Congo Red-Aβ (CR–Aβ) Spectrophotometric Assay. Anal. Biochem. 1999, 266, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Eisert, R.; Felau, L.; Brown, L.R. Methods for enhancing the accuracy and reproducibility of Congo red and thioflavin T assays. Anal. Biochem. 2006, 353, 144–146. [Google Scholar] [CrossRef] [PubMed]
- Howie, A.J.; Brewer, D.B. Optical properties of amyloid stained by Congo red: History and mechanisms. Micron 2009, 40, 285–301. [Google Scholar] [CrossRef]
- Sen, S.; Basdemir, G. Diagnosis of renal amyloidosis using Congo red fluorescence. Pathol. Int. 2003, 53, 534–538. [Google Scholar] [CrossRef]
- Giorgadze, T.A.; Shiina, N.; Baloch, Z.W.; Tomaszewski, J.E.; Gupta, P.K. Improved detection of amyloid in fat pad aspiration: An evaluation of Congo red stain by fluorescent microscopy. Diagn. Cytopathol. 2004, 31, 300–306. [Google Scholar] [CrossRef]
- Levine, H. Thioflavine T interaction with synthetic Alzheimer’s disease beta-amyloid peptides: Detection of amyloid aggregation in solution. Protein Sci. 1993, 2, 404–410. [Google Scholar] [CrossRef]
- Porat, Y.; Abramowitz, A.; Gazit, E. Inhibition of Amyloid Fibril Formation by Polyphenols: Structural Similarity and Aromatic Interactions as a Common Inhibition Mechanism. Chem. Biol. Drug Des. 2006, 67, 27–37. [Google Scholar] [CrossRef]
- Turnell, W.G.; Finch, J.T. Binding of the dye congo red to the amyloid protein pig insulin reveals a novel homology amongst amyloid-forming peptide sequences. J. Mol. Biol. 1992, 227, 1205–1223. [Google Scholar] [CrossRef]
- Kim, Y.-S.; Randolph, T.W.; Manning, M.C.; Stevens, F.J.; Carpenter, J.F. Congo red populates partially unfolded states of an amyloidogenic protein to enhance aggregation and amyloid fibril formation. J. Biol. Chem. 2003, 278, 10842–10850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caughey, B.; Ernst, D.; Race, R.E. Congo red inhibition of scrapie agent replication. J. Virol. 1993, 67, 6270–6272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenzo, A.; Yankner, B.A. Beta-amyloid neurotoxicity requires fibril formation and is inhibited by congo red. Proc. Natl. Acad. Sci. USA 1994, 91, 12243–12247. [Google Scholar] [CrossRef] [Green Version]
- Chander, H.; Chauhan, A.; Chauhan, V. Binding of Proteases to Fibrillar Amyloid-β Protein and its Inhibition by Congo Red. J. Alzheimer’s Dis. 2007, 12, 261–269. [Google Scholar] [CrossRef]
- Vassar, P.S.; Culling, C.F. Fluorescent stains, with special reference to amyloid and connective tissues. Arch. Pathol. 1959, 68, 487–498. [Google Scholar]
- Kelényi, G. Thioflavin S fluorescent and Congo red anisotropic stainings in the histologic demonstration of amyloid. Acta Neuropathol. 1967, 7, 336–348. [Google Scholar] [CrossRef]
- Younan, N.D.; Viles, J.H. A Comparison of Three Fluorophores for the Detection of Amyloid Fibers and Prefibrillar Oligomeric Assemblies. ThT (Thioflavin T); ANS (1-Anilinonaphthalene-8-sulfonic Acid); and bisANS (4,4′-Dianilino-1,1′-binaphthyl-5,5′-disulfonic Acid). Biochemistry 2015, 54, 4297–4306. [Google Scholar] [CrossRef]
- Nagarajan, S.; Lapidus, L.J. Fluorescent Probe DCVJ Shows High Sensitivity for Characterization of Amyloid β-Peptide Early in the Lag Phase. ChemBioChem 2017, 18, 2205–2211. [Google Scholar] [CrossRef] [Green Version]
- Mishra, R.; Sjölander, D.; Hammarström, P. Spectroscopic characterization of diverse amyloid fibrils in vitro by the fluorescent dye Nile red. Mol. Biosyst. 2011, 7, 1232–1240. [Google Scholar] [CrossRef]
- Kovalska, V.; Chernii, S.; Losytskyy, M.; Tretyakova, I.; Dovbii, Y.; Gorski, A.; Chernii, V.; Czerwieniec, R.; Yarmoluk, S. Design of functionalized β-ketoenole derivatives as efficient fluorescent dyes for detection of amyloid fibrils. New J. Chem. 2018, 42, 13308–13318. [Google Scholar] [CrossRef] [Green Version]
- Fanni, A.M.; Monge, F.A.; Lin, C.-Y.; Thapa, A.; Bhaskar, K.; Whitten, D.G.; Chi, E.Y. High Selectivity and Sensitivity of Oligomeric p -Phenylene Ethynylenes for Detecting Fibrillar and Prefibrillar Amyloid Protein Aggregates. ACS Chem. Neurosci. 2019, 10, 1813–1825. [Google Scholar] [CrossRef] [PubMed]
- Abbasbeigi, S.; Adibi, H.; Moradi, S.; Ghadami, S.A.; Khodarahmi, R. Detection/quantification of amyloid aggregation in solution using the novel fluorescent benzofuranone-derivative compounds as amyloid fluorescent probes: Synthesis and in vitro characterization. J. Iran. Chem. Soc. 2019, 16, 1225–1237. [Google Scholar] [CrossRef]
- Crystal, A.S.; Giasson, B.I.; Crowe, A.; Kung, M.-P.; Zhuang, Z.-P.; Trojanowski, J.Q.; Lee, V.M.-Y. A comparison of amyloid fibrillogenesis using the novel fluorescent compound K114. J. Neurochem. 2003, 86, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.L.; Schuck, T.; Sheridan, S.; Kung, M.P.; Kung, H.; Zhuang, Z.P.; Bergeron, C.; Lamarche, J.S.; Skovronsky, D.; Giasson, B.I.; et al. The fluorescent Congo red derivative, (trans, trans)-1-bromo-2,5-bis-(3-hydroxycarbonyl-4-hydroxy)styrylbenzene (BSB), labels diverse beta-pleated sheet structures in postmortem human neurodegenerative disease brains. Am. J. Pathol. 2001, 159, 937–943. [Google Scholar] [CrossRef]
- Styren, S.D.; Hamilton, R.L.; Styren, G.C.; Klunk, W.E. X-34, A Fluorescent Derivative of Congo Red: A Novel Histochemical Stain for Alzheimer’s Disease Pathology. J. Histochem. Cytochem. 2000, 48, 1223–1232. [Google Scholar] [CrossRef] [Green Version]
- He, X.-P.; Deng, Q.; Cai, L.; Wang, C.-Z.; Zang, Y.; Li, J.; Chen, G.-R.; Tian, H. Fluorogenic Resveratrol-Confined Graphene Oxide for Economic and Rapid Detection of Alzheimer’s Disease. ACS Appl. Mater. Interfaces 2014, 6, 5379–5382. [Google Scholar] [CrossRef]
- Volkova, K.D.; Kovalska, V.B.; Balanda, A.O.; Losytskyy, M.Y.; Golub, A.G.; Vermeij, R.J.; Subramaniam, V.; Tolmachev, O.I.; Yarmoluk, S.M. Specific fluorescent detection of fibrillar α-synuclein using mono- and trimethine cyanine dyes. Bioorganic Med. Chem. 2008, 16, 1452–1459. [Google Scholar] [CrossRef]
- Volkova, K.D.; Kovalska, V.B.; Balanda, A.O.; Vermeij, R.J.; Subramaniam, V.; Slominskii, Y.L.; Yarmoluk, S.M. Cyanine dye–protein interactions: Looking for fluorescent probes for amyloid structures. J. Biochem. Biophys. Methods 2007, 70, 727–733. [Google Scholar] [CrossRef]
- Luna-Muñoz, J.; Peralta-Ramirez, J.; Chávez-Macías, L.; Harrington, C.R.; Wischik, C.M.; Mena, R. Thiazin red as a neuropathological tool for the rapid diagnosis of Alzheimer’s disease in tissue imprints. Acta Neuropathol. 2008, 116, 507–515. [Google Scholar] [CrossRef]
- Sulatskaya, A.I.; Sulatsky, M.I.; Antifeeva, I.A.; Kuznetsova, I.M.; Turoverov, K.K. Structural Analogue of Thioflavin T, DMASEBT, as a Tool for Amyloid Fibrils Study. Anal. Chem. 2019, 91, 3131–3140. [Google Scholar] [CrossRef] [PubMed]
- Biancalana, M.; Koide, S. Molecular mechanism of Thioflavin-T binding to amyloid fibrils. Biochim. Et Biophys. Acta (BBA)-Proteins Proteom. 2010, 1804, 1405–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yakupova, E.I.; Bobyleva, L.G.; Vikhlyantsev, I.M.; Bobylev, A.G. Congo Red and amyloids: History and relationship. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, M.D.; Buxbaum, J.N.; Eisenberg, D.S.; Merlini, G.; Saraiva, M.J.M.; Sekijima, Y.; Sipe, J.D.; Westermark, P. Amyloid nomenclature 2018: Recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid 2018, 25, 215–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geddes, A.J.; Parker, K.D.; Atkins, E.D.T.; Beighton, E. “Cross-β” conformation in proteins. J. Mol. Biol. 1968, 32, 343–358. [Google Scholar] [CrossRef]
- Sunde, M.; Blake, C. The Structure of Amyloid Fibrils by Electron Microscopy and X-Ray Diffraction. Adv. Protein Chem. 1997, 50, 123–159. [Google Scholar]
- Kodali, R.; Wetzel, R. Polymorphism in the intermediates and products of amyloid assembly. Curr. Opin. Struct. Biol. 2007, 17, 48–57. [Google Scholar] [CrossRef]
- Eanes, E.D.; Glennner, G.G. X-ray Diffraction Studies on Amyloid Filaments. J. Histochem. Cytochem. 1968, 16, 673–677. [Google Scholar] [CrossRef]
- Bonar, L.; Cohen, A.S.; Skinner, M.M. Characterization of the Amyloid Fibril as a Cross- Protein. Exp. Biol. Med. 1969, 131, 1373–1375. [Google Scholar] [CrossRef]
- Sunde, M.; Serpell, L.C.; Bartlam, M.; Fraser, P.E.; Pepys, M.B.; Blake, C.C. Common core structure of amyloid fibrils by synchrotron X-ray diffraction. J. Mol. Biol. 1997, 273, 729–739. [Google Scholar] [CrossRef] [Green Version]
- Nelson, R.; Eisenberg, D. Recent atomic models of amyloid fibril structure. Curr. Opin. Struct. Biol. 2006, 16, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, M.I.; Thompson, M.J.; Eisenberg, D. A systematic screen of β2-microglobulin and insulin for amyloid-like segments. Proc. Natl. Acad. Sci. USA 2006, 103, 4079–4082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenberg, D.S.; Sawaya, M.R. Structural Studies of Amyloid Proteins at the Molecular Level. Annu. Rev. Biochem. 2017, 86, 69–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sambashivan, S.; Liu, Y.; Sawaya, M.R.; Gingery, M.; Eisenberg, D. Amyloid-like fibrils of ribonuclease A with three-dimensional domain-swapped and native-like structure. Nature 2005, 437, 266–269. [Google Scholar] [CrossRef]
- Fändrich, M.; Meinhardt, J.; Grigorieff, N. Structural polymorphism of Alzheimer Aβ and other amyloid fibrils. Prion 2009, 3, 89–93. [Google Scholar] [CrossRef] [Green Version]
- Meinhardt, J.; Sachse, C.; Hortschansky, P.; Grigorieff, N.; Fändrich, M. Aβ(1-40) Fibril Polymorphism Implies Diverse Interaction Patterns in Amyloid Fibrils. J. Mol. Biol. 2009, 386, 869–877. [Google Scholar] [CrossRef]
- Zapadka, K.L.; Becher, F.J.; Gomes dos Santos, A.L.; Jackson, S.E. Factors affecting the physical stability (aggregation) of peptide therapeutics. Interface Focus 2017, 7, 20170030. [Google Scholar] [CrossRef] [Green Version]
- Conchillo-Solé, O.; de Groot, N.S.; Avilés, F.X.; Vendrell, J.; Daura, X.; Ventura, S. AGGRESCAN: A server for the prediction and evaluation of “hot spots” of aggregation in polypeptides. BMC Bioinform. 2007, 8, 65. [Google Scholar] [CrossRef] [Green Version]
- de Groot, N.S.; Castillo, V.; Graña-Montes, R.; Ventura, S. AGGRESCAN: Method, Application, and Perspectives for Drug Design. In Methods in Molecular Biology (Clifton, N.J.); Springer: New York, NY, USA, 2012; Volume 819, pp. 199–220. [Google Scholar]
- Zambrano, R.; Jamroz, M.; Szczasiuk, A.; Pujols, J.; Kmiecik, S.; Ventura, S. AGGRESCAN3D (A3D): Server for prediction of aggregation properties of protein structures. Nucleic Acids Res. 2015, 43, W306–W313. [Google Scholar] [CrossRef]
- Pujols, J.; Peña-Díaz, S.; Ventura, S. AGGRESCAN3D: Toward the Prediction of the Aggregation Propensities of Protein Structures. In Methods in Molecular Biology (Clifton, N.J.); Springer: New York, NY, USA, 2018; Volume 1762, pp. 427–443. [Google Scholar]
- Wozniak, P.P.; Kotulska, M. AmyLoad: Website dedicated to amyloidogenic protein fragments. Bioinformatics 2015, 31, 3395–3397. [Google Scholar] [CrossRef] [Green Version]
