Structural Optimization and Structure–Activity Relationship of 4-Thiazolidinone Derivatives as Novel Inhibitors of Human Dihydroorotate Dehydrogenase


Abstract
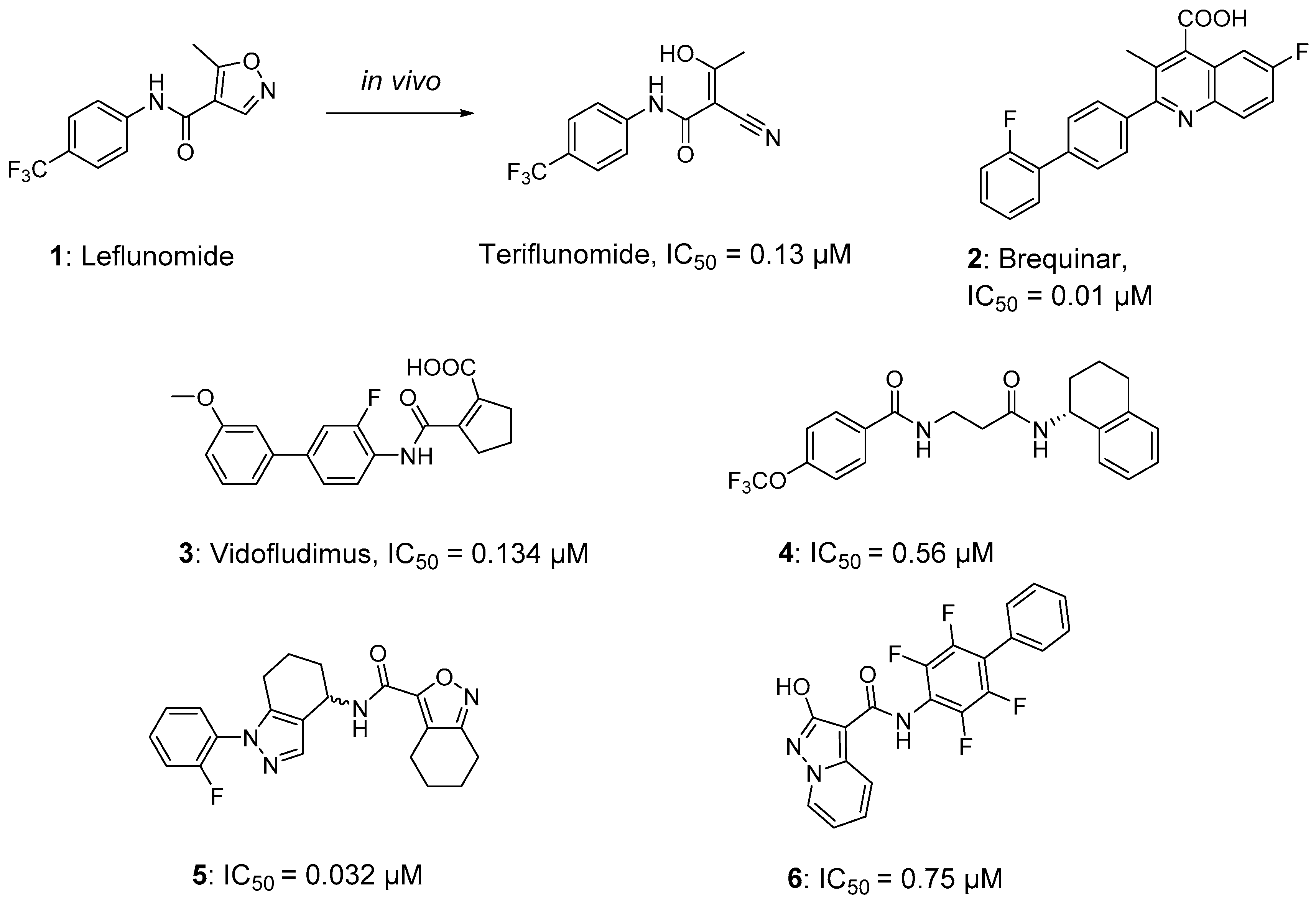
:1. Introduction
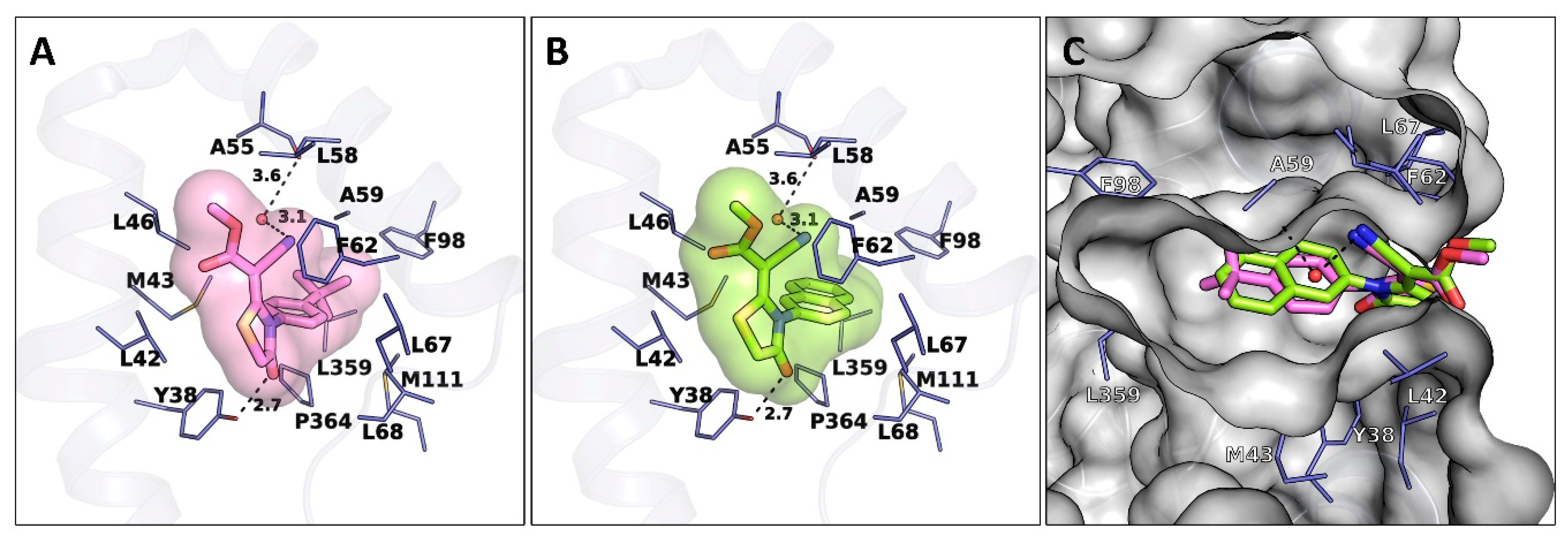
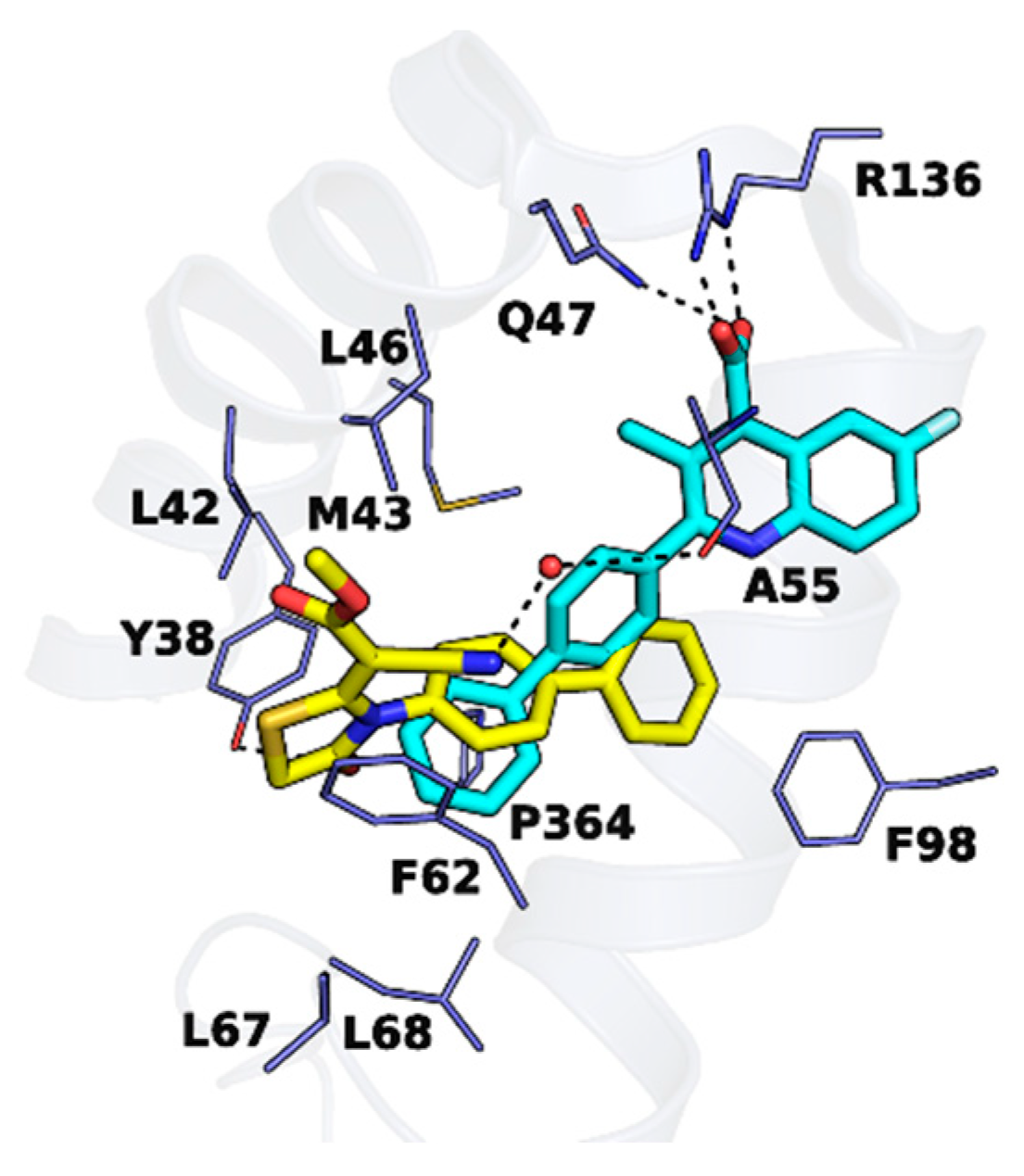
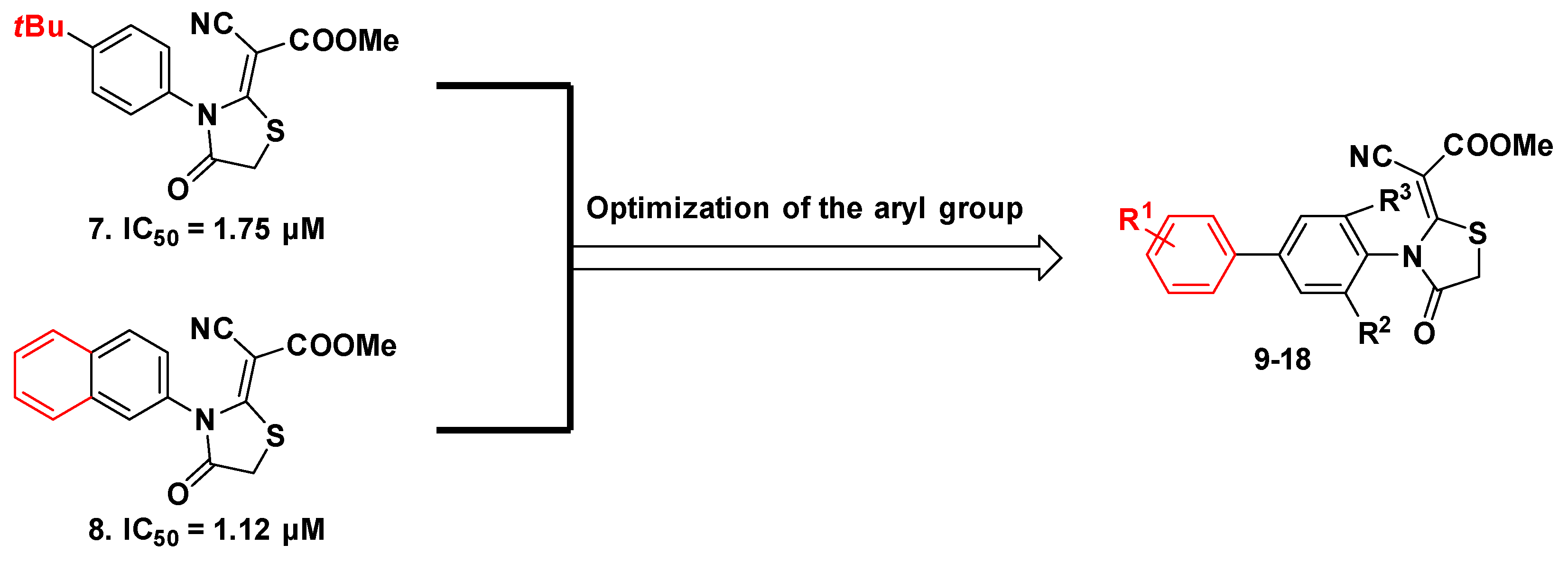
2. Results and Discussion
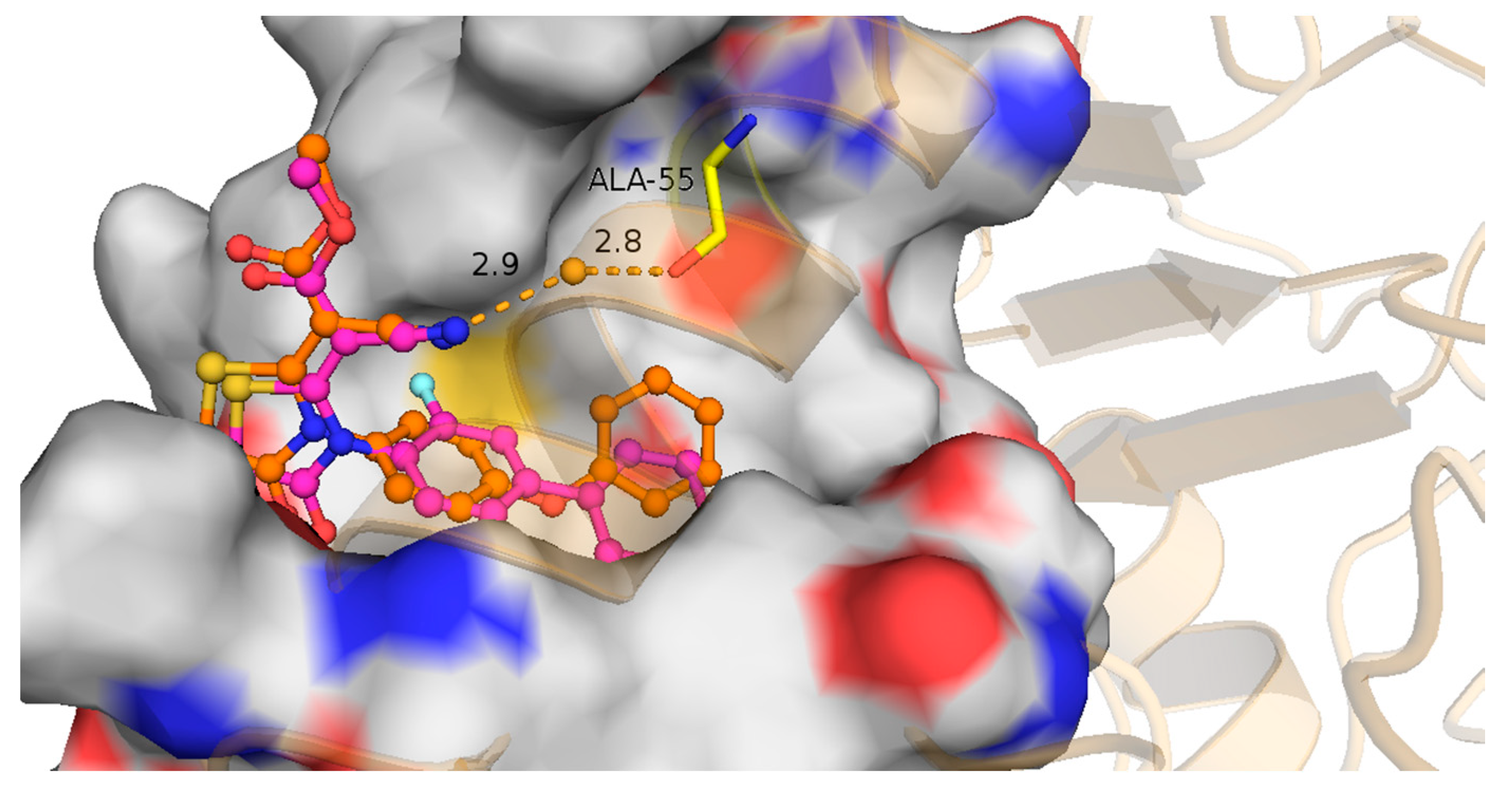
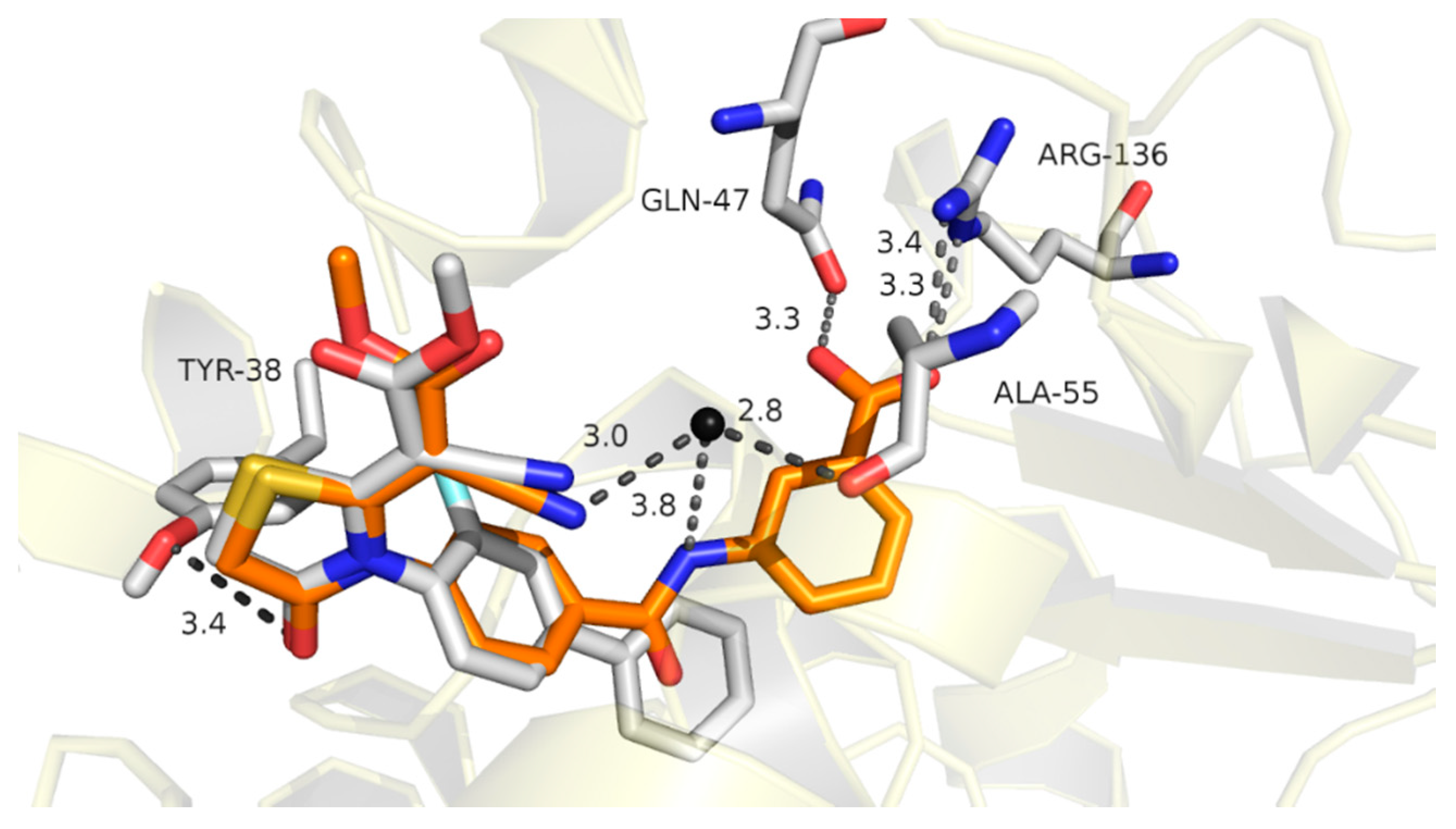
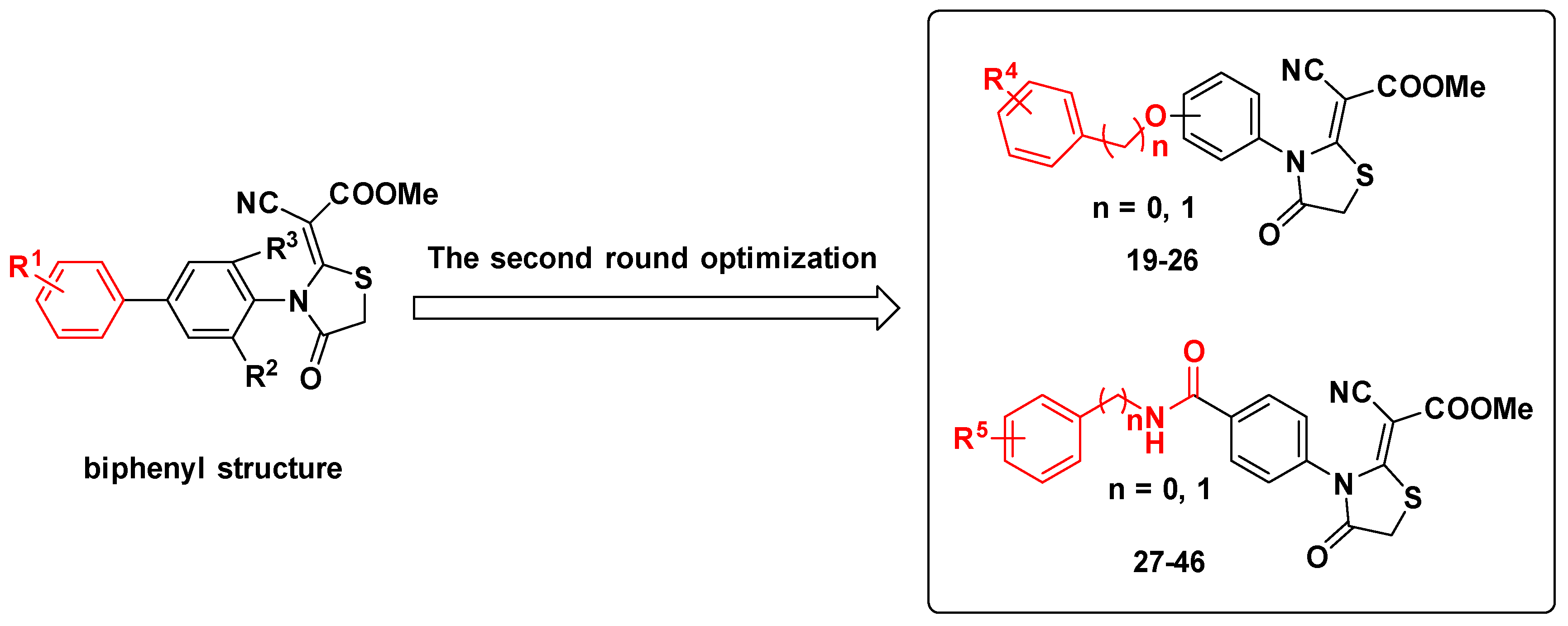
2.1. Molecular Design Strategies
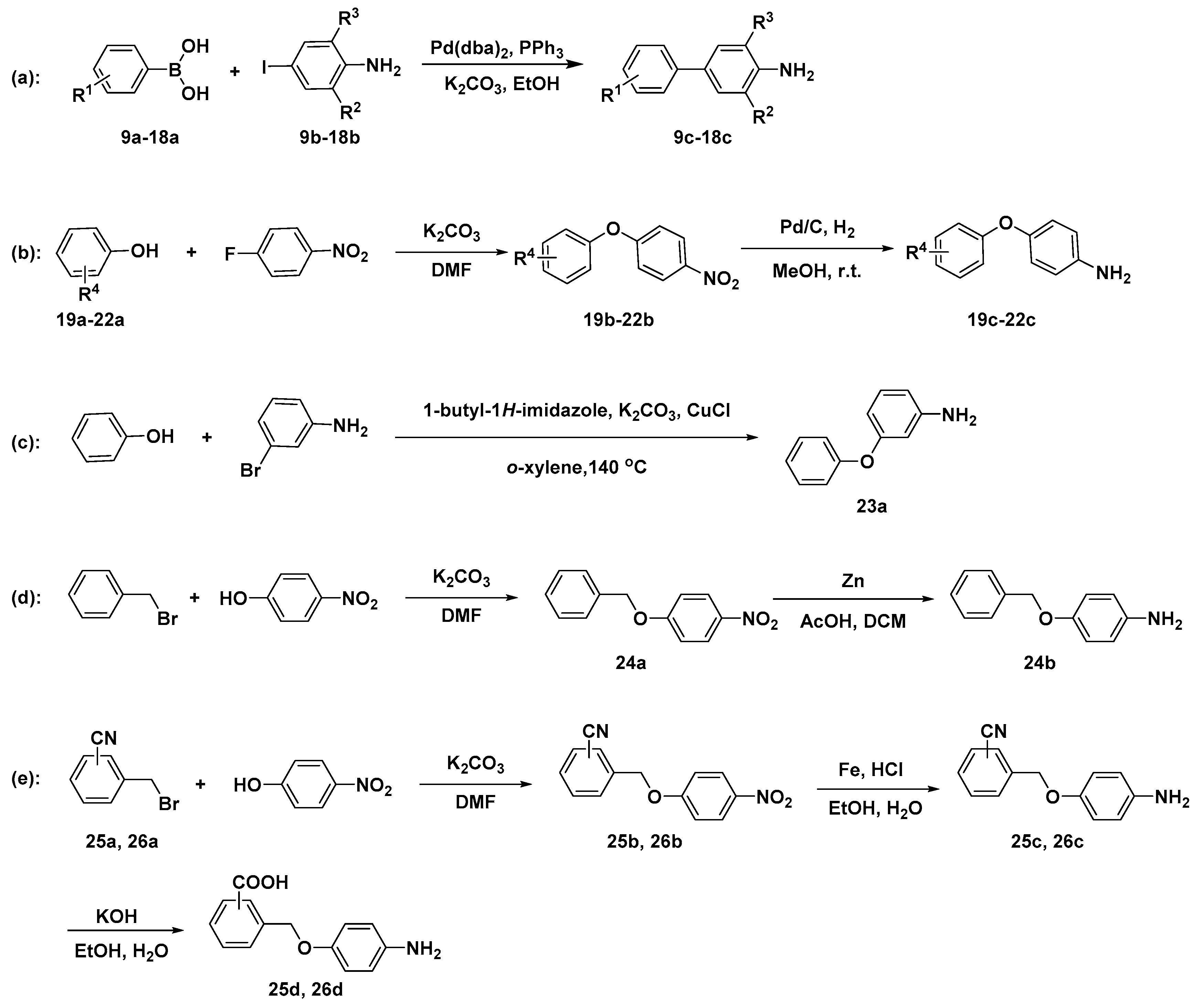
2.2. Chemistry
2.3. Inhibitory Activities against hDHODH and SAR Study
3. Materials and Methods
3.1. General Information
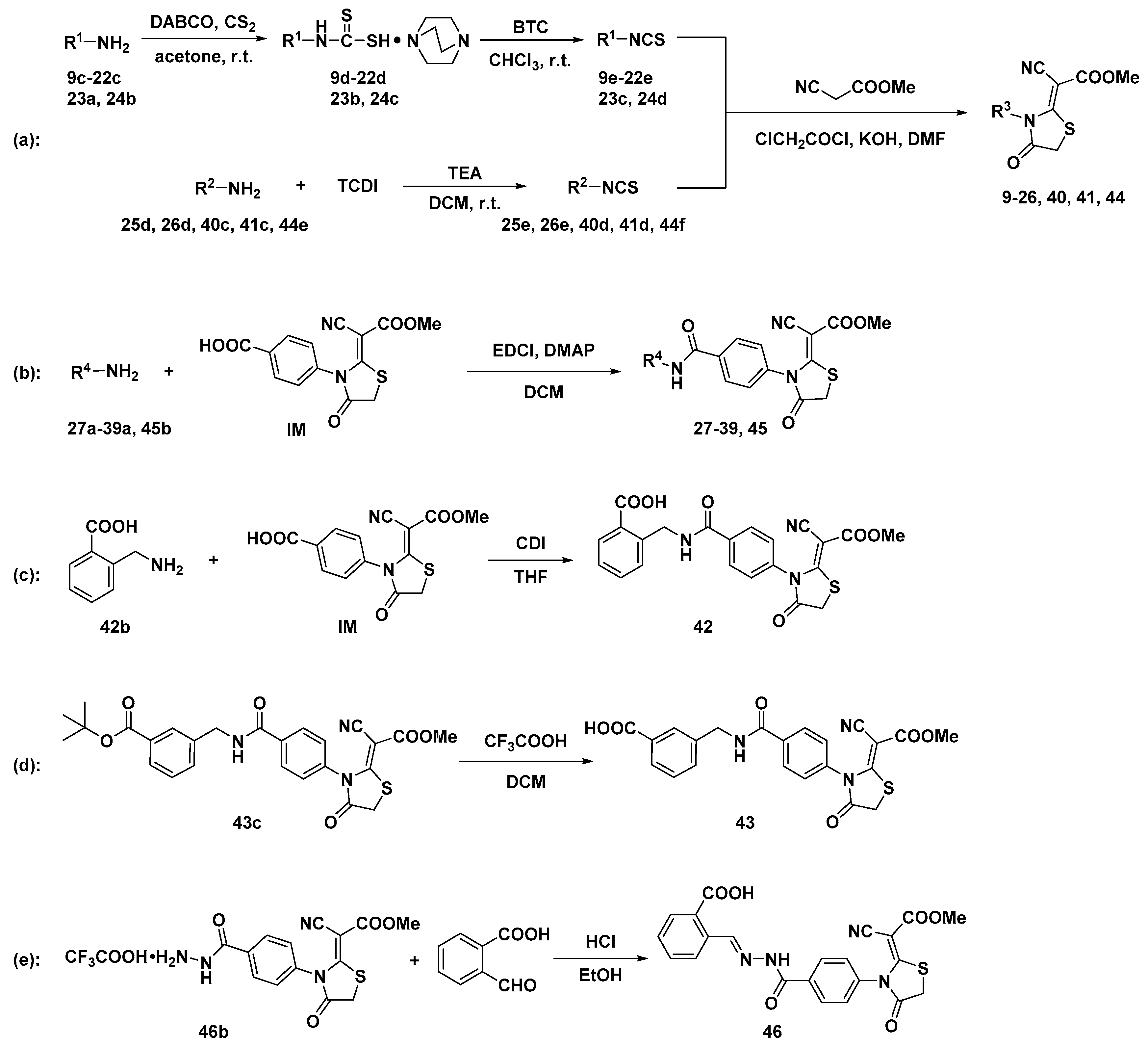
3.2. Chemistry
3.2.1. Synthesis of Key Intermediates of Compounds 9–26.
General Procedure for the Synthesis of Intermediates 9c–18c
General Procedure for the Synthesis of Intermediates 19b–22b
General Procedure for the Synthesis of Intermediates 19c–22c
Synthesis of Intermediate 23a
Synthesis of Intermediate 24a
Synthesis of Intermediate 24b
General Procedure for the Synthesis of Intermediates 25b and 26b
General Procedure for Synthesis of Intermediates 25c and 26c
General Procedure for Synthesis of Intermediates 25d and 26d
Synthesis of Intermediate IM
General Procedure for the Synthesis of Intermediates 40b and 41b
General Procedure for the Synthesis of Intermediates 40c and 41c
Synthesis of Intermediate 42a
Synthesis of Intermediate 42b
Synthesis of Intermediate 43a
Synthesis of Intermediate 43b
Synthesis of Intermediate 43c
Synthesis of Intermediate 44a
Synthesis of Intermediate 44b
