
Weinreb Amides as Directing Groups for Transition Metal-Catalyzed C-H Functionalizations


Department of Chemistry-BMC, Uppsala University, BOX 576, 75-123 Uppsala, Sweden
*
Author to whom correspondence should be addressed.
Molecules 2019, 24(5), 830; https://doi.org/10.3390/molecules24050830
Submission received: 28 January 2019
/
Revised: 15 February 2019
/
Accepted: 15 February 2019
/
Published: 26 February 2019
(This article belongs to the Special Issue Amide Bond Activation)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
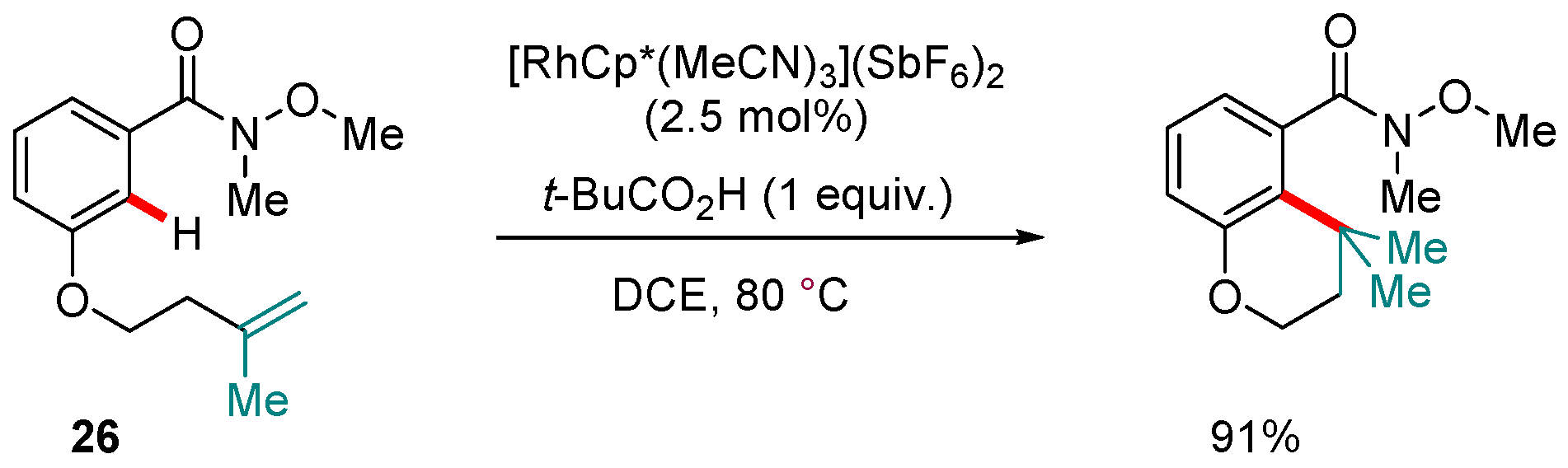{kind=link}
{kind=link}
{kind=link}
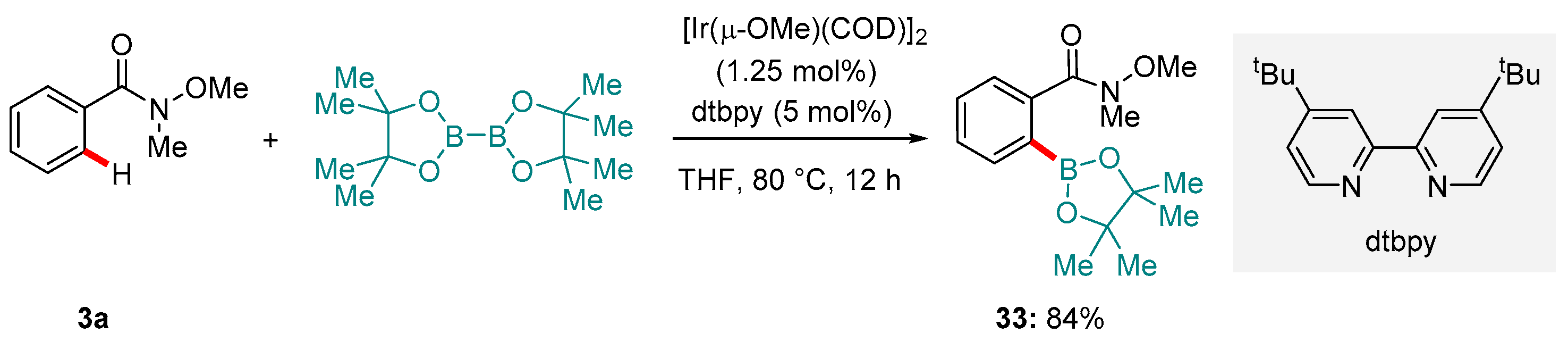{kind=link}
{kind=link}
{kind=link}
Abstract
:Weinreb amides are a privileged, multi-functional group with well-established utility in classical synthesis. Recently, several studies have demonstrated the use of Weinreb amides as interesting substrates in transition metal-catalyzed C-H functionalization reactions. Herein, we review this part of the literature, including the metal catalysts, transformations explored so far and specific insights from mechanistic studies.
1. Introduction
The pursuit of efficient methods for the direct, catalytic substitution of otherwise inert C-H bonds in organic molecules has become a major area of research focus in recent years [1]. Tremendous advances have been made in the development of previously impossible transformations [2,3,4,5], mechanistic understanding [6,7,8,9], milder and safer protocols [10], and selectivity [11,12,13,14]. The ubiquity of C-H bonds in organic molecules makes their regioselective activation and substitution particularly attractive, but also challenging. To this end, the use of directing groups–parts of an organic substrate that can coordinate to and position a metal center over the desired C-H bond–has met with enormous success [15]. The use of ortho-directing groups especially has provided the basis of many new homogeneous catalytic C-H functionalization reactions [16]. Directing groups able to deliver meta [17,18] and para [19] selectivity in C-H functionalization catalysis have also been described, although the generality of that approach is still some way off in the future.
Ideally, directing groups should not require separate and/or laborious installation/removal or otherwise be inutile in later steps of a synthesis. It is preferable that they should either be part of the desired target compound or that they could be converted to another useful group once their role during the C-H functionalization step is over. A plethora of strategies to realize the latter objective has been pursued. The invention of various removable or modifiable [20,21,22], traceless [23], or otherwise transient [24,25,26,27,28,29] directing groups has formed a sizeable category in and of itself within the field of C-H functionalization catalysis.
Amides are an incomparably important class of compounds. The development of methods for their selective synthesis and derivatization is, therefore, a key pursuit in synthetic methodology. That the amide group can serve as a ligand for transition metals enables various new approaches to this via catalytic C-H functionalization [30]. Most commonly, the amide group has been called upon to direct the catalytic substitution of neighboring Ar-H bonds but, as discussed below, they also enable C(sp3)-H bond manipulation.
N-methoxy-N-methyl amides (1, Figure 1), or Weinreb amides [31], are a valuable branch of the amide family. They provide the ‘textbook’ route to mono-addition products (especially ketones and aldehydes) via nucleophilic attack on the carbonyl group. Such attacks give rise to stable five-membered tetrahedral cyclic intermediates (2) to which a second addition (“over-addition”) is precluded. Weinreb amides may be prepared with ease from carboxylic acids or their chlorides, esters, aldehydes or ketones [32].