- Burdukiewicz, M.; Sobczyk, P.; Rödiger, S.; Duda-Madej, A.; Mackiewicz, P.; Kotulska, M. Amyloidogenic motifs revealed by n-gram analysis. Sci. Rep. 2017, 7, 12961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donnell, C.W.; Waldispühl, J.; Lis, M.; Halfmann, R.; Devadas, S.; Lindquist, S.; Berger, B. A method for probing the mutational landscape of amyloid structure. Bioinformatics 2011, 27, i34–i42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frousios, K.K.; Iconomidou, V.A.; Karletidi, C.-M.; Hamodrakas, S.J. Amyloidogenic determinants are usually not buried. BMC Struct. Biol. 2009, 9, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsolis, A.C.; Papandreou, N.C.; Iconomidou, V.A.; Hamodrakas, S.J. A Consensus Method for the Prediction of ‘Aggregation-Prone’ Peptides in Globular Proteins. PLoS ONE 2013, 8, e54175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Família, C.; Dennison, S.R.; Quintas, A.; Phoenix, D.A. Prediction of Peptide and Protein Propensity for Amyloid Formation. PLoS ONE 2015, 10, e0134679. [Google Scholar] [CrossRef] [Green Version]
- Bryan, A.W.; Menke, M.; Cowen, L.J.; Lindquist, S.L.; Berger, B. BETASCAN: Probable β-amyloids Identified by Pairwise Probabilistic Analysis. PLoS Comput. Biol. 2009, 5, e1000333. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; et al. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016, 44, D862–D868. [Google Scholar] [CrossRef] [Green Version]
- Tabatabaei Ghomi, H.; Topp, E.M.; Lill, M.A. Fibpredictor: A computational method for rapid prediction of amyloid fibril structures. J. Mol. Modeling 2016, 22, 206. [Google Scholar] [CrossRef]
- Gasior, P.; Kotulska, M. FISH Amyloid—A new method for finding amyloidogenic segments in proteins based on site specific co-occurence of aminoacids. BMC Bioinform. 2014, 15, 54. [Google Scholar] [CrossRef] [Green Version]
- Garbuzynskiy, S.O.; Lobanov, M.Y.; Galzitskaya, O.V. FoldAmyloid: A method of prediction of amyloidogenic regions from protein sequence. Bioinformatics 2010, 26, 326–332. [Google Scholar] [CrossRef]
- Thangakani, A.M.; Kumar, S.; Nagarajan, R.; Velmurugan, D.; Gromiha, M.M. GAP: Towards almost 100 percent prediction for β-strand-mediated aggregating peptides with distinct morphologies. Bioinformatics 2014, 30, 1983–1990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, T.Z.; Jernigan, R.L.; Garnier, J.; Kloczkowski, A. GOR V server for protein secondary structure prediction. Bioinformatics 2005, 21, 2787–2788. [Google Scholar] [CrossRef] [PubMed]
- Kouza, M.; Faraggi, E.; Kolinski, A.; Kloczkowski, A. The GOR Method of Protein Secondary Structure Prediction and Its Application as a Protein Aggregation Prediction Tool. In Methods in Molecular Biology (Clifton, N.J.); Humana Press: New York, NY, USA, 2017; Volume 1484, pp. 7–24. [Google Scholar]
- Emily, M.; Talvas, A.; Delamarche, C. MetAmyl: A METa-Predictor for AMYLoid Proteins. PLoS ONE 2013, 8, e79722. [Google Scholar] [CrossRef] [PubMed]
- Munir, F.; Gull, S.; Asif, A.; Minhas, F.u.A.A. MILAMP: Multiple Instance Prediction of Amyloid Proteins. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.; Choi, J.; Lee, S.J.; Welsh, W.J.; Yoon, S. NetCSSP: Web application for predicting chameleon sequences and amyloid fibril formation. Nucleic Acids Res. 2009, 37, W469–W473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trovato, A.; Chiti, F.; Maritan, A.; Seno, F. Insight into the Structure of Amyloid Fibrils from the Analysis of Globular Proteins. PLoS Comput. Biol. 2006, 2, e170. [Google Scholar] [CrossRef] [PubMed]
- Trovato, A.; Seno, F.; Tosatto, S.C.E. The PASTA server for protein aggregation prediction. Protein Eng. Des. Sel. 2007, 20, 521–523. [Google Scholar] [CrossRef] [Green Version]
- Walsh, I.; Seno, F.; Tosatto, S.C.E.; Trovato, A. PASTA 2.0: An improved server for protein aggregation prediction. Nucleic Acids Res. 2014, 42, W301–W307. [Google Scholar] [CrossRef]
- Niu, M.; Li, Y.; Wang, C.; Han, K. RFAmyloid: A Web Server for Predicting Amyloid Proteins. Int. J. Mol. Sci. 2018, 19, 2071. [Google Scholar] [CrossRef] [Green Version]
- De Baets, G.; Van Durme, J.; Reumers, J.; Maurer-Stroh, S.; Vanhee, P.; Dopazo, J.; Schymkowitz, J.; Rousseau, F. SNPeffect 4.0: On-line prediction of molecular and structural effects of protein-coding variants. Nucleic Acids Res. 2012, 40, D935–D939. [Google Scholar] [CrossRef]
- Bryan, A.W.; O’Donnell, C.W.; Menke, M.; Cowen, L.J.; Lindquist, S.; Berger, B. STITCHER: Dynamic assembly of likely amyloid and prion β-structures from secondary structure predictions. Proteins Struct. Funct. Bioinform. 2012, 80, 410–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Escamilla, A.-M.; Rousseau, F.; Schymkowitz, J.; Serrano, L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat. Biotechnol. 2004, 22, 1302–1306. [Google Scholar] [CrossRef] [PubMed]
- Maurer-Stroh, S.; Debulpaep, M.; Kuemmerer, N.; de la Paz, M.L.; Martins, I.C.; Reumers, J.; Morris, K.L.; Copland, A.; Serpell, L.; Serrano, L.; et al. Exploring the sequence determinants of amyloid structure using position-specific scoring matrices. Nat. Methods 2010, 7, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Beerten, J.; Van Durme, J.; Gallardo, R.; Capriotti, E.; Serpell, L.; Rousseau, F.; Schymkowitz, J. WALTZ-DB: A benchmark database of amyloidogenic hexapeptides. Bioinformatics 2015, 31, 1698–1700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louros, N.; Konstantoulea, K.; De Vleeschouwer, M.; Ramakers, M.; Schymkowitz, J.; Rousseau, F. WALTZ-DB 2.0: An updated database containing structural information of experimentally determined amyloid-forming peptides. Nucleic Acids Res. 2019, 1. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.J.; Sievers, S.A.; Karanicolas, J.; Ivanova, M.I.; Baker, D.; Eisenberg, D. The 3D profile method for identifying fibril-forming segments of proteins. Proc. Natl. Acad. Sci. USA 2006, 103, 4074–4078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tartaglia, G.G.; Vendruscolo, M. The Zyggregator method for predicting protein aggregation propensities. Chem. Soc. Rev. 2008, 37, 1395–1401. [Google Scholar] [CrossRef]
- GroB, M. Proteins that Convert from a Helix to b Sheet Implications for Folding and Disease. Curr. Protein Pept. Sci. 2000, 1, 339–347. [Google Scholar] [CrossRef]
- Hauser, C.A.E.; Deng, R.; Mishra, A.; Loo, Y.; Khoe, U.; Zhuang, F.; Cheong, D.W.; Accardo, A.; Sullivan, M.B.; Riekel, C.; et al. Natural tri- to hexapeptides self-assemble in water to amyloid beta-type fiber aggregates by unexpected alpha-helical intermediate structures. Proc. Natl. Acad. Sci. USA 2011, 108, 1361–1366. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, D.; Singh, P.K.; Sahay, S.; Jha, N.N.; Jacob, R.S.; Sen, S.; Kumar, A.; Riek, R.; Maji, S.K. Structure based aggregation studies reveal the presence of helix-rich intermediate during α-Synuclein aggregation. Sci. Rep. 2015, 5, 9228. [Google Scholar] [CrossRef] [Green Version]
- Cieślik-Boczula, K. Alpha-helix to beta-sheet transition in long-chain poly-l-lysine: Formation of alpha-helical fibrils by poly-l-lysine. Biochimie 2017, 137, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Ni, M.; Zhuo, S.; Iliescu, C.; So, P.T.C.; Mehta, J.S.; Yu, H.; Hauser, C.A.E. Self-assembling amyloid-like peptides as exogenous second harmonic probes for bioimaging applications. J. Biophotonics 2019, 12, e201900065. [Google Scholar] [CrossRef] [PubMed]
- Tayeb-Fligelman, E.; Tabachnikov, O.; Moshe, A.; Goldshmidt-Tran, O.; Sawaya, M.R.; Coquelle, N.; Colletier, J.-P.; Landau, M. The cytotoxic Staphylococcus aureus PSMα3 reveals a cross-α amyloid-like fibril. Science 2017, 355, 831–833. [Google Scholar] [CrossRef] [PubMed]
- Bousset, L.; Thomson, N.H.; Radford, S.E.; Melki, R. The yeast prion Ure2p retains its native alpha-helical conformation upon assembly into protein fibrils in vitro. EMBO J. 2002, 21, 2903–2911. [Google Scholar] [CrossRef] [Green Version]
- Taylor, K.S.; Lou, M.Z.; Chin, T.M.; Yang, N.C.; Garavito, R.M. A novel, multilayer structure of a helical peptide. Protein Sci. A Publ. Protein Soc. 1996, 5, 414–421. [Google Scholar] [CrossRef] [Green Version]
- Privé, G.G.; Anderson, D.H.; Wesson, L.; Cascio, D.; Eisenberg, D. Packed protein bilayers in the 0.90 å resolution structure of a designed alpha helical bundle. Protein Sci. 1999, 8, 1400–1409. [Google Scholar] [CrossRef] [Green Version]
- Mondal, S.; Adler-Abramovich, L.; Lampel, A.; Bram, Y.; Lipstman, S.; Gazit, E. Formation of functional super-helical assemblies by constrained single heptad repeat. Nat. Commun. 2015, 6, 1–8. [Google Scholar] [CrossRef]
- Brunette, T.; Parmeggiani, F.; Huang, P.-S.; Bhabha, G.; Ekiert, D.C.; Tsutakawa, S.E.; Hura, G.L.; Tainer, J.A.; Baker, D. Exploring the repeat protein universe through computational protein design. Nature 2015, 528, 580–584. [Google Scholar] [CrossRef] [Green Version]
- Pham, C.L.; Shanmugam, N.; Strange, M.; O’Carroll, A.; Brown, J.W.; Sierecki, E.; Gambin, Y.; Steain, M.; Sunde, M. Viral M45 and necroptosis-associated proteins form heteromeric amyloid assemblies. EMBO Rep. 2019, 20. [Google Scholar] [CrossRef]
- Mompeán, M.; Li, W.; Li, J.; Laage, S.; Siemer, A.B.; Bozkurt, G.; Wu, H.; McDermott, A.E. The Structure of the Necrosome RIPK1-RIPK3 Core, a Human Hetero-Amyloid Signaling Complex. Cell 2018, 173, 1244–1253.e10. [Google Scholar] [CrossRef] [Green Version]
- O’Nuallain, B.; Williams, A.D.; Westermark, P.; Wetzel, R. Seeding specificity in amyloid growth induced by heterologous fibrils. J. Biol. Chem. 2004, 279, 17490–17499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oskarsson, M.E.; Paulsson, J.F.; Schultz, S.W.; Ingelsson, M.; Westermark, P.; Westermark, G.T. In Vivo Seeding and Cross-Seeding of Localized Amyloidosis: A Molecular Link between Type 2 Diabetes and Alzheimer Disease. Am. J. Pathol. 2015, 185, 834–846. [Google Scholar] [CrossRef] [PubMed]
- Morales, R.; Estrada, L.D.; Diaz-Espinoza, R.; Morales-Scheihing, D.; Jara, M.C.; Castilla, J.; Soto, C. Molecular cross talk between misfolded proteins in animal models of Alzheimer’s and prion diseases. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 4528–4535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köppen, J.; Schulze, A.; Machner, L.; Wermann, M.; Eichentopf, R.; Guthardt, M.; Hähnel, A.; Klehm, J.; Kriegeskorte, M.-C.; Hartlage-Rübsamen, M.; et al. Amyloid-Beta Peptides Trigger Aggregation of Alpha-Synuclein In Vitro. Molecules 2020, 25, 580. [Google Scholar] [CrossRef] [Green Version]
- Gotz, J.; Chen, F.; van Dorpe, J.; Nitsch, R.M. Formation of Neurofibrillary Tangles in P301L Tau Transgenic Mice Induced by Abeta 42 Fibrils. Science 2001, 293, 1491–1495. [Google Scholar] [CrossRef]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Guerrero-Muñoz, M.J.; Jackson, G.R.; Kayed, R. Preparation and Characterization of Neurotoxic Tau Oligomers. Biochemistry 2010, 49, 10039–10041. [Google Scholar] [CrossRef]
- Bhasne, K.; Sebastian, S.; Jain, N.; Mukhopadhyay, S. Synergistic Amyloid Switch Triggered by Early Heterotypic Oligomerization of Intrinsically Disordered α-Synuclein and Tau. J. Mol. Biol. 2018, 430, 2508–2520. [Google Scholar] [CrossRef]
