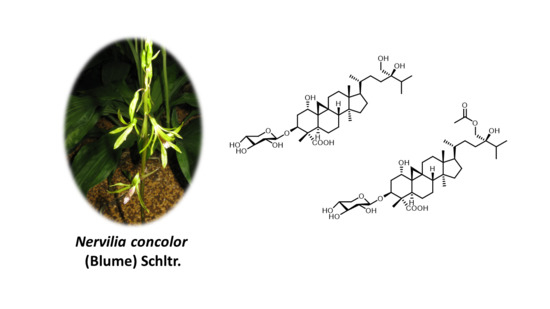
Nervisides I–J: Unconventional Side-Chain-Bearing Cycloartane Glycosides from Nervilia concolor

 , , , and
, , , and
Abstract
:
1. Introduction
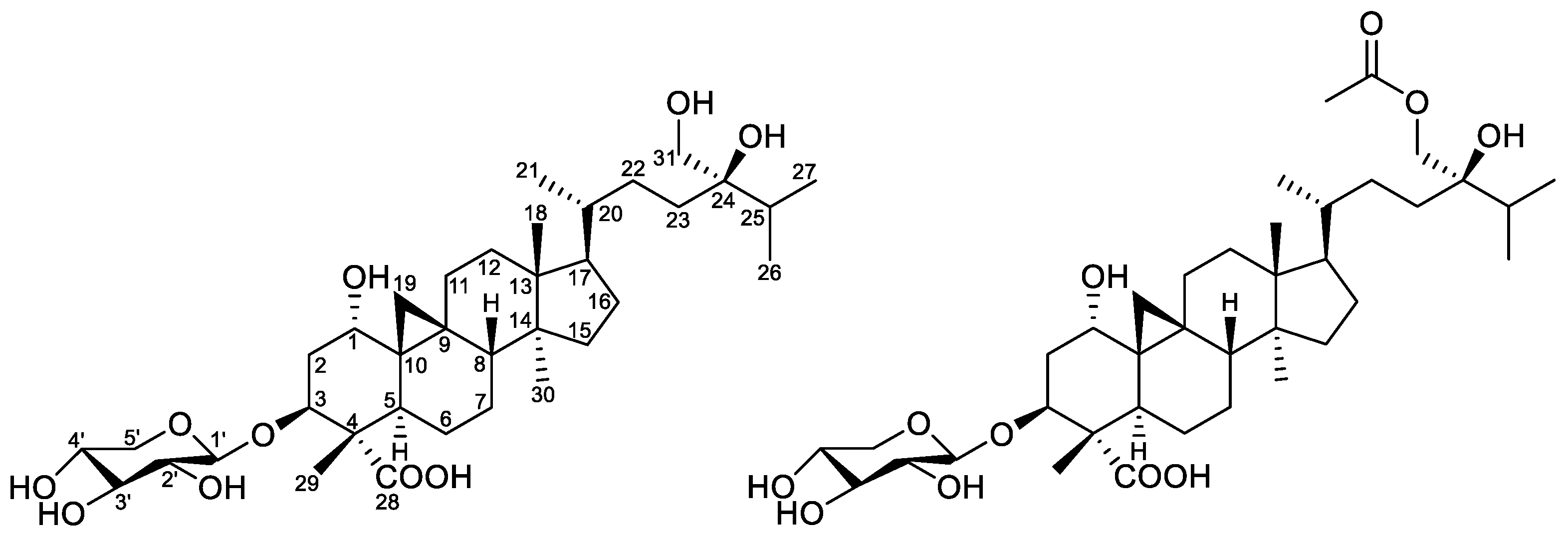
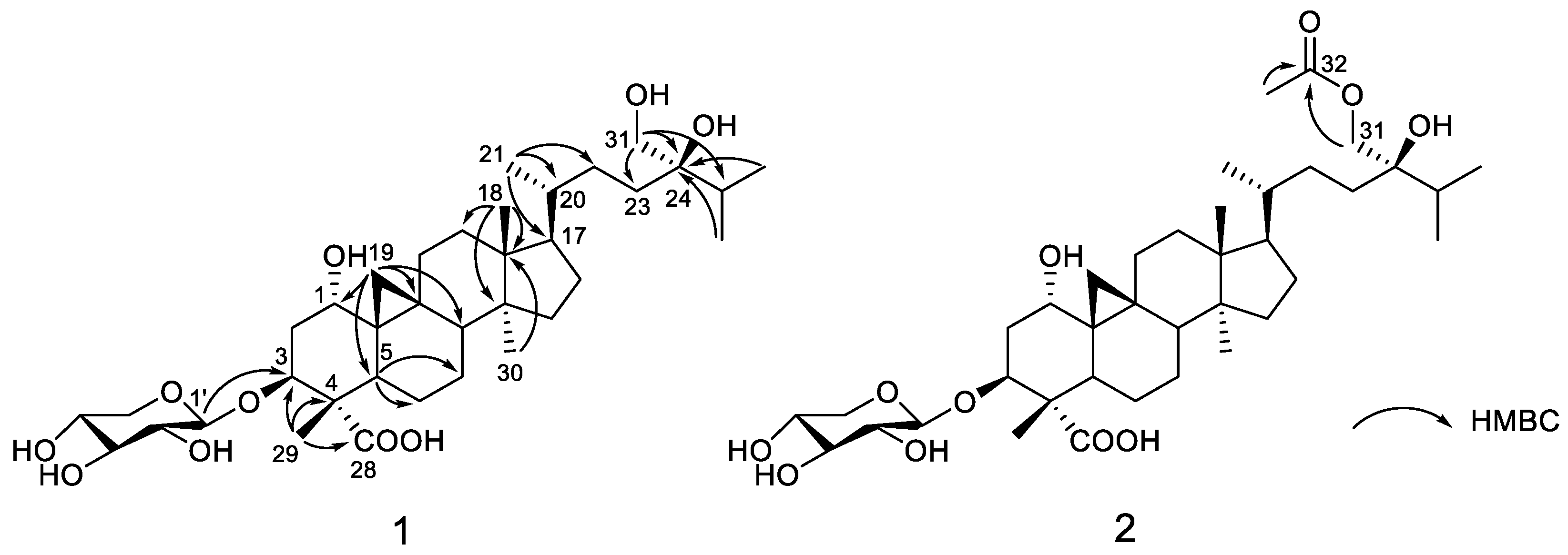
2. Results and Discussion
3. Materials and Methods
3.1. General
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Computational Chemistry
3.5. Biological Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gale, S.; Yukawa, T.; Kuroiwa, N. Studies in Asian Nervilia (Orchidaceae) I: Neotypification and Circumscription of N. nipponica in Japan. Kew Bull. 2007, 62, 85–94. [Google Scholar]
- Zhou, G.-X.; Lu, C.-L.; Wang, H.-S.; Yao, X.-S. An acetyl flavonol from Nervilia fordii (Hance) Schltr. J. Asian Nat. Prod. Res. 2009, 11, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-M.; Wei, L.-B.; Lu, C.-L.; Zhou, G.-X. A flavonoid 8- C -glycoside and a triterpenoid cinnamate from Nervilia fordii. J. Asian Nat. Prod. Res. 2013, 15, 1088–1093. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.-W.; Pei, Y.; Zhang, Y.-J.; Wang, Y.-F.; Yang, C.-R. 7-O-Methylkaempferol and quercetin Glycosides from the Whole Plant of Nervilia fordii. J. Nat. Prod. 2009, 72, 1057–1060. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhao, Z.-X.; Lin, C.-Z.; Zhu, C.-C.; Gao, L. Three new flavonol glycosides from Nervilia fordii. Phytochem. Lett. 2012, 5, 104–107. [Google Scholar] [CrossRef]
- Qiu, L.; Jiao, Y.; Xie, J.-Z.; Huang, G.-K.; Qiu, S.-L.; Miao, J.-H.; Yao, X.-S. Five new flavonoid glycosides from Nervilia fordii. J. Asian Nat. Prod. Res. 2013, 15, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.-B.; Chen, J.-M.; Ye, W.-C.; Yao, X.-S.; Zhou, G.-X. Three new cycloartane glycosides from Nervilia fordii. J. Asian Nat. Prod. Res. 2012, 14, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.-B.; Yuan, H.-E.; Chen, J.-M.; Wang, Q.-Q.; Wang, H.; Ye, W.-C.; Zhou, G.-X. Five new Cycloartane-type triterpenoid saponins from Nervilia fordii. Helv. Chim. Acta 2013, 96, 150–157. [Google Scholar] [CrossRef]