That Weinreb amides can also steer the regioselectivity of transition metal-catalyzed C-H functionalizations (Figure 2) qualifies them as noteworthy multi-functional directing groups. Remarkably, and despite the various synthetic advantages of this (for example that it can obviate the need for lithiation strategies and make available previously impossible reactions), Weinreb amides have only recently attracted attention as substrates for C-H functionalization. In part, this is due to the amide oxygen’s weaker coordination ability to most transition metal centers. The latter has presented a challenge to the development of their use in C-H functionalization reactions, although many carbonyl-directed reactions are now known [33].
This review describes progress in the use of Weinreb amides as directing groups in catalytic C-H functionalization. A variety of reactions falls under this category. We have chosen to group these according to the transition metal center responsible for the C-H functionalization catalysis, rather than the overall transformation, in order to maximize the ease of comparison between researchers working in similar areas, and to track the different rates at which progress has occurred and insights in to the underlying mechanisms. Overwhelmingly, the focus of the studies reviewed herein falls on the utility of Weinreb amides as directing groups for the C-H functionalization step. Therefore, whilst several publications describe subsequent manipulation of the Weinreb amide group, this is usually to illustrate the possibility, rather than a key development. We have opted therefore not to include many examples of the latter; we take the possibility of a posteriori conversion of Weinreb amides using conventional approaches (e.g., to ketones or aldehydes), for the most part, to be a safe assumption.
2. Ru-catalyzed Reactions
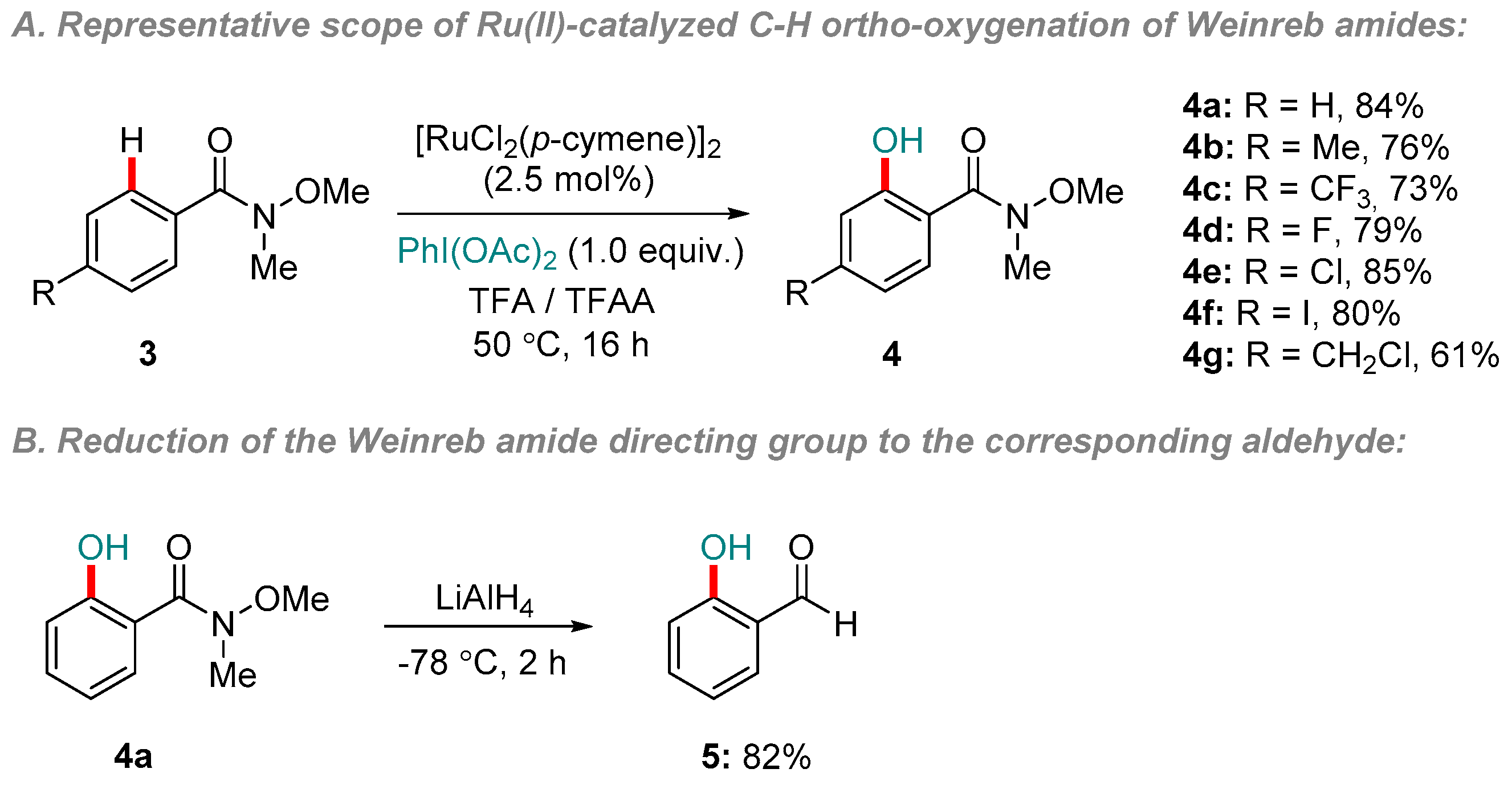
The versatility of Ru, as well as its considerably lower price compared to other 2nd and 3rd-row transition metals, make it an appealing candidate around which to develop economical C-H functionalization methodology [34,35]. Moreover, considerable advances have been made in Ru-catalyzed C-H functionalization directed by weakly coordinating groups, including amides [33]. In 2013, Ackermann and co-workers described the Weinreb amide-directed C-H ortho-oxygenation of arenes 3 using a Ru(II)-based system [36]. [RuCl2(p-cymene)]2 served as the catalyst precursor and PhI(OAc)2 as the most effective oxidant. Representative results from the study are shown in Scheme 1a.
The reaction showed a high selectivity for mono-oxygenated products 4, a preference for electron-rich substrates in competition experiments, and a kinetic isotope effect (KIE) of kH/kD = 3.0 [6]. The authors proposed that an irreversible C-H activation event was a key step en route to the products. It is notable that the N-alkyl substituent could also be varied substantially without any loss of reaction efficiency. The Weinreb amide group could be reduced in high yield (4a to 5) to reveal the corresponding aldehyde (Scheme 1b). A single Weinreb amide substrate was also shown to work as part of a study by Jeganmohan and co-workers on the ortho-directed C-H benzoyloxylation of various amides. The reaction system closely resembled that reported above by Ackermann, except for the use of (NH4)S2O8 as the terminal oxidant, higher temperatures and use of 1,2-dichloroethane as solvent [37].