Synthesis of Intermediate 44c
Synthesis of Intermediate 44d
Synthesis of Intermediate 44e
Synthesis of Intermediate 45a
Synthesis of Intermediates 45b
Synthesis of Intermediates 46a
Synthesis of Intermediate 46b
Synthesis of Aryl Isothiocyanates 9e–22e, 23c, 24d
Synthesis of Aryl Isothiocyanates 25e, 26e, 40d, 41d, 44f
3.2.2. Synthesis of Compounds 9–26, 40, 41, 44
3.2.3. Synthesis of Compounds 27–39, 45
3.2.4. Synthesis of Compounds 42, 43 and 46
Synthesis of (Z)-2-((4-(2-(1-cyano-2-methoxy-2-oxoethylidene)-4-oxothiazolidin-3-yl)benzamido)methyl)-benzoic acid (42)
Synthesis of (Z)-3-((4-(2-(1-cyano-2-methoxy-2-oxoethylidene)-4-oxothiazolidin-3-yl)benzamido)methyl) benzoic acid (43)
Synthesis of 2-(((E)-(4-((Z)-2-(1-cyano-2-methoxy-2-oxoethylidene)-4-oxothiazolidin-3-yl)benzamido)- methylene)amino)benzoic acid (46)
3.3. In Vitro Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Surekha, K.; Prabhu, D.; Richard, M.; Nachiappan, M.; Biswal, J.; Jeyakanthan, J. Investigation of vital pathogenic target orotate phosphoribosyltransferases (OPRTase) from Thermus thermophilus HB8: Phylogenetic and molecular modeling approach. Gene 2016, 583, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Roon, D.E.N.V.; Jansen, T.L.T.A.; Houtman, N.M.; Spoelstra, P.; Brouwers, J.R.B.J. Leflunomide for the Treatment of Rheumatoid Arthritis in Clinical Practice. Am. J. Med. Sci. 2002, 323, 190–193. [Google Scholar]