- Yoshikawa, K.; Katsuta, S.; Mizumori, J.; Arihara, S. New cycloartane triterpenoids from Passiflora edulis. J. Nat. Prod. 2000, 63, 1377–1380. [Google Scholar] [CrossRef]
- Yoshikawa, K.; Katsuta, S.; Mizumori, J.; Arihara, S. Four cycloartane triterpenoids and six related saponins from Passiflora edulis. J. Nat. Prod. 2000, 63, 1229–1234. [Google Scholar] [CrossRef]
- Xu, F.-Q.; Wang, N.; Fan, W.-W.; Zi, C.-T.; Zhao, H.-S.; Hu, J.-M.; Zhou, J. Protective effects of cycloartane triterpenoids from Passiflora edulis Sims against glutamate-induced neurotoxicity in PC12 cell. Fitoterapia 2016, 115, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Della Greca, M.; Fiorention, A.; Monaco, P.; Previtera, L. Cycloartane triterpenes from Juncus effusus. Phytochemistry 1994, 35, 1017–1022. [Google Scholar] [CrossRef]
- Linnek, J.; Mitaine-Offer, A.-C.; Miyamoto, T.; Tanaka, C.; Paululat, T.; Avunduk, S.; Alankuş-Çalişkan, Ö.; Lacaille-Dubois, M.-A. Cycloartane glycosides from three species of Astragalus (Fabaceae). Helv. Chim. Acta 2011, 94, 230–237. [Google Scholar] [CrossRef]
- Imai, A.; Lankin, D.C.; Nikolić, D.; Ahn, S.; van Breemen, R.B.; Farnsworth, N.R.; McAlpine, J.B.; Chen, S.-N.; Pauli, G.F. Cycloartane triterpenes from the aerial parts of Actaea racemosa. J. Nat. Prod. 2016, 79, 541–554. [Google Scholar] [CrossRef] [PubMed]
- Banskota, A.H.; Tezuka, Y.; Tran, K.Q.; Tanaka, K.; Saiki, I.; Kadota, S. Thirteen novel cycloartane-type triterpenes from Combretum quadrangulare. J. Nat. Prod. 2000, 63, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Lugo, M.-T.; Singh, M.P.; Maiese, W.M.; Timmermann, B.N. New antimicrobial cycloartane triterpenes from Acalypha communis. J. Nat. Prod. 2002, 65, 872–875. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.A. A diterpene xyloside from Conyza steudellii. Phytochemistry 1991, 30, 611–612. [Google Scholar] [CrossRef]
- Piacente, S.; Balderrama, L.; Tommasi, N.D.; Morales, L.; Vargas, L.; Pizza, C. Anadanthoside: A flavanol-3-O-b-d-xylopyranoside from Anadenanthera macrocarpa. Phytochemistry 1999, 51, 709–711. [Google Scholar] [CrossRef]