Subsequently, Das and Kapur produced a report demonstrating the Ru-catalyzed olefination of various amides, including Weinreb amide-decorated arenes [38]. The protocol closely resembles the Pd-catalyzed oxidative Heck reaction [39] (also known as the Fujiwara-Moritani reaction) and other related Ru-catalyzed C-H alkenylations [34]. However, for the majority of their entries (selected examples are shown in Scheme 2a), the authors observed cleavage of the Weinreb amide N-O bond; N-methyl amides were obtained as the main products. It is instructive to consider the proposed mechanism, an adapted version of which is shown in Scheme 2b. Presumably, the N-O bond serves as an oxidant with Cu(OAc)2∙H2O as the carboxylate source to facilitate repeated C-H functionalization [40] The use of similar “internal oxidant” strategies in C-H functionalization, including using N-methoxy amides, has been recently reviewed by Cui and co-workers [41]. Exceptions wherein the N-O bond was preserved presumably resulted from Cu(II) (or Cu(III) species arising via disproportionation) outcompeting N-O as an internal oxidant. The importance of such examples is under-appreciated, in our view. That an exogenous oxidant may divert reactivity away from damaging a group under otherwise identical conditions is a key aspect of modulating functional group tolerance; a factor that will govern the extent to which C-H activation–and other catalytic methods–gain acceptance as generalizable routes to construct molecular complexity.
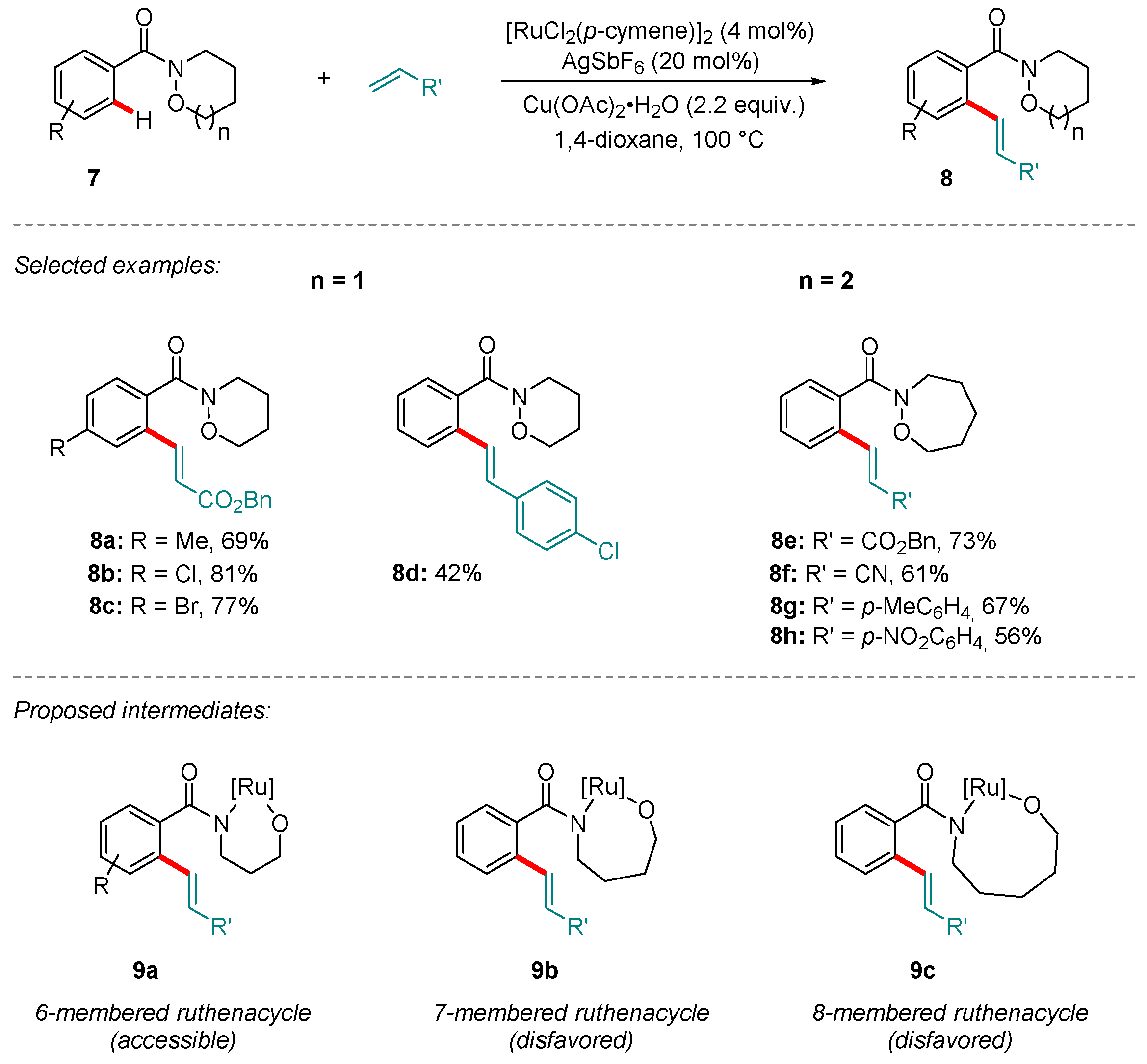
Das and Kapur reported that five-membered cyclic amides of the type 7 (n = 0) typically underwent N-O bond cleavage, affording ring-opened products. To prevent this, and thereby retain the Weinreb amide functionality, they subsequently sought to show that N-O bond cleavage could be prevented through judicious substrate design [42]. Increasing the Weinreb amide size to six- and seven-membered rings (7, n = 1 or 2, respectively) rendered the N-O bond cleavage energetically unfavorable. Under otherwise unchanged conditions, the oxidative C-H olefination afforded products 8 (Scheme 3), restoring Cu(II) to its role as the terminal oxidant. Further manipulation of the cyclic Weinreb amide moieties in 8, for example in their conversion to the corresponding aldehyde or ketones, was demonstrated to proceed in high yield.
An explanation for the greater relative stability of the 6- and 7-membered cyclic Weinreb amide groups in this protocol was proposed: In the course of the reaction, oxidative addition of the Ru center to the N-O bond would furnish intermediates of type 9. Of these, only the formation of 9a would be favorable; the 7- and 8-membered ruthenacycles (9b and 9c, respectively) are presumably too high in energy to be accessed.
3. Co-catalyzed Reactions
The expense and eventual scarcity of second- (4d) and third-row (5d) transition metals has, in recent years, motivated a shift towards exploring the potential of their more abundant first-row (3d) cousins for catalytic C-H functionalization [43]. Cobalt stands out in this context as a comparatively cheap, abundant option that comes with a proven track record in broad areas of homogenous catalysis, including several industrially important reactions [44,45,46].