- Lundmark, K.; Westermark, G.T.; Olsén, A.; Westermark, P. Protein fibrils in nature can enhance amyloid protein A amyloidosis in mice: Cross-seeding as a disease mechanism. Proc. Natl. Acad. Sci. USA 2005, 102, 6098–6102. [Google Scholar] [CrossRef] [Green Version]
- Derkatch, I.L.; Uptain, S.M.; Outeiro, T.F.; Krishnan, R.; Lindquist, S.L.; Liebman, S.W. Effects of Q/N-rich, polyQ, and non-polyQ amyloids on the de novo formation of the [PSI+] prion in yeast and aggregation of Sup35 in vitro. Proc. Natl. Acad. Sci. USA 2004, 101, 12934–12939. [Google Scholar] [CrossRef] [Green Version]
- Hammer, N.D.; Schmidt, J.C.; Chapman, M.R. The curli nucleator protein, CsgB, contains an amyloidogenic domain that directs CsgA polymerization. Proc. Natl. Acad. Sci. USA 2007, 104, 12494–12499. [Google Scholar] [CrossRef] [Green Version]
- Morris, K.L.; Serpell, L.C. X-Ray Fibre Diffraction Studies of Amyloid Fibrils. In Amyloid Proteins; Humana Press: Totowa, NJ, USA, 2012; pp. 121–135. [Google Scholar]
- Tycko, R. Solid-State NMR Studies of Amyloid Fibril Structure. Annu. Rev. Phys. Chem. 2011, 62, 279–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serpell, L.C.; Smith, J.M. Direct visualisation of the β-sheet structure of synthetic Alzheimer’s amyloid. J. Mol. Biol. 2000, 299, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.W.; Kelly, J.E.; Kelz, J.I. Advances in instrumentation and methodology for solid-state NMR of biological assemblies. J. Struct. Biol. 2019, 206, 73–89. [Google Scholar] [CrossRef] [PubMed]
- Meier, B.H.; Riek, R.; Böckmann, A. Emerging Structural Understanding of Amyloid Fibrils by Solid-State NMR. Trends Biochem. Sci. 2017, 42, 777–787. [Google Scholar] [CrossRef] [Green Version]
- Bai, X.; McMullan, G.; Scheres, S.H. How cryo-EM is revolutionizing structural biology. Trends Biochem. Sci. 2015, 40, 49–57. [Google Scholar] [CrossRef]
- COHEN, A.S.; CALKINS, E. Electron Microscopic Observations on a Fibrous Component in Amyloid of Diverse Origins. Nature 1959, 183, 1202–1203. [Google Scholar] [CrossRef]
- Kühlbrandt, W. The Resolution Revolution. Science 2014, 343, 1443–1444. [Google Scholar] [CrossRef]
- Milanesi, L.; Sheynis, T.; Xue, W.-F.; Orlova, E.V.; Hellewell, A.L.; Jelinek, R.; Hewitt, E.W.; Radford, S.E.; Saibil, H.R. Direct three-dimensional visualization of membrane disruption by amyloid fibrils. Proc. Natl. Acad. Sci. USA 2012, 109, 20455–20460. [Google Scholar] [CrossRef] [Green Version]
- Tipping, K.W.; van Oosten-Hawle, P.; Hewitt, E.W.; Radford, S.E. Amyloid fibres: Inert end-stage aggregates or key players in disease? Trends Biochem. Sci. 2015, 40, 719–727. [Google Scholar] [CrossRef]
- Verma, M.; Vats, A.; Taneja, V. Toxic species in amyloid disorders: Oligomers or mature fibrils. Ann. Indian Acad. Neurol. 2015, 18, 138–145. [Google Scholar]
- Evangelisti, E.; Cascella, R.; Becatti, M.; Marrazza, G.; Dobson, C.M.; Chiti, F.; Stefani, M.; Cecchi, C. Binding affinity of amyloid oligomers to cellular membranes is a generic indicator of cellular dysfunction in protein misfolding diseases. Sci. Rep. 2016, 6, 32721. [Google Scholar] [CrossRef] [PubMed]
- Vivoli Vega, M.; Cascella, R.; Chen, S.W.; Fusco, G.; De Simone, A.; Dobson, C.M.; Cecchi, C.; Chiti, F. The Toxicity of Misfolded Protein Oligomers Is Independent of Their Secondary Structure. ACS Chem. Biol. 2019, 14, 1593–1600. [Google Scholar] [CrossRef]
- Olzscha, H.; Schermann, S.M.; Woerner, A.C.; Pinkert, S.; Hecht, M.H.; Tartaglia, G.G.; Vendruscolo, M.; Hayer-Hartl, M.; Hartl, F.U.; Vabulas, R.M. Amyloid-like Aggregates Sequester Numerous Metastable Proteins with Essential Cellular Functions. Cell 2011, 144, 67–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannini, B.; Mulvihill, E.; Sgromo, C.; Cascella, R.; Khodarahmi, R.; Ramazzotti, M.; Dobson, C.M.; Cecchi, C.; Chiti, F. Toxicity of Protein Oligomers Is Rationalized by a Function Combining Size and Surface Hydrophobicity. ACS Chem. Biol. 2014, 9, 2309–2317. [Google Scholar] [CrossRef] [PubMed]
- Mannini, B.; Cascella, R.; Zampagni, M.; van Waarde-Verhagen, M.; Meehan, S.; Roodveldt, C.; Campioni, S.; Boninsegna, M.; Penco, A.; Relini, A.; et al. Molecular mechanisms used by chaperones to reduce the toxicity of aberrant protein oligomers. Proc. Natl. Acad. Sci. USA 2012, 109, 12479–12484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bucciantini, M.; Giannoni, E.; Chiti, F.; Baroni, F.; Formigli, L.; Zurdo, J.; Taddei, N.; Ramponi, G.; Dobson, C.M.; Stefani, M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 2002, 416, 507–511. [Google Scholar] [CrossRef]
- McLaurin, J.; Chakrabartty, A. Membrane Disruption by Alzheimer β-Amyloid Peptides Mediated through Specific Binding to Either Phospholipids or Gangliosides. J. Biol. Chem. 1996, 271, 26482–26489. [Google Scholar] [CrossRef] [Green Version]
- Benilova, I.; Karran, E.; De Strooper, B. The toxic Aβ oligomer and Alzheimer’s disease: An emperor in need of clothes. Nat. Neurosci. 2012, 15, 349–357. [Google Scholar] [CrossRef]
- Goodchild, S.C.; Sheynis, T.; Thompson, R.; Tipping, K.W.; Xue, W.-F.; Ranson, N.A.; Beales, P.A.; Hewitt, E.W.; Radford, S.E. β2-Microglobulin Amyloid Fibril-Induced Membrane Disruption Is Enhanced by Endosomal Lipids and Acidic pH. PLoS ONE 2014, 9, e104492. [Google Scholar] [CrossRef] [Green Version]
- Winklhofer, K.F.; Haass, C. Mitochondrial dysfunction in Parkinson’s disease. Biochim. Et Biophys. Acta (BBA)-Mol. Basis Dis. 2010, 1802, 29–44. [Google Scholar] [CrossRef]
- Roberts, H.; Brown, D. Seeking a Mechanism for the Toxicity of Oligomeric α-Synuclein. Biomolecules 2015, 5, 282–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaels, T.C.T.; Šarić, A.; Habchi, J.; Chia, S.; Meisl, G.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. Chemical Kinetics for Bridging Molecular Mechanisms and Macroscopic Measurements of Amyloid Fibril Formation. Ann. Rev. Phys. Chem. 2018, 69, 273–298. [Google Scholar] [CrossRef] [PubMed]
- Frieden, C. Protein aggregation processes: In search of the mechanism. Protein Sci. 2007, 16, 2334–2344. [Google Scholar] [CrossRef] [PubMed]
- Buell, A.K.; Dobson, C.M.; Knowles, T.P.J. The physical chemistry of the amyloid phenomenon: Thermodynamics and kinetics of filamentous protein aggregation. Essays Biochem. 2014, 56, 11–39. [Google Scholar]
- Jarrett, J.T.; Lansbury, P.T. Amyloid fibril formation requires a chemically discriminating nucleation event: Studies of an amyloidogenic sequence from the bacterial protein OsmB. Biochemistry 1992, 31, 12345–12352. [Google Scholar] [CrossRef]
- Gosal, W.S.; Morten, I.J.; Hewitt, E.W.; Smith, D.A.; Thomson, N.H.; Radford, S.E. Competing Pathways Determine Fibril Morphology in the Self-assembly of β2-Microglobulin into Amyloid. J. Mol. Biol. 2005, 351, 850–864. [Google Scholar] [CrossRef]
- Soldi, G.; Bemporad, F.; Torrassa, S.; Relini, A.; Ramazzotti, M.; Taddei, N.; Chiti, F. Amyloid Formation of a Protein in the Absence of Initial Unfolding and Destabilization of the Native State. Biophys. J. 2005, 89, 4234–4244. [Google Scholar] [CrossRef] [Green Version]
- Plakoutsi, G.; Bemporad, F.; Calamai, M.; Taddei, N.; Dobson, C.M.; Chiti, F. Evidence for a Mechanism of Amyloid Formation Involving Molecular Reorganisation within Native-like Precursor Aggregates. J. Mol. Biol. 2005, 351, 910–922. [Google Scholar] [CrossRef]
- Bemporad, F.; Vannocci, T.; Varela, L.; Azuaga, A.I.; Chiti, F. A model for the aggregation of the acylphosphatase from Sulfolobus solfataricus in its native-like state. Biochim. Et Biophys. Acta (BBA)-Proteins Proteom. 2008, 1784, 1986–1996. [Google Scholar] [CrossRef]
- Garcia-Pardo, J.; Graña-Montes, R.; Fernandez-Mendez, M.; Ruyra, A.; Roher, N.; Aviles, F.X.; Lorenzo, J.; Ventura, S. Amyloid formation by human carboxypeptidase D transthyretin-like domain under physiological conditions. J. Biol. Chem. 2014, 289, 33783–33796. [Google Scholar] [CrossRef] [Green Version]
- Raso, S.W.; Abel, J.; Barnes, J.M.; Maloney, K.M.; Pipes, G.; Treuheit, M.J.; King, J.; Brems, D.N. Aggregation of granulocyte-colony stimulating factor in vitro involves a conformationally altered monomeric state. Protein Sci. 2005, 14, 2246–2257. [Google Scholar] [CrossRef] [PubMed]
- Kendrick, B.S.; Carpenter, J.F.; Cleland, J.L.; Randolph, T.W. A transient expansion of the native state precedes aggregation of recombinant human interferon-gamma. Proc. Natl. Acad. Sci. USA 1998, 95, 14142–14146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintas, A.; Saraiva, M.J.M.; Brito, R.M.M. The Tetrameric Protein Transthyretin Dissociates to a Non-native Monomer in Solution a novel model for amyloidogenesis. J. Biol. Chem. 1999, 274, 32943–32949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintas, A.; Vaz, D.C.; Cardoso, I.; Saraiva, M.J.; Brito, R.M. Tetramer dissociation and monomer partial unfolding precedes protofibril formation in amyloidogenic transthyretin variants. J. Biol. Chem. 2001, 276, 27207–27213. [Google Scholar] [CrossRef] [Green Version]
- Booth, D.R.; Sunde, M.; Bellotti, V.; Robinson, C.V.; Hutchinson, W.L.; Fraser, P.E.; Hawkins, P.N.; Dobson, C.M.; Radford, S.E.; Blake, C.C.F.; et al. Instability, unfolding and aggregation of human lysozyme variants underlying amyloid fibrillogenesis. Nature 1997, 385, 787–793. [Google Scholar] [CrossRef]
- Guijarro, J.I.; Sunde, M.; Jones, J.A.; Campbell, I.D.; Dobson, C.M. Amyloid fibril formation by an SH3 domain. Proc. Natl. Acad. Sci. USA 1998, 95, 4224–4228. [Google Scholar] [CrossRef] [Green Version]
- Truscott, R.J.W.; Augusteyn, R.C. Oxidative changes in human lens proteins during senile nuclear cataract formation. Biochim. Et Biophys. Acta (BBA)-Protein Struct. 1977, 492, 43–52. [Google Scholar] [CrossRef]
- Guptasarma, P.; Balasubramanian, D.; Matsugo, S.; Saito, I. Hydroxyl radical mediated damage to proteins, with special reference to the crystallins. Biochemistry 1992, 31, 4296–4303. [Google Scholar] [CrossRef]
- Hermeling, S.; Schellekens, H.; Maas, C.; Gebbink, M.F.B.G.; Crommelin, D.J.A.; Jiskoot, W. Antibody Response to Aggregated Human Interferon Alpha2b in Wild-type and Transgenic Immune Tolerant Mice Depends on Type and Level of Aggregation. J. Pharm. Sci. 2006, 95, 1084–1096. [Google Scholar] [CrossRef]
- Mirzaei, H.; Regnier, F. Protein:protein aggregation induced by protein oxidation. J. Chromatogr. B 2008, 873, 8–14. [Google Scholar] [CrossRef]
- Landles, C.; Sathasivam, K.; Weiss, A.; Woodman, B.; Moffitt, H.; Finkbeiner, S.; Sun, B.; Gafni, J.; Ellerby, L.M.; Trottier, Y.; et al. Proteolysis of mutant huntingtin produces an exon 1 fragment that accumulates as an aggregated protein in neuronal nuclei in Huntington disease. J. Biol. Chem. 2010, 285, 8808–8823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, G.K.L.; Witkowski, A.; Gantz, D.L.; Zhang, T.O.; Zanni, M.T.; Jayaraman, S.; Cavigiolio, G. Myeloperoxidase-mediated Methionine Oxidation Promotes an Amyloidogenic Outcome for Apolipoprotein A-I. J. Biol. Chem. 2015, 290, 10958–10971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenfeld, M.A.; Leonova, V.B.; Konstantinova, M.L.; Razumovskii, S.D. Self-assembly of fibrin monomers and fibrinogen aggregation during ozone oxidation. Biochem. (Mosc.) 2009, 74, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Roostaee, A.; Côté, S.; Roucou, X. Aggregation and amyloid fibril formation induced by chemical dimerization of recombinant prion protein in physiological-like conditions. J. Biol. Chem. 2009, 284, 30907–30916. [Google Scholar] [CrossRef] [Green Version]