- Ali, Z.; Khan, S.; Khan, I. Phytochemical study of Actaea rubra and biological screenings of isolates. Planta Med. 2006, 72, 1350–1352. [Google Scholar] [CrossRef] [PubMed]
- Thimmappa, R.; Geisler, K.; Louveau, T.; O’Maille, P.; Osbourn, A. Triterpene biosynthesis in plants. Annu. Rev. Plant Biol. 2014, 65, 225–257. [Google Scholar] [CrossRef]
- Escobedo-Martínez, C.; Concepción Lozada, M.; Hernández-Ortega, S.; Villarreal, M.L.; Gnecco, D.; Enríquez, R.G.; Reynolds, W. 1H and 13C NMR characterization of new cycloartane triterpenes from Mangifera indica. Magn. Reson. Chem. 2012, 50, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, T.; Kadota, S.; Suehara, H.; Namba, T. Occurrence of non-conventional side chain sterols in an orchidaceous plant, Nervilia purpurea Schlechter and structure of nervisterol. Chem. Pharm. Bull. 1982, 30, 370–373. [Google Scholar] [CrossRef]
- Kadota, S.; Shima, T.; Kikuchi, T. Studies on the constituents of orchidaceous plants. VII. The C-24 stereochemistry of cyclohomonervilol and 24-isopropenylcholesterol non-conventional side chain triterpene and sterol from Nervilia purpurea Schlechter. Chem. Pharm. Bull. 1987, 35, 11. [Google Scholar] [CrossRef]
- Thompson, M.J.; Patterson, G.W.; Dutky, S.R.; Svoboda, J.A.; Kaplanis, J.N. Techniques for the isolation and identification of steroids in insects and algae. Lipids 1980, 15, 719–733. [Google Scholar] [CrossRef] [PubMed]
- Ikekawa, N.; Fujimoto, Y.; Kadota, S.; Kikuchi, T. Effective separation of sterol C-24 epimers. J. Chromatogr. A 1989, 468, 91–98. [Google Scholar] [CrossRef]
- Kasai, R.; Sasaki, A.; Hashimoto, T.; Kaneko, T.; Ohtani, K.; Yamasaki, K. Cycloartane glycosides from Trichosanthes tricuspidata. Phytochemistry 1999, 51, 803–808. [Google Scholar] [CrossRef]
- Wang, W.; Jang, H.; Hong, J.; Lee, C.-O.; Im, K.S.; Bae, S.-J.; Jung, J.H. Additional cytotoxic sterols and saponins from the starfish Certonardoa semiregularis. J. Nat. Prod. 2004, 67, 1654–1660. [Google Scholar] [CrossRef]