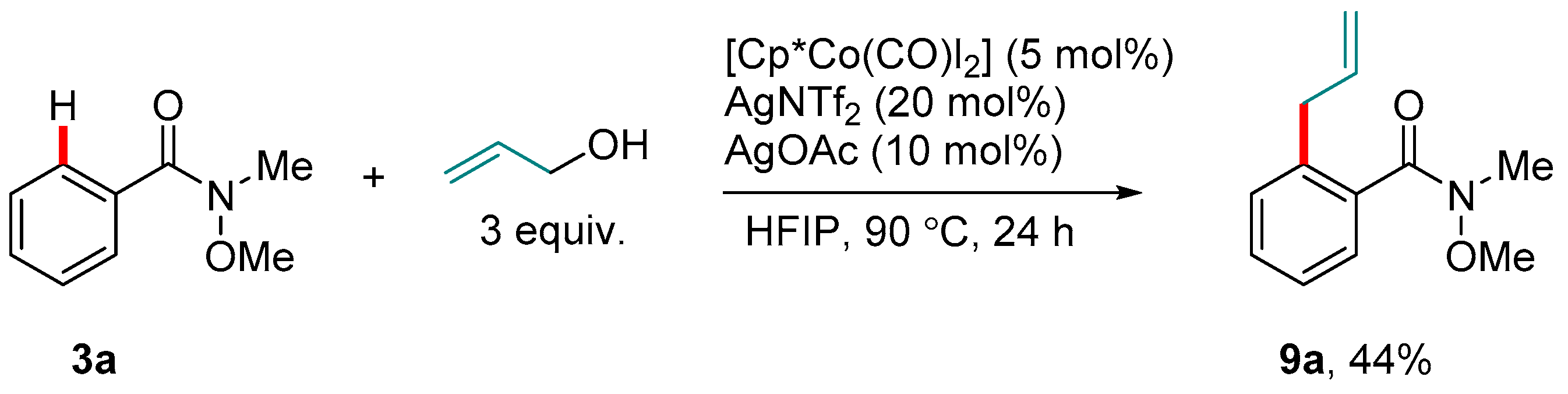
In 2013, Yoshino and Matsunaga disclosed the first C-H ortho-directed functionalization catalyzed by Cp*Co(III) species [47,48]. In a subsequent study on the Co-catalyzed C-H allylation of aryl purines and benzamides using allylic alcohol, they demonstrated the new reaction also on Weinreb amide 3a [49]. Here, the bench-stable complex Cp*Co(CO)I2, which was previously shown by Matsunaga and Kanai to be highly efficient for indole C2-H amidation [50], served as the pre-catalyst (Scheme 4). Hexafluoroisopropanol (HFIP) [51,52] was needed for the C-H allylation, as were acetate salts. The authors interpreted the latter as an indication that the C-H activation proceeded via a Concerted-Metalation-Deprotonation (CMD) mechanism [53], as has been elucidated in more detail for second row transition metals [8,40,54]. The moderate yield of product 9a in this reaction was attributed to the weaker coordinating ability of the Weinreb amide group compared to its simpler benzamide relatives or, indeed, to that of the purine Nsp2 centers.
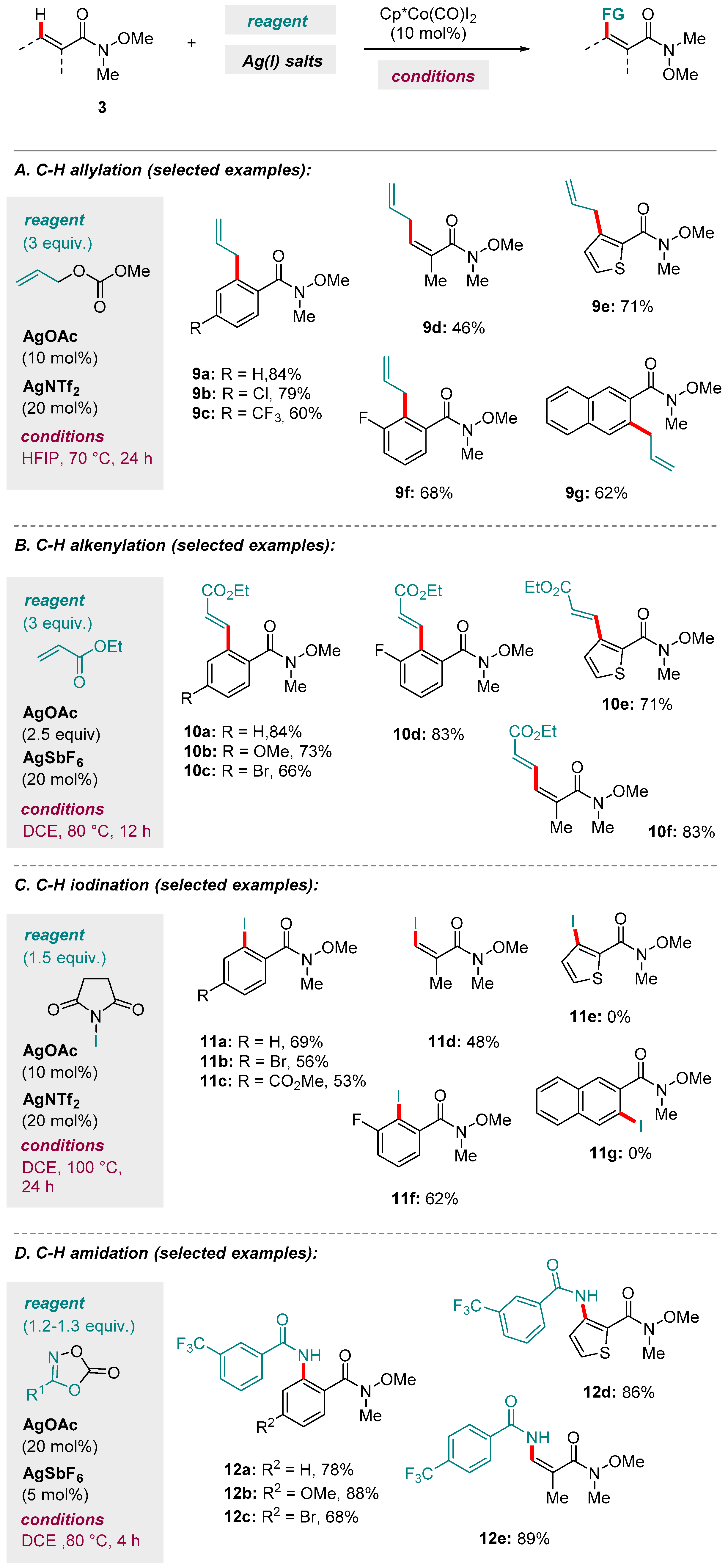
In a later study, the group published a considerably expanded range of Co-catalyzed C-H functionalizations using aromatic Weinreb amides (Scheme 5) [55]. These included C-H allylation using allylic carbonates (Scheme 5A), C-H alkenylation under oxidative conditions (Scheme 5B), C-H iodination using N-iodosuccinimide (Scheme 5C) [56] and C-H amidation using dioxazolones (Scheme 5D). These systems relied on similar combinations of [Cp*Co(CO)I2] pre-catalyst, a cationic silver additive (AgNTf2 or AgSbF6) and AgOAc. Unlike in the Ru-catalyzed case (see Scheme 2), the N-O bond of the Weinreb amide moiety was preserved. Competition and kinetic isotope exchange experiments showed that the C-H activation event in the case of the allylation reaction was both rate-limiting and all but irreversible, supplying further evidence for the CMD pathway Yoshino and Matsunaga proposed in their earlier study [49]. Their proposed mechanism for the Co(III)-catalyzed C-H allylation is shown in Figure 3. In related recent work, Whiteoak and Hamilton have elucidated the mechanistic details governing the oxidative Co-catalyzed C-H alkenylation of amides, and specifically whether and why alkyl or alkenyl products are obtained depending on the choice of alkene substrate [57].