- Takata, T.; Oxford, J.T.; Demeler, B.; Lampi, K.J. Deamidation destabilizes and triggers aggregation of a lens protein, betaA3-crystallin. Protein Sci. A Publ. Protein Soc. 2008, 17, 1565–1575. [Google Scholar] [CrossRef]
- Wei, Y.; Chen, L.; Chen, J.; Ge, L.; He, R.Q. Rapid glycation with D-ribose induces globular amyloid-like aggregations of BSA with high cytotoxicity to SH-SY5Y cells. BMC Cell Biol. 2009, 10, 10. [Google Scholar] [CrossRef] [Green Version]
- Redecke, L.; Binder, S.; Elmallah, M.I.Y.; Broadbent, R.; Tilkorn, C.; Schulz, B.; May, P.; Goos, A.; Eich, A.; Rübhausen, M.; et al. UV-light-induced conversion and aggregation of prion proteins. Free Radic. Biol. Med. 2009, 46, 1353–1361. [Google Scholar] [CrossRef]
- Roy, S.; Mason, B.D.; Schöneich, C.S.; Carpenter, J.F.; Boone, T.C.; Kerwin, B.A. Light-induced aggregation of type I soluble tumor necrosis factor receptor. J. Pharm. Sci. 2009, 98, 3182–3199. [Google Scholar] [CrossRef]
- Li, S.; Leblanc, R.M. Aggregation of Insulin at the Interface. J. Phys. Chem. B 2014, 118, 1181–1188. [Google Scholar] [CrossRef]
- Campioni, S.; Carret, G.; Jordens, S.; Nicoud, L.; Mezzenga, R.; Riek, R. The Presence of an Air–Water Interface Affects Formation and Elongation of α-Synuclein Fibrils. J. Am. Chem. Soc. 2014, 136, 2866–2875. [Google Scholar] [CrossRef]
- Trigg, B.J.; Lee, C.F.; Vaux, D.J.; Jean, L. The air–water interface determines the outcome of seeding during amyloidogenesis. Biochem. J. 2013, 456, 67–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jean, L.; Lee, C.F.; Vaux, D.J. Enrichment of Amyloidogenesis at an Air-Water Interface. Biophys. J. 2012, 102, 1154–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jean, L.; Lee, C.F.; Lee, C.; Shaw, M.; Vaux, D.J. Competing discrete interfacial effects are critical for amyloidogenesis. FASEB J. 2010, 24, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, A.; Cheng, C.-Y.; Kinnebrew, M.; Lew, J.; Dahlquist, F.W.; Han, S. Protein structural and surface water rearrangement constitute major events in the earliest aggregation stages of tau. Proc. Natl. Acad. Sci. USA 2016, 113, E127–E136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frachon, T.; Bruckert, F.; Le Masne, Q.; Monnin, E.; Weidenhaupt, M. Insulin Aggregation at a Dynamic Solid–Liquid–Air Triple Interface. Langmuir 2016, 32, 13009–13019. [Google Scholar] [CrossRef] [PubMed]
- Sluzky, V.; Tamada, J.A.; Klibanov, A.M.; Langer, R. Kinetics of insulin aggregation in aqueous solutions upon agitation in the presence of hydrophobic surfaces. Proc. Natl. Acad. Sci. USA 1991, 88, 9377–9381. [Google Scholar] [CrossRef] [Green Version]
- Duerkop, M.; Berger, E.; Dürauer, A.; Jungbauer, A. Impact of Cavitation, High Shear Stress and Air/Liquid Interfaces on Protein Aggregation. Biotechnol. J. 2018, 13, 1800062. [Google Scholar] [CrossRef] [Green Version]
- Jayaraman, M.; Buck, P.M.; Alphonse Ignatius, A.; King, K.R.; Wang, W. Agitation-induced aggregation and subvisible particulate formation in model proteins. Eur. J. Pharm. Biopharm. 2014, 87, 299–309. [Google Scholar] [CrossRef]
- Kiese, S.; Papppenberger, A.; Friess, W.; Mahler, H.-C. Shaken, Not Stirred: Mechanical Stress Testing of an IgG1 Antibody. J. Pharm. Sci. 2008, 97, 4347–4366. [Google Scholar] [CrossRef]
- Fesinmeyer, R.M.; Hogan, S.; Saluja, A.; Brych, S.R.; Kras, E.; Narhi, L.O.; Brems, D.N.; Gokarn, Y.R. Effect of Ions on Agitation- and Temperature-Induced Aggregation Reactions of Antibodies. Pharm. Res. 2009, 26, 903–913. [Google Scholar] [CrossRef]
- Zhang, J.; Topp, E.M. Protein G, Protein A and Protein A-Derived Peptides Inhibit the Agitation Induced Aggregation of IgG. Mol. Pharm. 2012, 9, 622–628. [Google Scholar] [CrossRef] [PubMed]
- Thirumangalathu, R.; Krishnan, S.; Ricci, M.S.; Brems, D.N.; Randolph, T.W.; Carpenter, J.F. Silicone oil- and agitation-induced aggregation of a monoclonal antibody in aqueous solution. J. Pharm. Sci. 2009, 98, 3167–3181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krielgaard, L.; Jones, L.S.; Randolph, T.W.; Frokjaer, S.; Flink, J.M.; Manning, M.C.; Carpenter, J.F. Effect of tween 20 on freeze-thawing- and agitation-induced aggregation of recombinant human factor XIII. J. Pharm. Sci. 1998, 87, 1597–1603. [Google Scholar] [CrossRef] [PubMed]
- Abdolvahabi, A.; Shi, Y.; Rasouli, S.; Croom, C.M.; Chuprin, A.; Shaw, B.F. How Do Gyrating Beads Accelerate Amyloid Fibrillization? Biophys. J. 2017, 112, 250–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kueltzo, L.A.; Wang, W.e.i.; Randolph, T.W.; Carpenter, J.F. Effects of Solution Conditions, Processing Parameters, and Container Materials on Aggregation of a Monoclonal Antibody during Freeze-Thawing. J. Pharm. Sci. 2008, 97, 1801–1812. [Google Scholar] [CrossRef] [PubMed]
- Perevozchikova, T.; Nanda, H.; Nesta, D.P.; Roberts, C.J. Protein adsorption, desorption, and aggregation mediated by solid-liquid interfaces. J. Pharm. Sci. 2015, 104, 1946–1959. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, A.; Mcgraw, N.R.; Schwartz, D.K.; Bee, J.S.; Carpenter, J.F.; Randolph, T.W. Protein Aggregation and Particle Formation in Prefilled Glass Syringes. J. Pharm. Sci. 2014, 103, 1601–1612. [Google Scholar] [CrossRef]
- Basu, P.; Thirumangalathu, R.; Randolph, T.W.; Carpenter, J.F. IgG1 aggregation and particle formation induced by silicone–water interfaces on siliconized borosilicate glass beads: A model for siliconized primary containers. J. Pharm. Sci. 2013, 102, 852–865. [Google Scholar] [CrossRef]
- Bekard, I.B.; Asimakis, P.; Bertolini, J.; Dunstan, D.E. The effects of shear flow on protein structure and function. Biopolymers 2011, 95, 733–745. [Google Scholar] [CrossRef]
- Cao, E.; Chen, Y.; Cui, Z.; Foster, P.R. Effect of freezing and thawing rates on denaturation of proteins in aqueous solutions. Biotechnol. Bioeng. 2003, 82, 684–690. [Google Scholar] [CrossRef]
- Miller, Y.; Ma, B.; Nussinov, R. Zinc ions promote Alzheimer Aβ aggregation via population shift of polymorphic states. Proc. Natl. Acad. Sci. USA 2010, 107, 9490–9495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagel, K.; Seri, T.; von Berlepsch, H.; Griebel, J.; Kirmse, R.; Böttcher, C.; Koksch, B. How metal ions affect amyloid formation: Cu2+-and Zn2+-sensitive peptides. ChemBioChem 2008, 9, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Hoernke, M.; Koksch, B.; Brezesinski, G. Amyloidogenic peptides at hydrophobic–hydrophilic interfaces: Coordination affinities and the chelate effect dictate the competitive binding of Cu2+ and Zn2+. ChemPhysChem 2011, 12, 2225–2229. [Google Scholar] [CrossRef] [PubMed]
- Hoernke, M.; Koksch, B.; Brezesinski, G. Influence of the hydrophobic interface and transition metal ions on the conformation of amyloidogenic model peptides. Biophys. Chem. 2010, 150, 64–72. [Google Scholar] [CrossRef] [Green Version]
- Hoernke, M.; Falenski, J.A.; Schwieger, C.; Koksch, B.; Brezesinski, G. Triggers for β-sheet formation at the hydrophobic–hydrophilic interface: High concentration, in-plane orientational order, and metal ion complexation. Langmuir 2011, 27, 14218–14231. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, X.Y. Effect of protein–protein interactions on protein aggregation kinetics. J. Chem. Phys. 2003, 119, 10972–10976. [Google Scholar] [CrossRef]
- Galvagnion, C.; Buell, A.K.; Meisl, G.; Michaels, T.C.T.; Vendruscolo, M.; Knowles, T.P.J.; Dobson, C.M. Lipid vesicles trigger α-synuclein aggregation by stimulating primary nucleation. Nat. Chem. Biol. 2015, 11, 229–234. [Google Scholar] [CrossRef] [Green Version]
- Gorbenko, G.P.; Ioffe, V.M.; Kinnunen, P.K.J. Binding of Lysozyme to Phospholipid Bilayers: Evidence for Protein Aggregation upon Membrane Association. Biophys. J. 2007, 93, 140–153. [Google Scholar] [CrossRef] [Green Version]
- Terzi, E.; Hölzemann, G.; Seelig, J. Self-association of β-Amyloid Peptide (1–40) in Solution and Binding to Lipid Membranes. J. Mol. Biol. 1995, 252, 633–642. [Google Scholar] [CrossRef]
- Zhao, H.; Tuominen, E.K.J.; Kinnunen, P.K.J. Formation of Amyloid Fibers Triggered by Phosphatidylserine-Containing Membranes. Biochemistry 2004, 43, 10302–10307. [Google Scholar] [CrossRef]
- Sparr, E.; Engel, M.F.M.; Sakharov, D.V.; Sprong, M.; Jacobs, J.; de Kruijff, B.; Höppener, J.W.M.; Antoinette Killian, J. Islet amyloid polypeptide-induced membrane leakage involves uptake of lipids by forming amyloid fibers. FEBS Lett. 2004, 577, 117–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, Z.; Hashemi, M.; Banerjee, S.; Zagorski, K.; Rochet, J.-C.; Lyubchenko, Y.L. Phospholipid membranes promote the early stage assembly of α-synuclein aggregates. bioRxiv 2018, 295782. [Google Scholar]
- Chauhan, A.; Ray, I.; Chauhan, V.P. Interaction of amyloid beta-protein with anionic phospholipids: Possible involvement of Lys28 and C-terminus aliphatic amino acids. Neurochem. Res. 2000, 25, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Ege, C.; Lee, K.Y.C. Insertion of Alzheimer’s Aβ40 peptide into lipid monolayers. Biophys. J. 2004, 87, 1732–1740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrone, F. [17] Analysis of protein aggregation kinetics. Methods Enzymol. 1999, 309, 256–274. [Google Scholar]
- Morris, A.M.; Watzky, M.A.; Finke, R.G. Protein aggregation kinetics, mechanism, and curve-fitting: A review of the literature. Biochim. Et Biophys. Acta (BBA)-Proteins Proteom. 2009, 1794, 375–397. [Google Scholar] [CrossRef]
- Arosio, P.; Knowles, T.P.J.; Linse, S. On the lag phase in amyloid fibril formation. Phys. Chem. Chem. Phys. 2015, 17, 7606–7618. [Google Scholar] [CrossRef] [Green Version]
- Harper, J.D.; Lansbury, P.T. Models of Amyloid Seeding in Alzheimer’s Disease and Scrapie: Mechanistic Truths and Physiological Consequences of the Time-Dependent Solubility of Amyloid Proteins. Annu. Rev. Biochem. 1997, 66, 385–407. [Google Scholar] [CrossRef]
- O’Nuallain, B.; Shivaprasad, S.; Kheterpal, I.; Wetzel, R. Thermodynamics of Aβ(1−40) Amyloid Fibril Elongation. Biochemistry 2005, 44, 12709–12718. [Google Scholar] [CrossRef]
- Watzky, M.A.; Morris, A.M.; Ross, E.D.; Finke, R.G. Fitting Yeast and Mammalian Prion Aggregation Kinetic Data with the Finke−Watzky Two-Step Model of Nucleation and Autocatalytic Growth †. Biochemistry 2008, 47, 10790–10800. [Google Scholar] [CrossRef]
- Morris, A.M.; Watzky, M.A.; Agar, J.N.; Finke, R.G. Fitting Neurological Protein Aggregation Kinetic Data via a 2-Step, Minimal/“Ockham’s Razor” Model: The Finke−Watzky Mechanism of Nucleation Followed by Autocatalytic Surface Growth †. Biochemistry 2008, 47, 2413–2427. [Google Scholar] [CrossRef] [PubMed]
- Watzky, M.A.; Finke, R.G. Transition Metal Nanocluster Formation Kinetic and Mechanistic Studies. A New Mechanism When Hydrogen Is the Reductant: Slow, Continuous Nucleation and Fast Autocatalytic Surface Growth. J. Am. Chem. Soc. 1997, 119, 10382–10400. [Google Scholar] [CrossRef]