- Knight, S.A. Carbon-13 NMR spectra of some tetra- and pentacyclic triterpenoids. Org. Magn. Reson. 1974, 6, 603–611. [Google Scholar] [CrossRef]
- Wright, J.L.C.; McInnes, A.G.; Shimizu, S.; Smith, D.G.; Walter, J.A.; Idler, D.; Khalil, W. Identification of C-24 alkyl epimers of marine sterols by 13C nuclear magnetic resonance spectroscopy. Can. J. Chem. 1978, 56, 1898–1903. [Google Scholar]
- Herz, W.; Watanabe, K.; Kulanthaivel, P.; Blount, J.F. Cycloartanes from Lindheimera texana. Phytochemistry 1985, 24, 2645–2654. [Google Scholar] [CrossRef]
- Ju, J.; Liu, D.; Lin, G.; Zhang, Y.; Yang, J.; Lu, Y.; Gong, N.; Zheng, Q. Beesiosides G, H, and J−N, seven new cycloartane triterpene glycosides from Beesia calthifolia. J. Nat. Prod. 2002, 65, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Li, X.-C.; Smillie, T.J.; Carvalho, P.; Mabusela, W.; Syce, J.; Johnson, Q.; Folk, W.; Avery, M.A.; Khan, I.A. Cycloartane glycosides from Sutherlandia frutescens. J. Nat. Prod. 2008, 71, 1749–1753. [Google Scholar] [CrossRef] [PubMed]
- Kolossváry, I.; Guida, W.C. Low-mode conformational search elucidated: Application to C39H80 and flexible docking of 9-deazaguanine inhibitors into PNP. J. Comput. Chem. 1999, 20, 14. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1995, 17, 490–519. [Google Scholar] [CrossRef]
- Kundrot, C.E.; Ponder, J.W.; Richards, F.M. Algorithms for calculating excluded volume and its derivatives as a function of molecular conformation and their use in energy minimization. J. Comput. Chem. 1991, 12, 402–409. [Google Scholar] [CrossRef] [Green Version]
- Dudek, M.J.; Ponder, J.W. Accurate modeling of the intramolecular electrostatic energy of proteins. J. Comput. Chem. 1995, 16, 791–816. [Google Scholar] [CrossRef] [Green Version]
- Kong, Y.; Ponder, J.W. Calculation of the reaction field due to off-center point multipoles. J. Chem. Phys. 1997, 107, 481–492. [Google Scholar] [CrossRef] [Green Version]