The relevance to wider synthetic contexts of the transformations reported by Yoshino and Matsunaga was demonstrated through the conversion of the Weinreb amide group in their various products to the corresponding ketones, aldehydes and alkenes. This thus delivers several motifs whose preparation might otherwise be longer and more demanding.
4. Pd-catalyzed Reactions
Palladium is perhaps the most firmly established metal for coupling chemistry [58,59,60,61,62], and this has translated to its high level of popularity for C-H functionalization reactions [16,63,64,65,66]. Many Pd-based catalysts are well-defined [67,68], permitting systematic tuning of their properties, and are understood, most usually, to work via Pd(0)/Pd(II) or Pd(II)/Pd(IV) manifolds [62,64,66]. Despite the prevalence of many ortho-directing groups, including several closely related amides [30], there have been relatively few examples of Pd-catalyzed reactions using Weinreb amides.
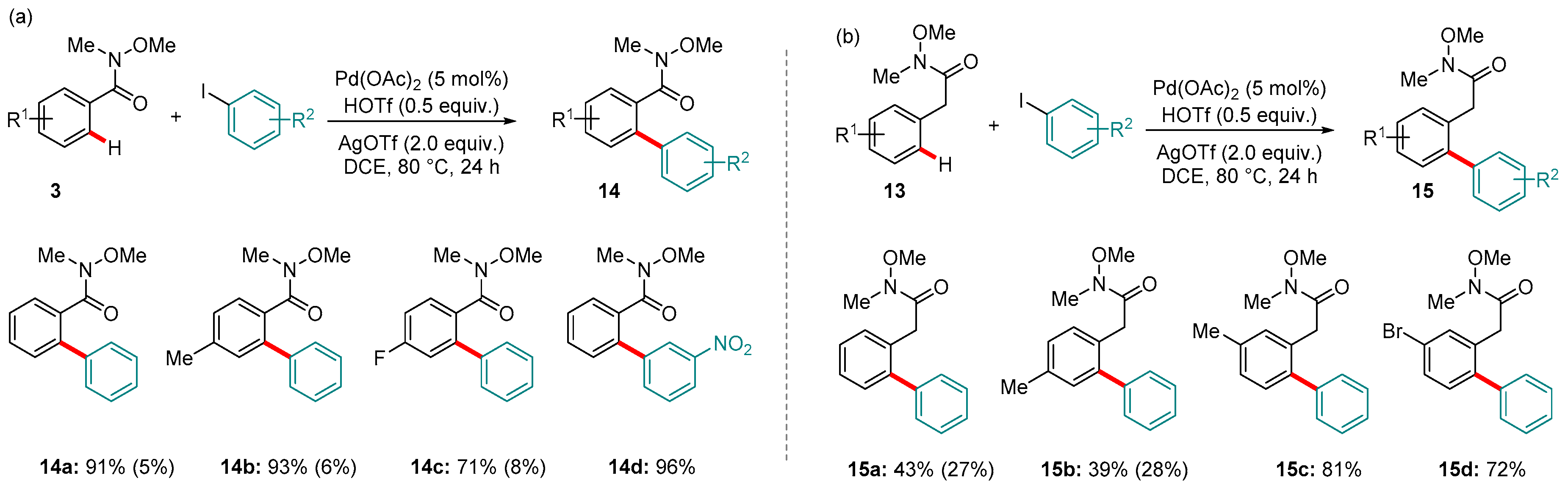
In 2015, Wang and co-workers reported the first Weinreb amide-directed C-H functionalization catalyzed by Pd. The reaction took place between either aryl Weinreb amides 3 (Scheme 6a) or benzyl amides 13 and iodoarenes using Pd(OAc)2 as the pre-catalyst in DCE solvent [69]. Both Weinreb types showed high tolerance towards electron-donating and electron-withdrawing substituents, as well as halogens (e.g., the bromide in 15d). However, benzyl amides showed considerably worse mono-selectivity, except if meta substituents were present (e.g., 15c and 15d) presumably to impart steric hindrance.
Figure 4 shows one of the proposed mechanisms, adapted for the specific case of substrate 3a. A kinetic isotope effect (KIE) of 1.1 suggested that the C-H activation step itself (forming 16 from 3) was not rate-determining. Oxidative addition of the aryl iodide to 16 was postulated to form 17 from which the product 14a is formed via reductive elimination. The authors reported that AgOTf, as well as acting as an oxidant, was crucial for the reaction and hypothesized that in situ generated Pd(OTf)2 is a key aspect of the catalytic cycle. (We note that the various roles Ag plays in Pd-catalyzed reactions have recently been reviewed by Pérez-Temprano and co-workers [70].)
To the best of our knowledge, the only other example of a Pd-catalyzed Weinreb amide-directed C-H arylation comes from a single entry in a study by Bhanage and workers on the use of anilines as arylating reagents for N-methoxybenzamides (Scheme 7) [71]. Conditions were largely analogous to those described for Wang’s reaction above, with the principal exception that aniline underwent in situ oxidation using tBuONO to generate the electrophilic coupling partner. However, the yield for the Weinreb amide specifically was low, indicating that although this approach holds some promise, it is harder to optimize for the Weinreb amide compared to the using more conventional coupling partners.