- Morel, B.; Carrasco, M.P.; Jurado, S.; Marco, C.; Conejero-Lara, F. Dynamic micellar oligomers of amyloid beta peptides play a crucial role in their aggregation mechanisms. Phys. Chem. Chem. Phys. 2018, 20, 20597–20614. [Google Scholar] [CrossRef] [PubMed]
- Schmit, J.D.; Ghosh, K.; Dill, K. What Drives Amyloid Molecules to Assemble into Oligomers and Fibrils? Biophys. J. 2011, 100, 450–458. [Google Scholar] [CrossRef] [Green Version]
- Hasecke, F.; Miti, T.; Perez, C.; Barton, J.; Schölzel, D.; Gremer, L.; Grüning, C.S.R.; Matthews, G.; Meisl, G.; Knowles, T.P.J.; et al. Origin of metastable oligomers and their effects on amyloid fibril self-assembly. Chem. Sci. 2018, 9, 5937–5948. [Google Scholar] [CrossRef] [Green Version]
- Powers, E.T.; Powers, D.L. The Kinetics of Nucleated Polymerizations at High Concentrations: Amyloid Fibril Formation Near and Above the “Supercritical Concentration”. Biophys. J. 2006, 91, 122–132. [Google Scholar] [CrossRef] [Green Version]
- Serio, T.R.; Cashikar, A.G.; Kowal, A.S.; Sawicki, G.J.; Moslehi, J.J.; Serpell, L.; Arnsdorf, M.F.; Lindquist, S.L. Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science 2000, 289, 1317–1321. [Google Scholar] [CrossRef] [Green Version]
- Fu, Z.; Aucoin, D.; Davis, J.; Van Nostrand, W.E.; Smith, S.O. Mechanism of Nucleated Conformational Conversion of Aβ42. Biochemistry 2015, 54, 4197–4207. [Google Scholar] [CrossRef]
- Lee, J.; Culyba, E.K.; Powers, E.T.; Kelly, J.W. Amyloid-β forms fibrils by nucleated conformational conversion of oligomers. Nat. Chem. Biol. 2011, 7, 602–609. [Google Scholar] [CrossRef]
- Chimon, S.; Shaibat, M.A.; Jones, C.R.; Calero, D.C.; Aizezi, B.; Ishii, Y. Evidence of fibril-like β-sheet structures in a neurotoxic amyloid intermediate of Alzheimer’s β-amyloid. Nat. Struct. Mol. Biol. 2007, 14, 1157–1164. [Google Scholar] [CrossRef]
- Ruzafa, D.; Morel, B.; Varela, L.; Azuaga, A.I.; Conejero-Lara, F. Characterization of Oligomers of Heterogeneous Size as Precursors of Amyloid Fibril Nucleation of an SH3 Domain: An Experimental Kinetics Study. PLoS ONE 2012, 7, e49690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruzafa, D.; Conejero-Lara, F.; Morel, B. Modulation of the stability of amyloidogenic precursors by anion binding strongly influences the rate of amyloid nucleation. Phys. Chem. Chem. Phys. 2013, 15, 15508–15517. [Google Scholar] [CrossRef] [PubMed]
- Morel, B.; Ruzafa, D.; Conejero-Lara, F. SH3 Domains as Suitable Models to Study Amyloid Aggregation. In SH Domains; Springer International Publishing: Cham, Switzerland; New York, NY, USA, 2015; pp. 1–15. [Google Scholar]
- Fay, N.; Inoue, Y.; Bousset, L.; Taguchi, H.; Melki, R. Assembly of the yeast prion Ure2p into protein fibrils. Thermodynamic and kinetic characterization. J. Biol. Chem. 2003, 278, 30199–30205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharyya, A.M.; Thakur, A.K.; Wetzel, R. polyglutamine aggregation nucleation: Thermodynamics of a highly unfavorable protein folding reaction. Proc. Natl. Acad. Sci. USA 2005, 102, 15400–15405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parmar, A.S.; Gottschall, P.E.; Muschol, M. Sub-micron lysozyme clusters distort kinetics of crystal nucleation in supersaturated lysozyme solutions. Biophys. Chem 2007, 129, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.E.; Robinson, J.; Matthews, G.; Muschol, M. Amyloid Protofibrils of Lysozyme Nucleate and Grow Via Oligomer Fusion. Biophys. J. 2009, 96, 3781–3790. [Google Scholar] [CrossRef] [Green Version]
- Jarrett, J.T.; Lansbury, P.T. Seeding “one-dimensional crystallization” of amyloid: A pathogenic mechanism in Alzheimer’s disease and scrapie? Cell 1993, 73, 1055–1058. [Google Scholar] [CrossRef]
- Cohen, S.I.; Linse, S.; Luheshi, L.M.; Hellstrand, E.; White, D.A.; Rajah, L.; Otzen, D.E.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P. Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, 9758–9763. [Google Scholar] [CrossRef] [Green Version]
- Meisl, G.; Yang, X.; Dobson, C.M.; Linse, S.; Knowles, T.P.J. Modulation of electrostatic interactions to reveal a reaction network unifying the aggregation behaviour of the Aβ42 peptide and its variants. Chem. Sci. 2017, 8, 4352–4362. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.I.A.; Vendruscolo, M.; Welland, M.E.; Dobson, C.M.; Terentjev, E.M.; Knowles, T.P.J. Nucleated polymerization with secondary pathways. I. Time evolution of the principal moments. J. Chem. Phys. 2011, 135, 065105. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.I.A.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. Nucleated polymerization with secondary pathways. II. Determination of self-consistent solutions to growth processes described by non-linear master equations. J. Chem. Phys. 2011, 135, 065106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knowles, T.P.J.; Waudby, C.A.; Devlin, G.L.; Cohen, S.I.A.; Aguzzi, A.; Vendruscolo, M.; Terentjev, E.M.; Welland, M.E.; Dobson, C.M. An analytical solution to the kinetics of breakable filament assembly. Science (New York N.Y.) 2009, 326, 1533–1537. [Google Scholar] [CrossRef] [Green Version]
- Linse, S. Monomer-dependent secondary nucleation in amyloid formation. Biophys. Rev. 2017, 9, 329–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Törnquist, M.; Michaels, T.C.T.; Sanagavarapu, K.; Yang, X.; Meisl, G.; Cohen, S.I.A.; Knowles, T.P.J.; Linse, S. Secondary nucleation in amyloid formation. Chem. Commun. 2018, 54, 8667–8684. [Google Scholar] [CrossRef] [Green Version]
- Meisl, G.; Yang, X.; Hellstrand, E.; Frohm, B.; Kirkegaard, J.B.; Cohen, S.I.A.; Dobson, C.M.; Linse, S.; Knowles, T.P.J. Differences in nucleation behavior underlie the contrasting aggregation kinetics of the Aβ40 and Aβ42 peptides. Proc. Natl. Acad. Sci. USA 2014, 111, 9384–9389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachandran, G.; Udgaonkar, J.B. Evidence for the existence of a secondary pathway for fibril growth during the aggregation of tau. J. Mol. Biol. 2012, 421, 296–314. [Google Scholar] [CrossRef]
- Buell, A.K.; Galvagnion, C.; Gaspar, R.; Sparr, E.; Vendruscolo, M.; Knowles, T.P.; Linse, S.; Dobson, C.M. Solution conditions determine the relative importance of nucleation and growth processes in α-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2014, 111, 7671–7676. [Google Scholar] [CrossRef] [Green Version]
- Gaspar, R.; Meisl, G.; Buell, A.K.; Young, L.; Kaminski, C.F.; Knowles, T.P.J.; Sparr, E.; Linse, S. Secondary nucleation of monomers on fibril surface dominates α -synuclein aggregation and provides autocatalytic amyloid amplification. Q. Rev. Biophys. 2017, 50, e6. [Google Scholar] [CrossRef] [Green Version]
- Padrick, S.B.; Miranker, A.D. Islet Amyloid: Phase Partitioning and Secondary Nucleation Are Central to the Mechanism of Fibrillogenesis. Biochemistry 2002, 41, 4694–4703. [Google Scholar] [CrossRef]
- Foderà, V.; Librizzi, F.; Groenning, M.; van de Weert, M.; Leone, M. Secondary Nucleation and Accessible Surface in Insulin Amyloid Fibril Formation. J. Phys. Chem. B 2008, 112, 3853–3858. [Google Scholar] [CrossRef]
- Garg, D.K.; Kundu, B. Clues for divergent, polymorphic amyloidogenesis through dissection of amyloid forming steps of bovine carbonic anhydrase and its critical amyloid forming stretch. Biochim. Et Biophys. Acta (BBA)-Proteins Proteom. 2016, 1864, 794–804. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.I.A.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. Nucleated Polymerisation in the Presence of Pre-Formed Seed Filaments. Int. J. Mol. Sci. 2011, 12, 5844–5852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales, R.; Moreno-Gonzalez, I.; Soto, C. Cross-Seeding of Misfolded Proteins: Implications for Etiology and Pathogenesis of Protein Misfolding Diseases. PLoS Pathog. 2013, 9, e1003537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, L.C.; Diamond, M.I.; Duff, K.E.; Hyman, B.T. Mechanisms of Protein Seeding in Neurodegenerative Diseases. JAMA Neurol. 2013, 70, 304–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Come, J.H.; Fraser, P.E.; Lansbury, P.T. A kinetic model for amyloid formation in the prion diseases: Importance of seeding. Proc. Natl. Acad. Sci. USA 1993, 90, 5959–5963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brundin, P.; Melki, R.; Kopito, R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat. Rev. Mol. Cell Biol. 2010, 11, 301–307. [Google Scholar] [CrossRef] [Green Version]
- Walker, L.C.; Jucker, M. Neurodegenerative Diseases: Expanding the Prion Concept. Annu. Rev. Neurosci. 2015, 38, 87–103. [Google Scholar] [CrossRef] [Green Version]
- Jucker, M.; Walker, L.C. Pathogenic protein seeding in alzheimer disease and other neurodegenerative disorders. Ann. Neurol. 2011, 70, 532–540. [Google Scholar] [CrossRef]
- Ono, K.; Takahashi, R.; Ikeda, T.; Yamada, M. Cross-seeding effects of amyloid β-protein and α-synuclein. J. Neurochem. 2012, 122, 883–890. [Google Scholar] [CrossRef] [Green Version]
- Oosawa, F.; Kasai, M. A theory of linear and helical aggregations of macromolecules. J. Mol. Biol. 1962, 4, 10–21. [Google Scholar] [CrossRef]
- Oosawa, F.; Asakura, S. Thermodynamics of the Polymerization of Protein; Academic Press: London, UK, 1975. [Google Scholar]
- Ruzafa, D.; Varela, L.; Azuaga, A.I.; Conejero-Lara, F.; Morel, B. Mapping the structure of amyloid nucleation precursors by protein engineering kinetic analysis. Phys. Chem. Chem. Phys. 2014, 16, 2989–3000. [Google Scholar] [CrossRef]
- Hori, Y.; Hashimoto, T.; Wakutani, Y.; Urakami, K.; Nakashima, K.; Condron, M.M.; Tsubuki, S.; Saido, T.C.; Teplow, D.B.; Iwatsubo, T. The Tottori (D7N) and English (H6R) familial Alzheimer disease mutations accelerate Abeta fibril formation without increasing protofibril formation. J. Biol. Chem. 2007, 282, 4916–4923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Udgaonkar, J.B. Conformational Conversion May Precede or Follow Aggregate Elongation on Alternative Pathways of Amyloid Protofibril Formation. J. Mol. Biol. 2009, 385, 1266–1276. [Google Scholar] [CrossRef] [PubMed]
- Hurshman, A.R.; White, J.T.; Powers, E.T.; Kelly, J.W. Transthyretin Aggregation under Partially Denaturing Conditions Is a Downhill Polymerization. Biochemistry 2004, 43, 7365–7381. [Google Scholar] [CrossRef] [PubMed]
- Faria, T.Q.; Almeida, Z.L.; Cruz, P.F.; Jesus, C.S.H.; Castanheira, P.; Brito, R.M.M. A look into amyloid formation by transthyretin: Aggregation pathway and a novel kinetic model. Phys. Chem. Chem. Phys. 2015, 17, 7255–7263. [Google Scholar] [CrossRef] [PubMed]
- O’Nuallain, B.; Freir, D.B.; Nicoll, A.J.; Risse, E.; Ferguson, N.; Herron, C.E.; Collinge, J.; Walsh, D.M. Amyloid beta-protein dimers rapidly form stable synaptotoxic protofibrils. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 14411–14419. [Google Scholar] [CrossRef] [PubMed]
- Rangachari, V.; Moore, B.D.; Reed, D.K.; Sonoda, L.K.; Bridges, A.W.; Conboy, E.; Hartigan, D.; Rosenberry, T.L. Amyloid-β (1− 42) rapidly forms protofibrils and oligomers by distinct pathways in low concentrations of sodium dodecylsulfate. Biochemistry 2007, 46, 12451–12462. [Google Scholar] [CrossRef]