- Liu, S.; Neidhardt, E.A.; Grossman, T.H.; Ocain, T.; Clardy, J. Structures of human dihydroorotate dehydrogenase in complex with antiproliferative agents. Structure 2000, 8, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Jöckel, J.; Wendt, B.; Löffler, M. Structural and functional comparison of agents interfering with dihydroorotate, succinate and NADH oxidation of rat liver mitochondria. Biochem. Pharmacol. 1998, 56, 1053–1060. [Google Scholar] [CrossRef]
- Breedveld, F.C.; Dayer, J.M. Leflunomide: Mode of action in the treatment of rheumatoid arthritis. Ann. Rheum. Dis. 2000, 59, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Löffler, M.; Fairbanks, L.D.; Zameitat, E.; Marinaki, A.M.; Simmonds, H.A. Pyrimidine pathways in health and disease. Trends Mol. Med. 2005, 11, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Rusai, K.; Schmaderer, C.; Baumann, M.; Chmielewski, S.; Prókai, Á.; Kis, E.; Szabó, A.J.; Leban, J.; Doblhofer, R.; Ammendola, A. Immunosuppression with 4SC-101, a novel inhibitor of dihydroorotate dehydrogenase, in a rat model of renal transplantation. Transplantation 2012, 93, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Vyas, V.K.; Ghate, M. Recent developments in the medicinal chemistry and therapeutic potential of dihydroorotate dehydrogenase (DHODH) inhibitors. Mini Rev. Med. Chem. 2011, 11, 1039–1055. [Google Scholar] [CrossRef]
- Sykes, D.B.; Kfoury, Y.S.; Mercier, F.E.; Wawer, M.J.; Law, J.M.; Haynes, M.K.; Lewis, T.A.; Schajnovitz, A.; Jain, E.; Lee, D. Inhibition of Dihydroorotate Dehydrogenase Overcomes Differentiation Blockade in Acute Myeloid Leukemia. Cell 2016, 167, 171–186. [Google Scholar] [CrossRef]