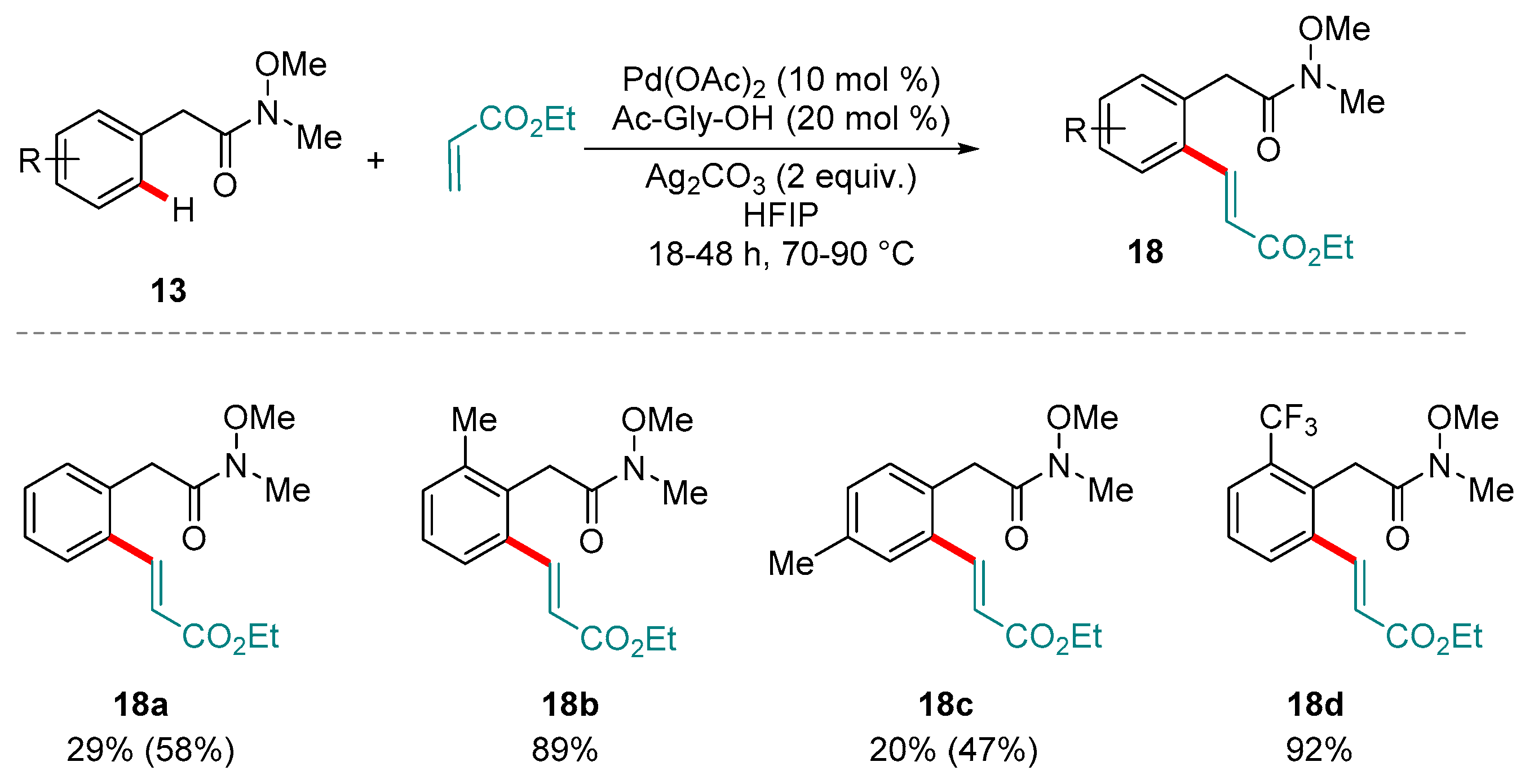
Also in 2015, Yu and co-workers described conditions for the efficient alkenylation and acetoxylation of Weinreb amides (Scheme 8) [72], overcoming the additional entropic penalty imposed by the extra methylene unit present in the directing group (and thus the challenge of forming seven-membered palladacyclic intermediates). In keeping with related protocols reported by Yu and co-workers, the presence of Ac-Gly-OH was found to be crucial to facilitate the C-H functionalization step [73].
Yu and co-workers found the olefination reaction to have moderate to excellent yields, exclusively mono C-H olefination with ortho and meta-substituted benzyl Weinreb amides, but reduced selectivity if the substituents were positioned para with respect to the directing group (Scheme 8).
The acetoxylation of benzyl amides (Scheme 9) used PhI(OAc)2 as the oxidant in the presence of either Ac2O or tert-butyl peroxyacetate (MeC(O)OOtBu). Yields and tolerance towards various substituents were good (examples 19a–c). Furthermore, it was possible to expand the scope by an extra methylene unit (i.e., the reaction can proceed with good efficiency via an 8-membered palladacycle) to give product 19d.
The introduction of halogens to organic substrates is an inherently valuable pursuit in methodology development, owing the great versatility of C-halogen bonds in synthesis [74]. In 2017, the group of Kapur disclosed the Pd-catalyzed C-H halogenation (iodination, bromination and chlorination) of Weinreb amides (Scheme 10) [75]. The reaction relied on a combination of Pd(OAc)2 and Cu(OTf)2 with N-halo-succinimide (NXS; X = I, Br or Cl) as the halogen source. The reaction worked well with a variety of substituents, except for nitro groups, which switch off the reaction from either of the ortho or para position.
Mechanistic studies suggested that the C-H activation step was irreversible, but not rate-limiting. The catalytic cycle (Figure 5) was proposed to proceed via a route closely related to that of the corresponding arylation reaction (Figure 4, above): carbonyl directed cyclometallation to give 22 precedes oxidative addition into NXS, the latter of which produces Pd(IV) species 23 (a bimetallic Pd(III) species [76,77,78,79]—not shown—is also possible). Thereafter, reductive elimination releases the desired product.
2018 saw Yu and co-workers extend the utility of the Weinreb amide directing group further [80]. This time, the Yu group used it as the basis for directing Pd-catalyzed Csp3-H arylation of alkyl groups (Scheme 11). The discovery of general and efficient methods that allow selective substitution of C(sp3)-H at transition metal centers is a long-standing challenge. For Pd, progress has been notably rapid in in the past few years [63,81,82,83], and its various aspects have been reviewed recently [65,84,85].
Yu and co-workers found that the inclusion of 3-pyridinesulfonic acid was crucial to the reaction, both Weinreb and other amides. Computational studies at the DFT level revealed that 3-pyridinesulfonic acid stabilizes cationic Pd intermediates during the reaction and that it promotes the dissociation of acetate ligands, which is required for the C(sp3)-H bond cleavage to occur [80].
5. Rh-catalyzed Reactions
The first two decades of this century have seen an explosion of interest in the use of Rh(III) catalysts for selective C-H functionalization. Substantial progress has been made in both scope [86,87] and attendant mechanistic understanding [7,88,89,90]. As part of their efforts to answer key questions relating to the regioselectivity of Rh(III)-catalyzed transformations, Rovis and co-workers reported in 2013 an intramolecular alkene hydroarylation directed by Weinreb amide 26 in excellent yield (Scheme 12) [91].
Wang and co-workers reported a closely-related but more elaborate system, involving Cu(OAc)2 as a catalytic oxidant regenerated under air to retain olefin functionality at the end of a cycle coupling aromatic Weinreb amides with alkenes (Scheme 13) [92]. An ample scope demonstrated the reaction’s tolerance towards various functional groups, including halogens, various ortho-substituents and a range of electron-donating and electron-withdrawing groups. Competition experiments revealed that electron-rich arenes reacted faster than their electron-poor counterparts.
The mechanism proposed by Wang and co-workers (Figure 6) involved coordination of the Rh(III) center to the carbonyl moiety of the Weinreb amide, insertion of the alkene to the rhodacyclic intermediate (27), -hydride elimination and regeneration of the active catalyst by Cu(OAc)2.
Whilst, strictly speaking, it represents a slight departure from the current focus on the Weinreb amide group, it is noteworthy that a very closely related class of substrates, has been used extensively wherein their C(O)NH-OMe bond as an “internal oxidant” for transition metal centers. Thus, C-H functionalization reactions may be performed without exogenous oxidants. This strategy has worked with a number of transition metals, Rh(I/III) catalytic cycles have dominated in this area [41].
In 2018, Qin and co-workers disclosed a near identical set of conditions to those used by Wang above, but using ethenesulfonyl fluoride (ESF) as the alkene coupling partner and 1,4-dioxane as the solvent [93]. However, the Qin group further observed that increasing the loading of AgSbF6 to 1 equivalent favored the formation of cyclic lactones. Residual water introduced from the hygroscopic AgSbF6 was proposed to promote the in situ hydrolysis of the Weinreb amide group to account for the lactone formation, though mechanistic work proved inconclusive. In switching away from Weinreb amides to N-methoxybenazmides (ArC(O)NH-OMe), Qin and co-workers were able to cause a similar oxidative cyclization involving insertion of the ESF double bond into the amide N-H unit.