- Ramachandran, G.; Udgaonkar, J.B. Understanding the kinetic roles of the inducer heparin and of rod-like protofibrils during amyloid fibril formation by Tau protein. J. Biol. Chem. 2011, 286, 38948–38959. [Google Scholar] [CrossRef] [Green Version]
- Juárez, J.; Taboada, P.; Mosquera, V. Existence of Different Structural Intermediates on the Fibrillation Pathway of Human Serum Albumin. Biophys. J. 2009, 96, 2353–2370. [Google Scholar] [CrossRef] [Green Version]
- Holm, N.K.; Jespersen, S.K.; Thomassen, L.V.; Wolff, T.Y.; Sehgal, P.; Thomsen, L.A.; Christiansen, G.; Andersen, C.B.; Knudsen, A.D.; Otzen, D.E. Aggregation and fibrillation of bovine serum albumin. Biochim. Et Biophys. Acta (BBA)-Proteins Proteom. 2007, 1774, 1128–1138. [Google Scholar] [CrossRef]
- Campioni, S.; Mossuto, M.F.; Torrassa, S.; Calloni, G.; de Laureto, P.P.; Relini, A.; Fontana, A.; Chiti, F. Conformational properties of the aggregation precursor state of HypF-N. J. Mol. Biol. 2008, 379, 554–567. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, P.; Navarro, S.; Baño-Polo, M.; Morel, B.; Graña-Montes, R.; Sabe, A.; Canals, F.; Fernandez, M.R.; Conejero-Lara, F.; Ventura, S. Global Protein Stabilization Does Not Suffice to Prevent Amyloid Fibril Formation. ACS Chem. Biol. 2018, 13, 2094–2105. [Google Scholar] [CrossRef] [PubMed]
- Calamai, M.; Taddei, N.; Stefani, M.; Ramponi, G.; Chiti, F. Relative Influence of Hydrophobicity and Net Charge in the Aggregation of Two Homologous Proteins. Biochemistry 2003, 42, 15078–15083. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Griffin, M.D.W.; Binger, K.J.; Schuck, P.; Howlett, G.J. An Equilibrium Model for Linear and Closed-Loop Amyloid Fibril Formation. J. Mol. Biol. 2012, 421, 364–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffin, M.D.W.; Mok, M.L.Y.; Wilson, L.M.; Pham, C.L.L.; Waddington, L.J.; Perugini, M.A.; Howlett, G.J. Phospholipid Interaction Induces Molecular-level Polymorphism in Apolipoprotein C-II Amyloid Fibrils via Alternative Assembly Pathways. J. Mol. Biol. 2008, 375, 240–256. [Google Scholar] [CrossRef]
- Morel, B.; Varela, L.; Azuaga, A.I.; Conejero-Lara, F. Environmental Conditions Affect the Kinetics of Nucleation of Amyloid Fibrils and Determine Their Morphology. Biophys. J. 2010, 99, 3801–3810. [Google Scholar] [CrossRef] [Green Version]
- Zurdo, J.; Guijarro, J.; Jiménez, J.L.; Saibil, H.R.; Dobson, C.M. Dependence on solution conditions of aggregation and amyloid formation by an SH3 domain. J. Mol. Biol. 2001, 311, 325–340. [Google Scholar] [CrossRef]
- Varela, L.; Morel, B.; Azuaga, A.I.; Conejero-Lara, F. A single mutation in an SH3 domain increases amyloid aggregation by accelerating nucleation, but not by destabilizing thermodynamically the native state. FEBS Lett. 2009, 583, 801–806. [Google Scholar] [CrossRef] [Green Version]
- Watters, A.L.; Deka, P.; Corrent, C.; Callender, D.; Varani, G.; Sosnick, T.; Baker, D. The highly cooperative folding of small naturally occurring proteins is likely the result of natural selection. Cell 2007, 128, 613–624. [Google Scholar] [CrossRef] [Green Version]
- Vendruscolo, M.; Paci, E.; Karplus, M.; Dobson, C. Structures and relative free energies of partially folded states of proteins. Proc. Natl. Acad. Sci. USA 2003, 100, 14817–14821. [Google Scholar] [CrossRef] [Green Version]
- Brockwell, D.J.; Radford, S.E. Intermediates: Ubiquitous species on folding energy landscapes? Curr. Opin. Struct. Biol. 2007, 17, 30–37. [Google Scholar] [CrossRef] [Green Version]
- Jahn, T.R.; Radford, S.E. Folding versus aggregation: Polypeptide conformations on competing pathways. Arch. Biochem. Biophys. 2008, 469, 100–117. [Google Scholar] [CrossRef] [Green Version]
- Turoverov, K.K.; Kuznetsova, I.M.; Uversky, V.N. The protein kingdom extended: Ordered and intrinsically disordered proteins, their folding, supramolecular complex formation, and aggregation. Prog. Biophys. Mol. Biol. 2010, 102, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Sekijima, Y.; Wiseman, R.L.; Matteson, J.; Hammarström, P.; Miller, S.R.; Sawkar, A.R.; Balch, W.E.; Kelly, J.W. The Biological and Chemical Basis for Tissue-Selective Amyloid Disease. Cell 2005, 121, 73–85. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.I.A.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. From Macroscopic Measurements to Microscopic Mechanisms of Protein Aggregation. J. Mol. Biol. 2012, 421, 160–171. [Google Scholar] [CrossRef]
- Routledge, K.E.; Tartaglia, G.G.; Platt, G.W.; Vendruscolo, M.; Radford, S.E. Competition between Intramolecular and Intermolecular Interactions in an Amyloid-Forming Protein. J. Mol. Biol. 2009, 389, 776–786. [Google Scholar] [CrossRef] [Green Version]
- Modler, A.; Gast, K.; Lutsch, G.; Damaschun, G. Assembly of Amyloid Protofibrils via Critical Oligomers—A Novel Pathway of Amyloid Formation. J. Mol. Biol. 2003, 325, 135–148. [Google Scholar] [CrossRef]
- Thirumalai, D.; Reddy, G. Protein thermodynamics: Are native proteins metastable? Nat. Chem. 2011, 3, 910. [Google Scholar] [CrossRef]
- Chiti, F.; Taddei, N.; Baroni, F.; Capanni, C.; Stefani, M.; Ramponi, G.; Dobson, C.M. Kinetic partitioning of protein folding and aggregation. Nat. Struct. Biol. 2002, 9, 137–143. [Google Scholar] [CrossRef]
- Jiménez, J.L.; Nettleton, E.J.; Bouchard, M.; Robinson, C.V.; Dobson, C.M.; Saibil, H.R. The protofilament structure of insulin amyloid fibrils. Proc. Natl. Acad. Sci. USA 2002, 99, 9196–9201. [Google Scholar] [CrossRef] [Green Version]
- Eichner, T.; Radford, S.E. A Diversity of Assembly Mechanisms of a Generic Amyloid Fold. Mol. Cell 2011, 43, 8–18. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, A.J.; Knowles, T.P.J.; Tartaglia, G.G.; Fitzpatrick, A.W.; Devlin, G.L.; Shammas, S.L.; Waudby, C.A.; Mossuto, M.F.; Meehan, S.; Gras, S.L.; et al. Metastability of Native Proteins and the Phenomenon of Amyloid Formation. J. Am. Chem. Soc. 2011, 133, 14160–14163. [Google Scholar] [CrossRef]
- Chong, S.-H.; Ham, S. Distinct Role of Hydration Water in Protein Misfolding and Aggregation Revealed by Fluctuating Thermodynamics Analysis. Acc. Chem. Res. 2015, 48, 956–965. [Google Scholar] [CrossRef]
- Sunde, M.; Blake, C.C.F. From the globular to the fibrous state: Protein structure and structural conversion in amyloid formation. Q. Rev. Biophys. 1998, 31, 1–39. [Google Scholar] [CrossRef]
- Dorta-Estremera, S.M.; Li, J.; Cao, W. Rapid Generation of Amyloid from Native Proteins In vitro. J. Vis. Exp. 2013, e50869. [Google Scholar]
- Goldschmidt, L.; Teng, P.K.; Riek, R.; Eisenberg, D. Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc. Natl. Acad. Sci. USA 2010, 107, 3487–3492. [Google Scholar] [CrossRef] [Green Version]
- Wetzel, R. Kinetics and Thermodynamics of Amyloid Fibril Assembly. Acc. Chem. Res. 2006, 39, 671–679. [Google Scholar] [CrossRef]
Figure 1.
Schematic representation of the variety of conformational states that can be adopted by a polypeptide chain upon biosynthesis in the ribosome. The non-amyloidogenic pathway includes the formation of native sates and functional amyloids. The amyloidogenic pathway associated with pathological states can result from the formation of amorphous aggregates, amyloid aggregates and fibrils, and native-like aggregates. Adapted from reference [1].
Figure 1.
Schematic representation of the variety of conformational states that can be adopted by a polypeptide chain upon biosynthesis in the ribosome. The non-amyloidogenic pathway includes the formation of native sates and functional amyloids. The amyloidogenic pathway associated with pathological states can result from the formation of amorphous aggregates, amyloid aggregates and fibrils, and native-like aggregates. Adapted from reference [1].

Figure 2.
Chemical structures of Congo red, thioflavin-T, and some of their derivatives used in amyloid fibril detection.
Figure 2.
Chemical structures of Congo red, thioflavin-T, and some of their derivatives used in amyloid fibril detection.

Figure 3.
The ten possible amyloid symmetry classes of homo-steric zipper amyloid spines. Legend: face (F), back (B), up (U), down (D), N-terminal and C-terminal. Adapted from reference [65].
Figure 3.
The ten possible amyloid symmetry classes of homo-steric zipper amyloid spines. Legend: face (F), back (B), up (U), down (D), N-terminal and C-terminal. Adapted from reference [65].

Figure 4.
Representative general model for amyloid fibril formation by nucleation-dependent mechanisms (including primary and secondary nucleation) and nucleation-independent mechanisms (absence of nucleation). kn1, kn2, k−, kon, koff, and k+ represent rate constants. The stationary phase involves the assembly of protofibrils into mature amyloid fibrils with different morphological structures and a high level of polymorphism. Adapted from reference [148].
Figure 4.
Representative general model for amyloid fibril formation by nucleation-dependent mechanisms (including primary and secondary nucleation) and nucleation-independent mechanisms (absence of nucleation). kn1, kn2, k−, kon, koff, and k+ represent rate constants. The stationary phase involves the assembly of protofibrils into mature amyloid fibrils with different morphological structures and a high level of polymorphism. Adapted from reference [148].

Scheme 1.
The minimalistic Finke–Watzky mechanism for protein aggregation via a nucleation-dependent pathway, where M is the monomer, and A is the transient nucleus aggregate. The rate constants for the nucleation and growth steps are knucleation and kgrowth, respectively [216,217,218].

Scheme 2.
The sequential monomer (M) addition mechanism for protein aggregation with identical equilibrium constants (k) via a nucleation-independent pathway [257,258].

Figure 5.
Schematic representation of funnel-shaped energy landscapes for protein folding (non-amyloidogenic pathway, green) and aggregation (amyloidogenic pathway, red). The surface exhibits the roughness of the protein energy landscape showing the possible conformational states adopted by the polypeptide chain. Unfolded, partially unfolded and folded species may be implicated in the aggregation landscape, as long as they are prone to establish intermolecular interactions and aggregate, thereby resulting in the formation of amorphous aggregates, amyloid fibrils, and native-like aggregates, respectively. Adapted from references [1,279,280].
Figure 5.
Schematic representation of funnel-shaped energy landscapes for protein folding (non-amyloidogenic pathway, green) and aggregation (amyloidogenic pathway, red). The surface exhibits the roughness of the protein energy landscape showing the possible conformational states adopted by the polypeptide chain. Unfolded, partially unfolded and folded species may be implicated in the aggregation landscape, as long as they are prone to establish intermolecular interactions and aggregate, thereby resulting in the formation of amorphous aggregates, amyloid fibrils, and native-like aggregates, respectively. Adapted from references [1,279,280].