- Dexter, D.L.; Hesson, D.P.; Ardecky, R.J.; Rao, G.V.; Tippett, D.L.; Dusak, B.A.; Paull, K.D.; Plowman, J.; DeLarco, B.M.; Narayanan, V. Activity of a novel 4-quinolinecarboxylic acid, NSC 368390 [6-fluoro-2-(2′-fluoro-1, 1′-biphenyl-4-yl)-3-methyl-4-quinolinecarboxylic acid sodium salt], against experimental tumors. Cancer Res. 1985, 45, 5563–5568. [Google Scholar]
- Cramer, D.V.; Chapman, F.A.; Jaffee, B.D.; Jones, E.A.; Knoop, M.; Hreha-Eiras, G.; Makowka, L. The effect of a new immunosuppressive drug, brequinar sodium, on heart, liver, and kidney allograft rejection in the rat. Transplantation 1992, 53, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.L.; Wolfe, F.; Huizinga, T.W.J. Rheumatoid arthritis. Lancet 2010, 376, 1094–1108. [Google Scholar] [CrossRef]
- Dang, W.; Wang, W.; Chen, W.; Lian, F.; Li, L.; Ying, H.; Xu, Y.; Zhang, N.; Chen, Y.; Liu, M. Pharmacologic inhibition of dihydroorotate dehydrogenase induces apoptosis and differentiation in acute myeloid leukemia cells. Haematologica 2018, 103, 1472–1483. [Google Scholar]
- Sainas, S.; Pippione, A.C.; Lupino, E.; Giorgis, M.; Circosta, P.; Gaidano, V.; Goyal, P.; Bonanni, D.; Rolando, B.; Cignetti, A. Targeting Myeloid Differentiation Using Potent 2-Hydroxypyrazolo [1,5-a]pyridine Scaffold-Based Human Dihydroorotate Dehydrogenase Inhibitors. J. Med. Chem. 2018, 61, 6034–6055. [Google Scholar] [CrossRef] [PubMed]
- Munierlehmann, H.; Vidalain, P.O.; Tangy, F.; Janin, Y.L. On Dihydroorotate Dehydrogenases and Their Inhibitors and Uses. J. Med. Chem. 2013, 56, 3148–3167. [Google Scholar] [CrossRef] [PubMed]
- Finckh, A.; Dehler, S.; Gabay, C. The effectiveness of leflunomide as a co-therapy of tumour necrosis factor inhibitors in rheumatoid arthritis: A population-based study. Ann. Rheum. Dis. 2009, 68, 33. [Google Scholar] [CrossRef] [PubMed]