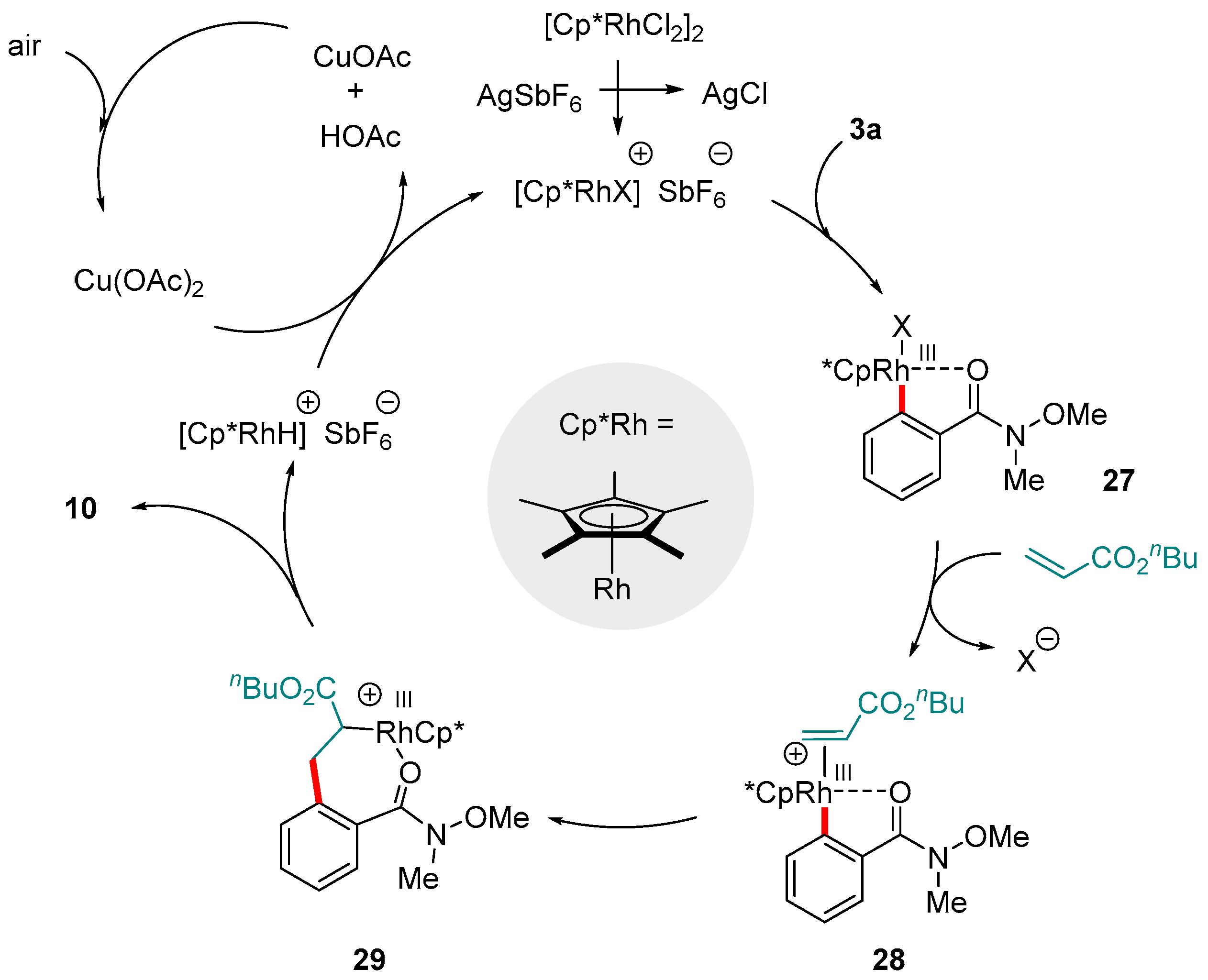
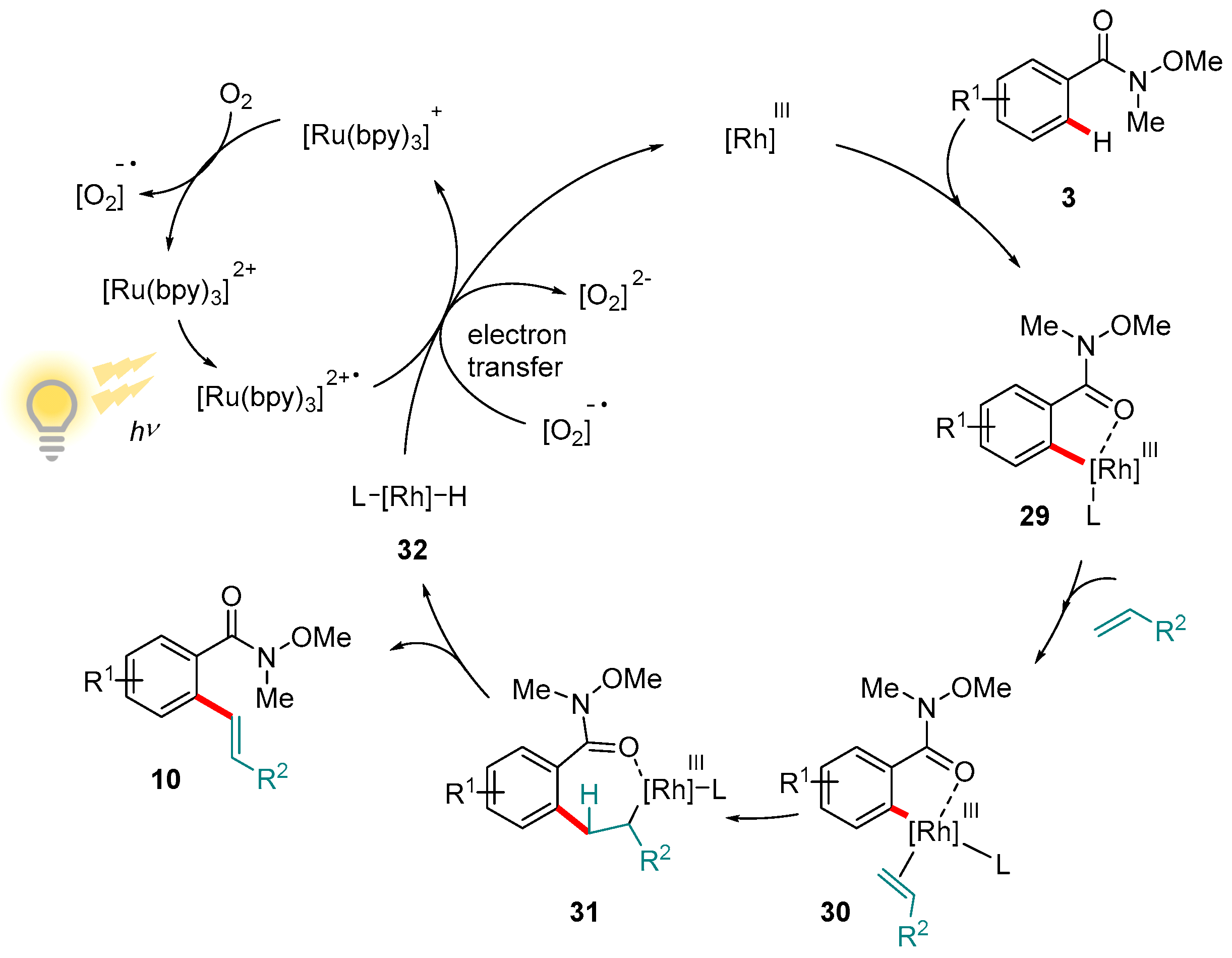
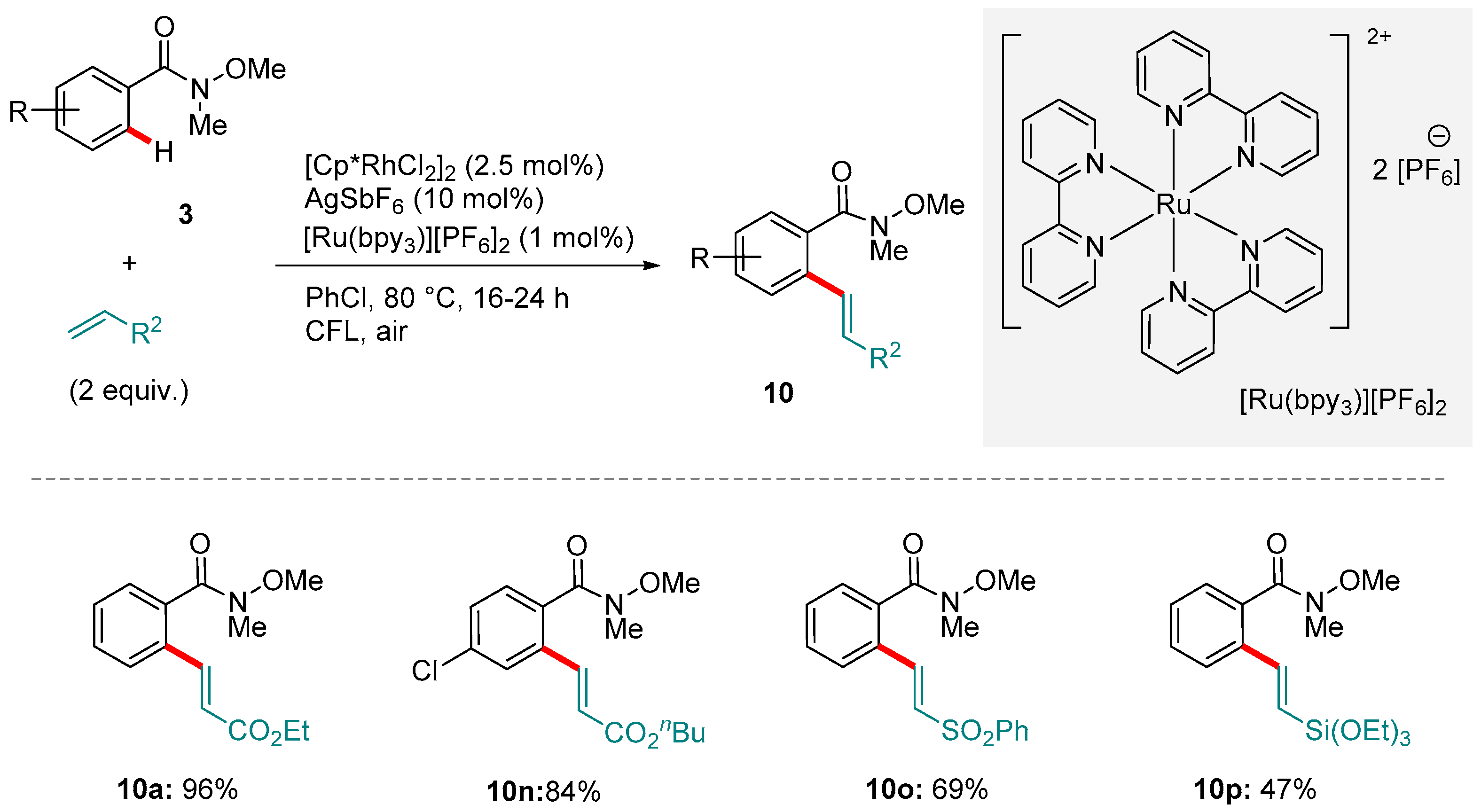
Recent years have witnessed the rediscovery and subsequent explosion of interest in functionalizations enabled by photoredox catalysis. The photoexcitation of transition metal complexes serves as a greener and more economically viable method of generating radical intermediates able to initiate a wide range of valuable reactions [94,95,96,97,98]. One application of this is the replacement of terminal oxidants with lighter loadings of a photocatalyst whose oxidative power is obtained via photoexcitation. Rueping and co-workers applied this strategy to demonstrate that the oxidative alkenylation of aromatic amides, including Weinreb amides, is viable using a manifold analogous to that described by Wang’s group (see above), but driven by a visible light-regenerated Ru-based photocatalyst, [Ru(bpy)3][PF6]2 (Scheme 14) [99,100]. The protocol is notable not just for its efficiency and high functional group tolerance, but also for the fact that it could be extended to other amides as well as a variety of olefin substrates (various groups R2).
The catalytic cycle proposed by Rueping and co-workers closely resembles to that of Wang: ortho-rhodation of 3 to give 29 coordination and insertion of the olefin (complex 30 to 31) and β-hydride elimination to form the hydride intermediate 32 and the product (Figure 7). Reductive loss of the hydride and re-oxidation to Rh(III), however, is mediated by the photo-excited [Ru(bpy)3]2+∙ species, of which only a 1 mol% loading is required.
6. Ir-catalyzed Reactions