- Hehre, W.J.; Radom, L.; Schleyer, P.V.R.; Pople, J.A. Ab initio Molecular Orbital Theory; John Wiley & Sons, Inc.: New York, NY, USA, 1986. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Becke’s three parameter hybrid method using the LYP correlation functional. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, H.B.; Schlegel, G.W.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision B.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Ditchfield, R. Self-consistent perturbation theory of diamagnetism: I. A gauge-invariant LCAO method for NMR chemical shifts. Mol. Phys. 1974, 27, 789–807. [Google Scholar] [CrossRef]
- Wolinski, K.; Hinton, J.F.; Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Smith, S.G.; Goodman, J.M. Assigning stereochemistry to single diastereoisomers by GIAO NMR calculation: The DP4 probability. J. Am. Chem. Soc. 2010, 132, 12946–12959. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Sample of compounds 1 and 2 are not available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | |||
|---|---|---|---|---|
| δC | δH (J, Hz) | δC | δH (J, Hz) | |
| 1 | 70.8 | 3.34, 1H, br s | 70.7 | 3.34, 1H, br s |
| 2 | 36.1 | 1.84, 1H, m 1.60, 1H, m | 36.0 | 1.84, 1H, m 1.59, 1H, m |
| 3 | 78.8 | 4.40, 1H, dd, 12.0, 4.5 | 78.8 | 4.40, 1H, dd, 12.0, 4.5 |
| 4 | 53.0 | _ | 52.9 | _ |
| 5 | 36.6 | 2.41, 1H, dd, 13.0, 4.5 | 36.5 | 2.42,1H, dd, 12.5, 3.5 |
| 6 | 22.0 | 1.14, 1H, m 0.84, 1H, m | 21.9 | 1.16, 1H, m 0.83, 1H, m |
| 7 | 25.1 | 1.02, 2H, m | 25.0 | 1.02, 2H, m |
| 8 | 47.5 | 1.43, 1H, m | 47.3 | 1.46, 1H, m |
| 9 | 20.1 | - | 19.9 | - |
| 10 | 28.9 | - | 28.7 | |
| 11 | 25.0 | 2.27, 1H, m 1.17, 1H, m | 24.9 | 2.27, 1H, m 1.19, 1H, m |
| 12 | 32.6 | 1.55–1.57, 2H, m | 32.8 | 1.56–1.60, 2H, m |
| 13 | 44.8 | - | 44.7 | - |
| 14 | 48.7 | - | 48.6 | - |
| 15 | 35.3 | 1.21–1.22, 2H, m | 35.2 | 1.26–1.29, 2H, m |
| 16 | 27.8 | 1.84, 1H, m 1.22, 1H, m | 27.6 | 1.84, 1H, m 1.23, 1H, m |
| 17 | 51.9 | 1.54, 1H, m | 51.6 | 1.57, 1H, m |
| 18 | 18.0 | 0.91, 3H, s | 17.8 | 0.90, 3H, m |
| 19 | 28.8 | 0.36, 1H, d, 4.0 0.56, 1H, d, 4.0 | 28.7 | 0.35, 1H, d, 4.0 0.57, 1H, d, 4.0 |