- Rückemann, K.; Fairbanks, L.D.; Carrey, E.A.; Hawrylowicz, C.M.; Richards, D.F.; Kirschbaum, B.; Simmonds, H.A. Leflunomide inhibits pyrimidine de novo synthesis in mitogen-stimulated T-lymphocytes from healthy humans. J. Biol. Chem. 1998, 273, 21682–21691. [Google Scholar] [CrossRef] [PubMed]
- Christian, C.; Li, D.K.; Freedman, M.S.; Philippe, T.; Hadj, B.; Dazhe, W.; Amit, B.O.; Traboulsee, A.L.; Reiman, L.E.; O’Connor, P.W. Long-term follow-up of a phase 2 study of oral teriflunomide in relapsing multiple sclerosis: Safety and efficacy results up to 8.5 years. Mult. Scler. 2012, 18, 1278–1289. [Google Scholar]
- Confavreux, C.; O’Connor, P.; Comi, G.; Freedman, M.S.; Miller, A.E.; Olsson, T.P.; Wolinsky, J.S.; Bagulho, T.; Delhay, J.L.; Dukovic, D. Oral teriflunomide for patients with relapsing multiple sclerosis (TOWER): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. 2014, 13, 247–256. [Google Scholar] [CrossRef]
- Claussen, M.C.; Korn, T. Immune mechanisms of new therapeutic strategies in MS: Teriflunomide. Clin. Immunol. 2012, 142, 49–56. [Google Scholar] [CrossRef]
- Maroun, J.; Ruckdeschel, J.; Natale, R.; Morgan, R.; Dallaire, B.; Sisk, R.; Gyves, J. Multicenter phase II study of brequinar sodium in patients with advanced lung cancer. Cancer Chemother. Pharmacol. 1993, 32, 64–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodion, P.F.; Wagener, T.; Stoter, G.; Drozd, A.; Lev, L.M.; Skovsgaard, T.; Renard, J.; Cavalli, F. Phase II trial with Brequinar (DUP-785, NSC 368390) in patients with metastatic colorectal cancer: A study of the Early Clinical Trials Group of the EORTC. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 1990, 1, 79. [Google Scholar] [CrossRef]