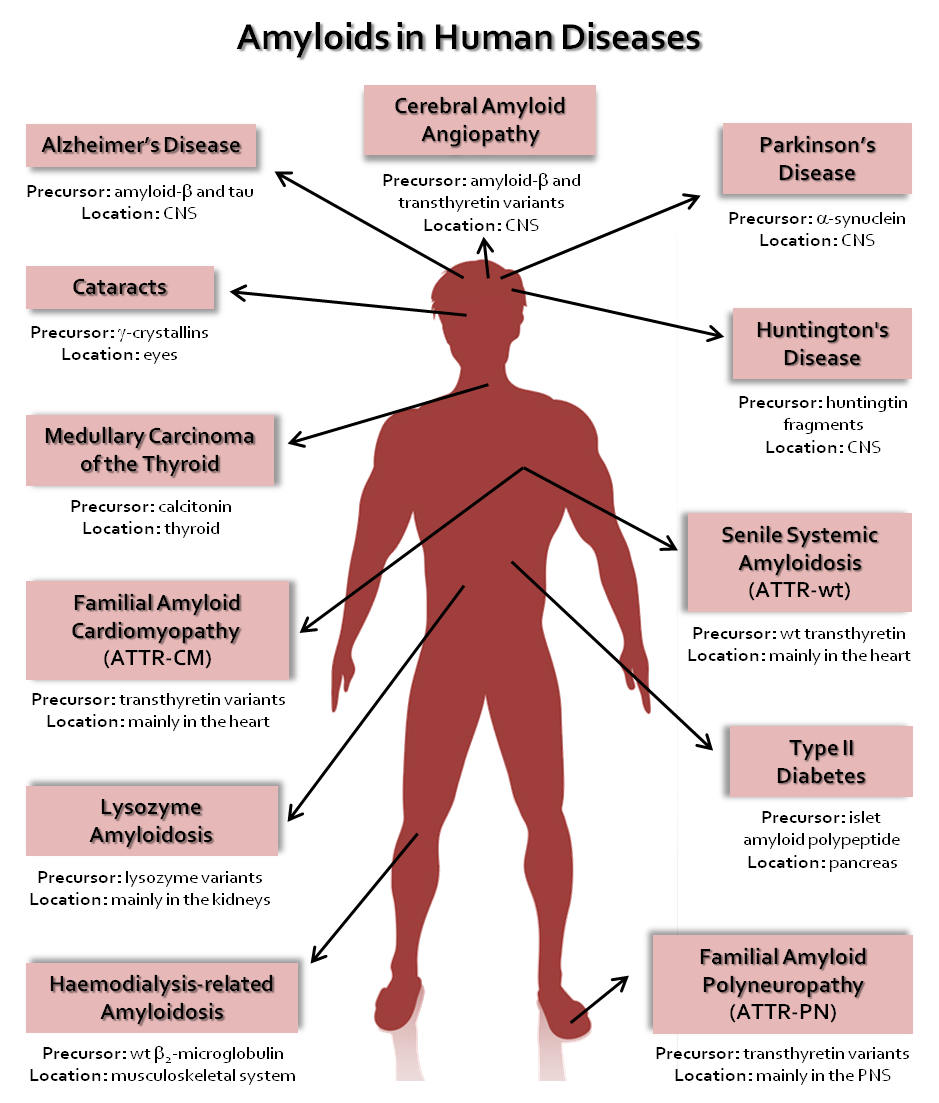{kind=link}
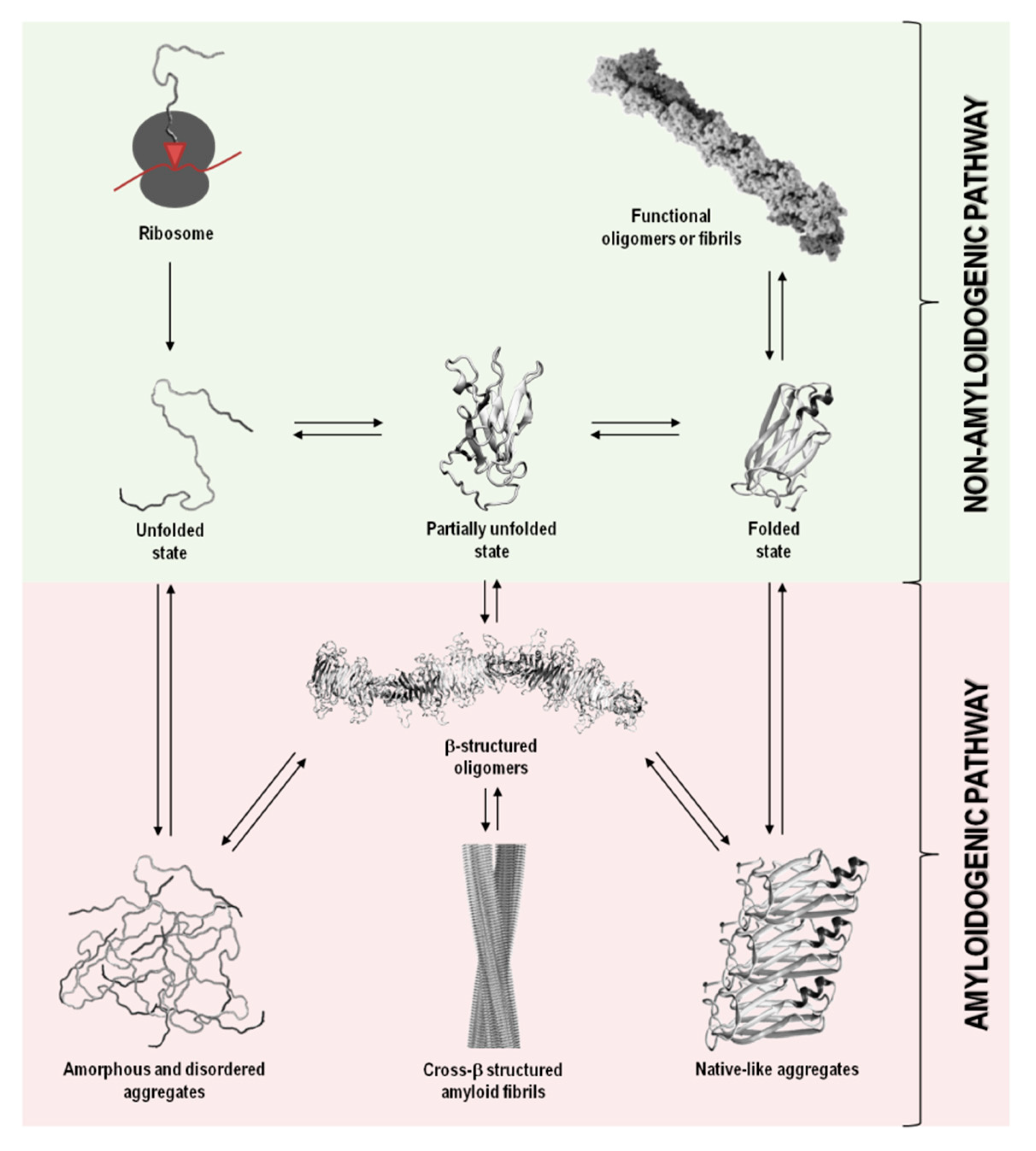{kind=link}
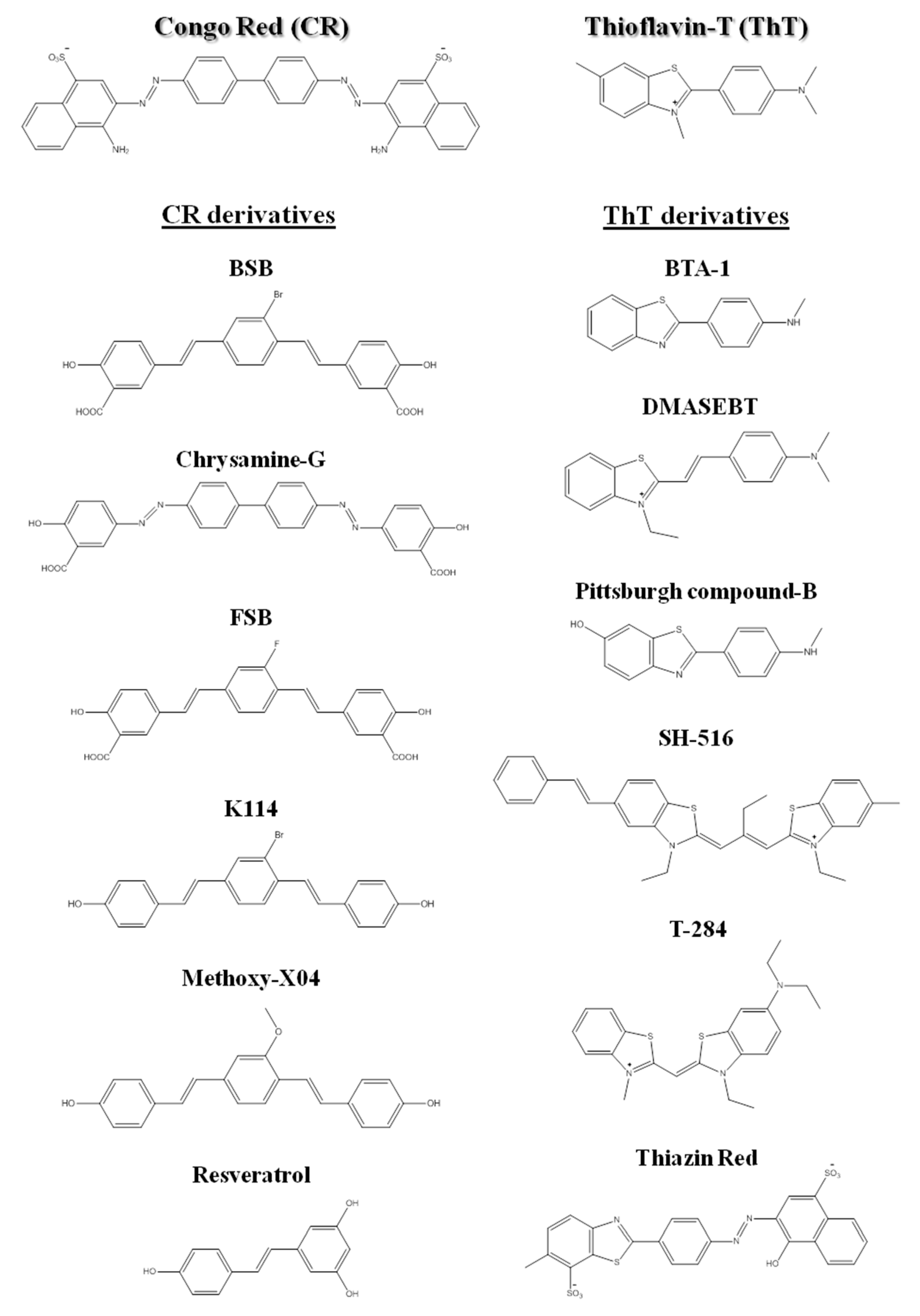{kind=link}
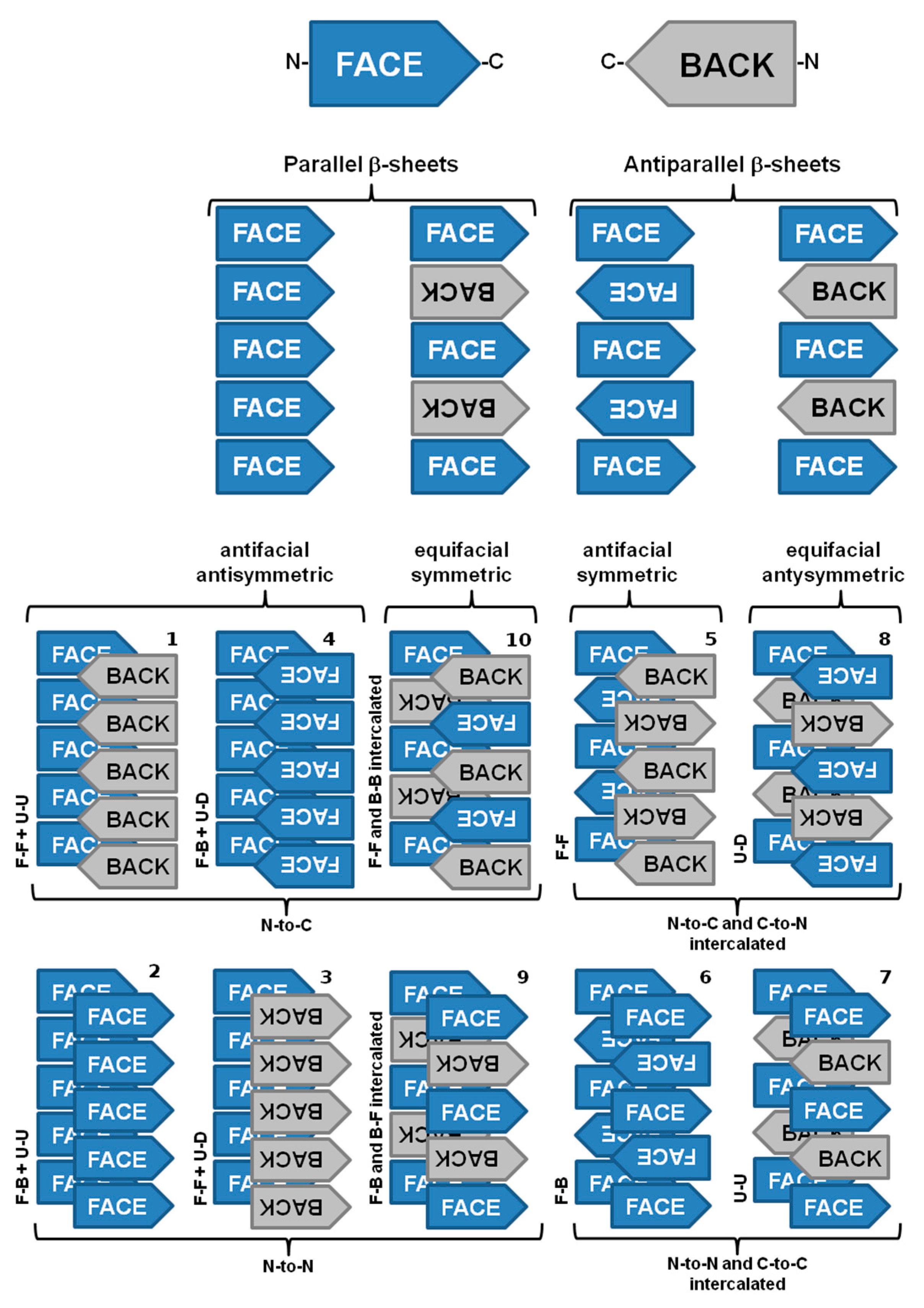{kind=link}
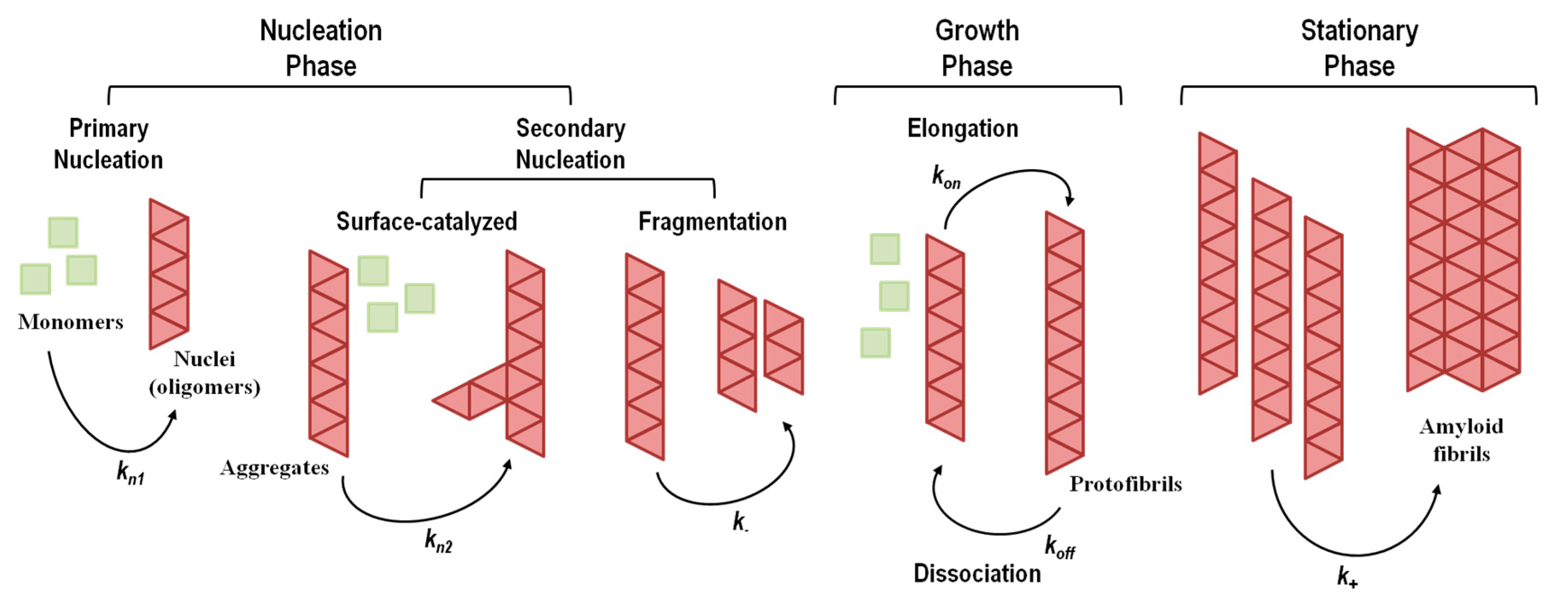{kind=link}
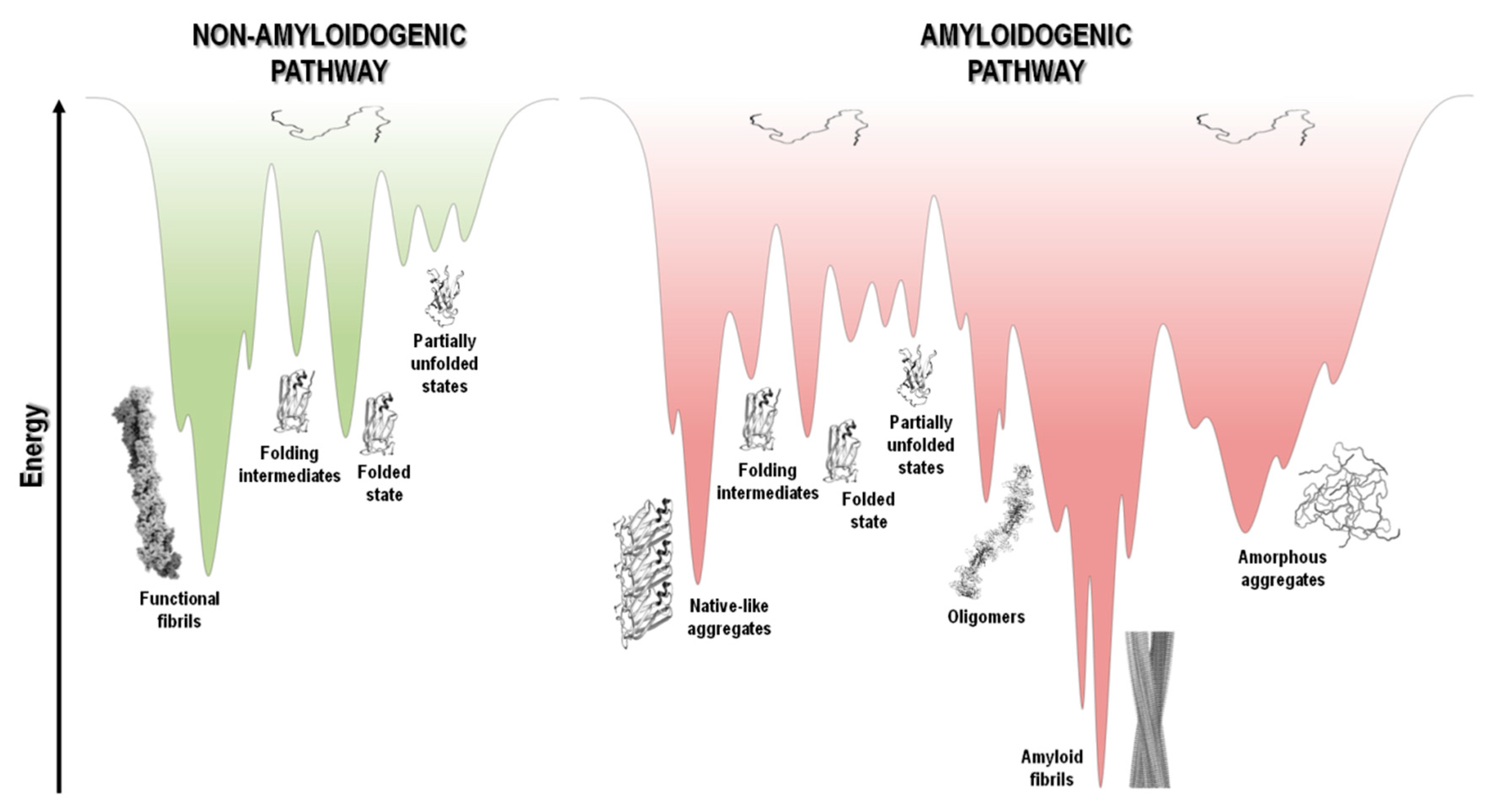{kind=link}
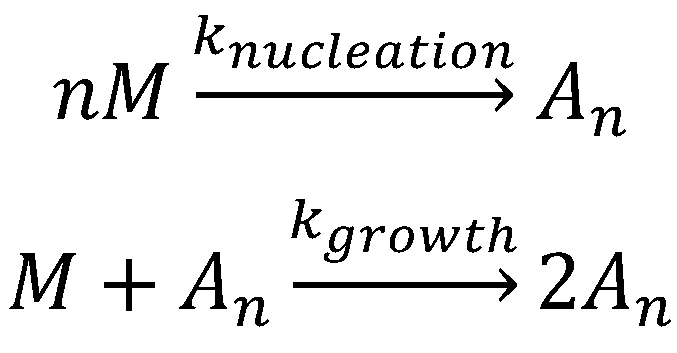{kind=link}
{kind=link}
| Disease | Precursor Protein | Polypeptide Length (n° of Residues) | Structural Organization of Precursor |
|---|---|---|---|
| Neurodegenerative Diseases | |||
| Alzheimer’s disease | Amyloid-β variants | 37–44 | IDP |
| Spongiform encephalopathies | Prion protein or its fragments | 208 | IDP and α-helical |
| Parkinson’s disease | α-synuclein | 140 | IDP |
| Frontotemporal dementia with Parkinsonism | Tau | 352–441 | IDP |
| Amyotrophic lateral sclerosis | Superoxide dismutase 1 | 153 | β-sheet |
| Huntington’s disease | Huntingtin with polyQ expansion | 3144 | Mostly IDP |
| Neuroferritinopathy | Ferritin | 175 or 183 | α-helical |
| Familial British dementia | ABri | 34 | IDP |
| Familial Danish dementia | ADan | 34 | IDP |
| Familial amyloid polyneuropathy | Transthyretin variants | 127 | β-sheet |
| Non-Neuropathic Systemic Amyloidosis | |||
| Amyloid light chain amyloidosis | Immunoglobulin light chains or its fragments | ~90 | β-sheet |
| Amyloid heavy chain amyloidosis | Immunoglobulin heavy chains or its fragments | ~220 | β-sheet |
| Amyloid A amyloidosis | Serum amyloid A protein fragments | 45–104 | α-helical and unknown fold |
| Familial Mediterranean fever | Serum amyloid A protein fragments | 45–104 | α-helical and unknown fold |
| Apolipoprotein A1 amyloidosis | Apo A-1 fragments | 80–93 | IDP |
| Senile systemic amyloidosis | Wild-type transthyretin | 127 | β-sheet |
| Familial amyloid cardiomyopathy | Transthyretin variants | 127 | β-sheet |
| Haemodialysis-related amyloidosis | β2-microglobulin | 99 | β-sheet |
| Lysozyme amyloidosis | Lysozyme variants | 130 | α-helical and β-sheet |
| Finnish hereditary amyloidosis | Fragments of gelsolin variants | 53 or 71 | IDP |
| Non-Neuropathic Localized Amyloidosis | |||
| Type II diabetes | Islet amyloid polypeptide | 37 | IDP |
| Injection-localized amyloidosis | Insulin | 21 and 30 | α-helical |
| Gelatinous drop-like corneal dystrophy | Lactoferrin | 691 | α-helical and β-sheet |
| Medullary carcinoma of the thyroid | Calcitonin | 32 | IDP |
| Localized cutaneous amyloidosis | Galectin 7 | 136 | β-sheet |
| Atrial amyloidosis | Atrial natriuretic factor | 28 | IDP |
| Cataracts | γ-crystallins | variable | β-sheet |
IDP: Intrinsically disordered protein.
Table 2.
Available online methods to predict amyloidogenic regions in amino acid sequences.
Table 3.
The pros and cons of the main methods that inform on amyloid structure at atomic level resolution.
Table 3.
The pros and cons of the main methods that inform on amyloid structure at atomic level resolution.
| Technique | Advances | Advantages | Disadvantages | References |
|---|---|---|---|---|
| X-ray and electron diffraction | 1. Discovery of short protein segments that can themselves form amyloid fibrils and closely related crystals; 2. Development of synchrotron X-ray microbeams sufficiently focused and intense to determine a structure from a single crystal. | 1. May yield atomic resolution; 2. Is not limited by the molecular weight of the specimen. | 1. Well-ordered microcrystals needed; 2. The fibrils formed by some segments may represent the spines of polymorphs of full fibrils, but others may not; 3. The crystallized segment is only a few residues in length, thus nothing is revealed about the fibril structure outside the spine; 4. The steric zippers structures only show homo-steric zippers. | [23,60,62,126] |
| ssNMR | 1. Innovations in high-field magnets, pulse sequences, high-resolution multi-channel magic-angle spinning (MAS) probes, ultrafast MAS, isotopic labeling schemes, use of quadrupolar nuclei as spectroscopic probes and solid-state dynamic nuclear polarization (DNP). | 1. No need for crystals; 2. Structural information obtained on: identity of residues, recognition of parallel versus antiparallel β-sheets, register of strands within a sheet, and inter-residue contacts of amino acid side chains; 3. ssNMR-determined models show the overall conformation of the well-ordered portion of the chain around the protofilament spine; 4. Can be used to determine dihedral angles and inter-atom distances in the fibril subunits. | 1. Amyloid-forming proteins are expressed recombinantly from media containing isotopically labeled amino acids; 2. Reliability of molecular models is highly dependent on the number of experimental constraints that have been collected; 3. The relative positions of atoms are not as accurately determined as in an atomic-resolution crystal structure; 4. The sensitivity of the experiments and spectral resolution decrease with the increase in molecular weight. | [127,129,130] |