| 20 | 36.4 | 1.26, 1H, m | 36.0 | 1.29, 1H, m |
| 21 | 18.3 | 0.82, 3H, s | 18.2 | 0.85, 3H, s |
| 22 | 29.0 | 1.43, 1H, m 0.92, 1H, m | 29.0 | 1.45, 1H, m 1.00, 1H, m |
| 23 | 30.8 | 1.43, 1H, m 1.23, 1H, m | 30.9 | 1.45, 1H, m 1.30, 1H, m |
| 24 | 74.6 | - | 73.4 | _ |
| 25 | 32.3 | 1.70, 1H, m | 32.4 | 1.72, 1H, m |
| 26 | 17.1 | 0.80–0.82, 3H, m | 16.8 | 0.80–0.85, 3H, m |
| 27 | 17.1 | 0.80–0.82, 3H, m | 16.7 | 0.80–0.85, 3H, m |
| 28 | 178.5 | 178.6 | ||
| 29 | 9.5 | 0.97, 3H, s | 9.5 | 0.97, 3H, s |
| 30 | 19.1 | 0.89, 3H, s | 19.0 | 0.91, 3H, s |
| 31 | 64.7 | 3.25, 2H, m | 66.9 | 3.89, 1H, d, 11.0 3.85, 2H, d, 11.0 |
| 1’ | 104.1 | 4.14, 1H, d, 7.5 | 104.0 | 4.15, 1H, d, 7.5 |
| 2’ | 73.7 | 2.88, 1H, dd, 9.0, 7.5 | 73.5 | 2.88, 1H, t, 8.5 |
| 3’ | 76.4 | 3.05, 1H, t, 9.0 | 76.3 | 3.06, 1H, t, 8.5 |
| 4’ | 69.6 | 3.23, 1H, m | 69.5 | 3.24, 1H, m |
| 5’ | 65.7 | 3.66, 1H, dd, 11.5, 5.0 | 65.6 | 3.66, 1H, dd, 11.0, 5.0 |
| 2.97, 1H, t, 11.5 | 2.97, 1H, t, 11.0 | |||
| OAc | 20.7 | 1.99, 3H, s | ||
| 170.3 | ||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tran, T.-N.-M.; Bernadat, G.; Mai, D.-T.; Nguyen, V.-K.; Sichaem, J.; Nguyen, T.-P.; Tran, C.-L.; Do, P.-V.; Tran, N.-M.-A.; Nguyen, H.-H.; et al. Nervisides I–J: Unconventional Side-Chain-Bearing Cycloartane Glycosides from Nervilia concolor. Molecules 2019, 24, 2599. https://doi.org/10.3390/molecules24142599
Tran T-N-M, Bernadat G, Mai D-T, Nguyen V-K, Sichaem J, Nguyen T-P, Tran C-L, Do P-V, Tran N-M-A, Nguyen H-H, et al. Nervisides I–J: Unconventional Side-Chain-Bearing Cycloartane Glycosides from Nervilia concolor. Molecules. 2019; 24(14):2599. https://doi.org/10.3390/molecules24142599
Chicago/Turabian StyleTran, Thi-Ngoc-Mai, Guillaume Bernadat, Dinh-Tri Mai, Van-Kieu Nguyen, Jirapast Sichaem, Tan-Phat Nguyen, Cong-Luan Tran, Phuong-Vy Do, Nguyen-Minh-An Tran, Huu-Hung Nguyen, and et al. 2019. "Nervisides I–J: Unconventional Side-Chain-Bearing Cycloartane Glycosides from Nervilia concolor" Molecules 24, no. 14: 2599. https://doi.org/10.3390/molecules24142599