- Cramer, D.V.; Chapman, F.A.; Makowka, L. The use of brequinar sodium for transplantation. Ann. N. Y. Acad. Sci. 2010, 696, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Min, Q.; Gang, Z.; Wang, Q.Y.; Hao, Y.X.; Dong, H.; Yuan, Z.; Shi, P.Y. Characterization of Dengue Virus Resistance to Brequinar in Cell Culture. Antimicrob. Agents Chemother. 2010, 54, 3686–3695. [Google Scholar] [Green Version]
- Schnellrath, L.C.; Damaso, C.R. Potent antiviral activity of brequinar against the emerging Cantagalo virus in cell culture. Int. J. Antimicrob. Agents 2011, 38, 435–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrlinger, K.R.; Diculescu, M.; Fellermann, K.; Hartmann, H.; Howaldt, S.; Nikolov, R.; Petrov, A.; Reindl, W.; Otte, J.M.; Stoynov, S. Efficacy, safety and tolerability of vidofludimus in patients with inflammatory bowel disease: The ENTRANCE study. Gastroenterology 2013, 7, 636–643. [Google Scholar] [Green Version]
- Fitzpatrick, L.R.; Ammendola, A. Vidofludimus Improves Colitis in Rats by a Dual Mode of Action. J. Pharmacol. Exp. Ther. 2012, 342, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, L.R.; Small, J.S.; Doblhofer, R.; Henning, S.W.; Ammendola, A. P012 Vidofludimus inhibits IL-17 and improves hapten-induced colitis in young rats by a unique dual mode of action. J. Pharmacol. Exp. Ther. 2012, 142, 850–860. [Google Scholar] [CrossRef]