| cryo-EM | 1. Introduction of high-field microscopes; 2. New generation of direct detectors record the incident electrons in a thin, sensitive layer so that the signal is not scattered into surrounding pixels resulting in an improvement in image processing. | 1. Near atomic-resolution structures of large molecular complexes without the need for crystals; 2. May yield the overall fibril structure: the number of protofilaments; the degree of twist; and, depending on the number of well-ordered specimens, information on the atomic structure of the fibril. | 1. Due to a lack of contrast, images often have a very low signal-to-noise ratio, requiring highly advanced detection hardware and image processing; 2. Sample preparation can be difficult, not only to optimize thickness, but also to optimize particle distribution; 3. The most advanced cryo-EM equipment is very expensive. | [131,132,133] |
Table 4.
Early events that may lead to amyloid fibril formation.
| Nature of Monomeric Species | Description | References |
| Native monomer | Normally folded proteins may retain a substantial tendency to aggregate through direct assembly of monomers in their native state when the native state exposes complementary surfaces. | [153,154,155,156] |
| Conformationally altered monomer | The native monomer has very low propensity to associate. Partial unfolding or conformational changes of the native monomer are required, resulting in a non-native species prone to aggregate. | [157,158,159,160,161,162] |
| Chemically modified monomer | Chemical modifications (deamidation, isomerization, hydrolysis, oxidation, photolysis, etc.) may cause conformational changes in native monomers, leading to species with high propensity to aggregate. | [163,164,165,166,167,168,169,170,171,172,173,174] |
| Nature of Aggregation Interfaces | Description | References |
| Gas-liquid interface | Hydrophobic–hydrophilic interfaces may induce aggregation reactions. | [175,176,177,178,179,180,181] |
| Mechanical stress (agitation, stirring, pumping, or shaking) has been associated with cavitation which generates air bubbles and, consequently, the formation of an air-water interface which facilitates protein denaturation and aggregation. | [176,182,183,184,185,186,187,188,189] | |
| The use of beads during agitation accelerates the aggregation process by enhancing cavitation. | [190] | |
| Solid-liquid interface | Solid-liquid interfaces may facilitate monomer encounters and initial monomer to monomer association and later further aggregation. | |
| In vitro, interaction with glass, silicone, graphite, polypropylene, Teflon, mica, gold, etc. might lead to protein partial unfolding and aggregation. | [181,191,192,193,194] | |
| In vitro and in vivo, flow through tubes and vessels produce shear forces that may lead to protein partial unfolding and aggregation. | [195] | |
| Freeze-thaw cycles create new ice-water interfaces which may induce protein partial unfolding and aggregation. | [189,191,196] | |
| Presence of metal ions, in particular, Cu2+ and Zn2+, may promote aggregation of protein monomers bearing metal-ion binding sites or binding residues (e.g., histidines). | [197,198,199,200,201] | |
| Monomer association at the surface of biomembranes or biomolecules may also enhance aggregation. | [202,203,204,205,206,207,208,209,210] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Almeida, Z.L.; Brito, R.M.M. Structure and Aggregation Mechanisms in Amyloids. Molecules 2020, 25, 1195. https://doi.org/10.3390/molecules25051195
AMA Style
Almeida ZL, Brito RMM. Structure and Aggregation Mechanisms in Amyloids. Molecules. 2020; 25(5):1195. https://doi.org/10.3390/molecules25051195
Chicago/Turabian StyleAlmeida, Zaida L., and Rui M. M. Brito. 2020. "Structure and Aggregation Mechanisms in Amyloids" Molecules 25, no. 5: 1195. https://doi.org/10.3390/molecules25051195