- Lewis, T.A.; Sykes, D.B.; Law, J.M.; Muñoz, B.; Rustiguel, J.K.; Nonato, M.C.; Scadden, D.T.; Schreiber, S.L. Development of ML390: A Human DHODH Inhibitor That Induces Differentiation in Acute Myeloid Leukemia. ACS Med. Chem. Lett. 2016, 7, 1112. [Google Scholar] [CrossRef]
- Mjgw, L.; Imm, V.L.; Drummond, C.J.; Chu, S.; Healy, A.R.; Popova, G.; Pastor, F.A.; Mollick, T.; Darekar, S.; Sedimbi, S.K. A DHODH inhibitor increases p53 synthesis and enhances tumor cell killing by p53 degradation blockage. Nat. Commun. 2018, 9, 1107. [Google Scholar]
- Zeng, F.; Qi, T.; Li, C.; Li, T.; Li, H.; Li, S.L.; Zhu, L.; Xu, X. Synthesis, structure-activity relationship and binding mode analysis of 4-thiazolidinone derivatives as novel inhibitors of human dihydroorotate dehydrogenase. Medchemcomm 2017, 8, 1297–1302. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Li, S.; Tong, Y.; Wang, J.; Quan, L.; Chen, Z.; Zhao, Z.; Xu, Y.; Zhu, L.; Qian, X. Structure-based design of potent human dihydroorotate dehydrogenase inhibitors as anticancer agents. Medchemcomm 2016, 7, 1441–1448. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 7–46 are available from the authors. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
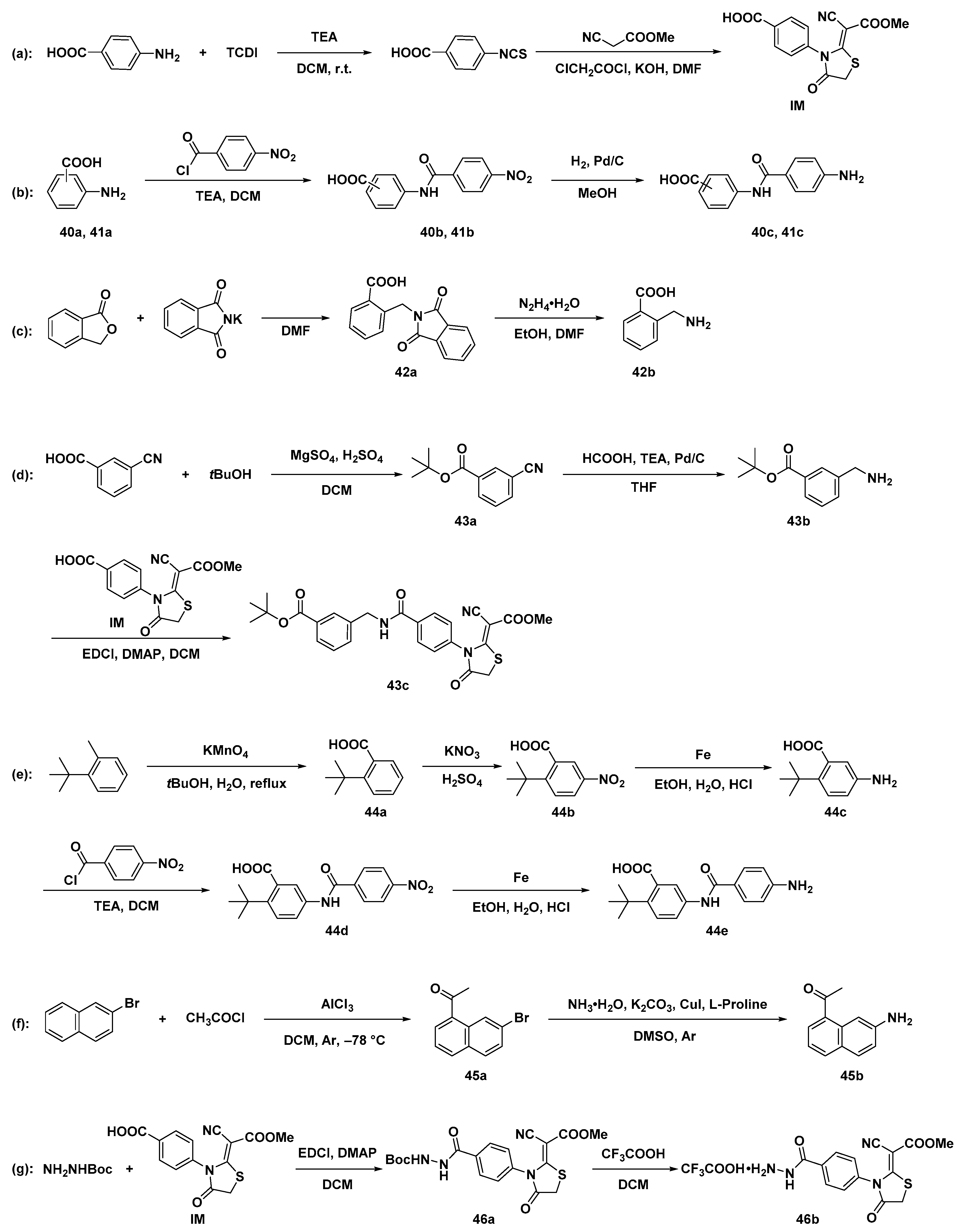{kind=link}
{kind=link}

| Compd | R | % Inhibition at 10 µM | hDHODH IC50a (μM) |
|---|---|---|---|
| 7 |  | 79.1 | 1.75 |
| 8 |  | 80.3 | 1.12 |
| 9 |  | 73.8 | 1.32 |
| 10 |  | 58.8 | 3.52 |
| 11 |  | 33.9 | >10 |
| 12 |  | 57.6 | 6.04 |
| 13 |  | 54.7 | 6.64 |
| 14 |  | 63.3 | 5.42 |
| 15 |  | 55.4 | 6.86 |
| 16 |  | 50.2 | 8.39 |
| 17 |  | 50.7 | 4.35 |
| 18 |  | 53.1 | 9.29 |
| Brequinar | 0.0084 |

| Compd | R | % Inhibition at 10 µM | hDHODH IC50 (μM) |
|---|---|---|---|
| 19 |  | 52.0 | 9.43 |
| 20 |  | 44.7 | >10 |
| 21 |  | 29.7 | >10 |
| 22 |  | 18.1 | >10 |
| 23 |  | 34.4 | >10 |
| 24 |  | 55.4 | 4.32 |
| 25 |  | 29.9 | >10 |
| 26 |  | 36.2 | >10 |
| 27 |  | 41.7 | >10 |
| 28 |  | 69.0 | 2.98 |
| 29 |  | 47.0 | >10 |
| 30 |  | 71.3 | 3.01 |
| 31 |  | 53.6 | 8.56 |
| 32 |  | 52.7 | 7.50 |
| 33 |  | 42.0 | >10 |
| 34 |  | 38.1 | >10 |
| 35 |  | 24.5 | >10 |
| 36 |  | 55.4 | 5.01 |
| 37 |  | 75.6 | 1.45 |
| 38 |  | 38.2 | >10 |
| 39 |  | 39.9 | >10 |
| 40 |  | 34.5 | >10 |
| 41 |  | 23.5 | >10 |
| 42 |  | 36.8 | >10 |
| 43 |  | 44.0 | >10 |
| 44 |  | 30.3 | >10 |
| 45 |  | 42.2 | >10 |
| 46 |  | 34.8 | >10 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, F.; Quan, L.; Yang, G.; Qi, T.; Zhang, L.; Li, S.; Li, H.; Zhu, L.; Xu, X. Structural Optimization and Structure–Activity Relationship of 4-Thiazolidinone Derivatives as Novel Inhibitors of Human Dihydroorotate Dehydrogenase. Molecules 2019, 24, 2780. https://doi.org/10.3390/molecules24152780
Zeng F, Quan L, Yang G, Qi T, Zhang L, Li S, Li H, Zhu L, Xu X. Structural Optimization and Structure–Activity Relationship of 4-Thiazolidinone Derivatives as Novel Inhibitors of Human Dihydroorotate Dehydrogenase. Molecules. 2019; 24(15):2780. https://doi.org/10.3390/molecules24152780
Chicago/Turabian StyleZeng, Fanxun, Lina Quan, Guantian Yang, Tiantian Qi, Letian Zhang, Shiliang Li, Honglin Li, Lili Zhu, and Xiaoyong Xu. 2019. "Structural Optimization and Structure–Activity Relationship of 4-Thiazolidinone Derivatives as Novel Inhibitors of Human Dihydroorotate Dehydrogenase" Molecules 24, no. 15: 2780. https://doi.org/10.3390/molecules24152780