Boronates rank amongst the most versatile of groups; they may be converted to an extraordinarily broad range of functionality through a many different mechanisms [101,102,103]. Moreover, recent years have seen boronate-based methodology emerge as the basis of automated synthesis, which holds enormous potential to streamline the synthesis of many complex (hetero)aromatic and olefinic molecules [104,105,106,107]. Such advantages render especially important methods that allow the regioselective introduction of boronates to organic substrates. Amongst these, Ir-catalyzed C-H borylation ranks as one of the mildest and most enabling, as it is amenable to regiocontrol through sterics [108,109], directing groups (both to the ortho [110,111,112,113,114,115] and para [116] positions) and/or the inherent electronic properties [117,118,119] of a substrate.
Krska, Maleczka and Smith have demonstrated [120] that Weinreb amides rank amongst groups that competently direct Ir catalysts towards ortho-C-H borylation (Scheme 15). Although only a single entry using a Weinreb amide was reported in their Communication, the success of the reaction (a high yield and regioselectivity) was representative, suggesting that other advantages of the method, such as the high functional group tolerance, could easily be paired with the Weinreb amide functionality. Conditions for this transformation deviated little from those commonly used for Ir-catalyzed C-H borylation more generally: [Ir(μ-OMe)(COD)]2 as the pre-catalyst with dtbtpy as a ligand and B2(pin)2 as the boron source. The corresponding mono-borylated product, 33 was reported to form in 84% yield.
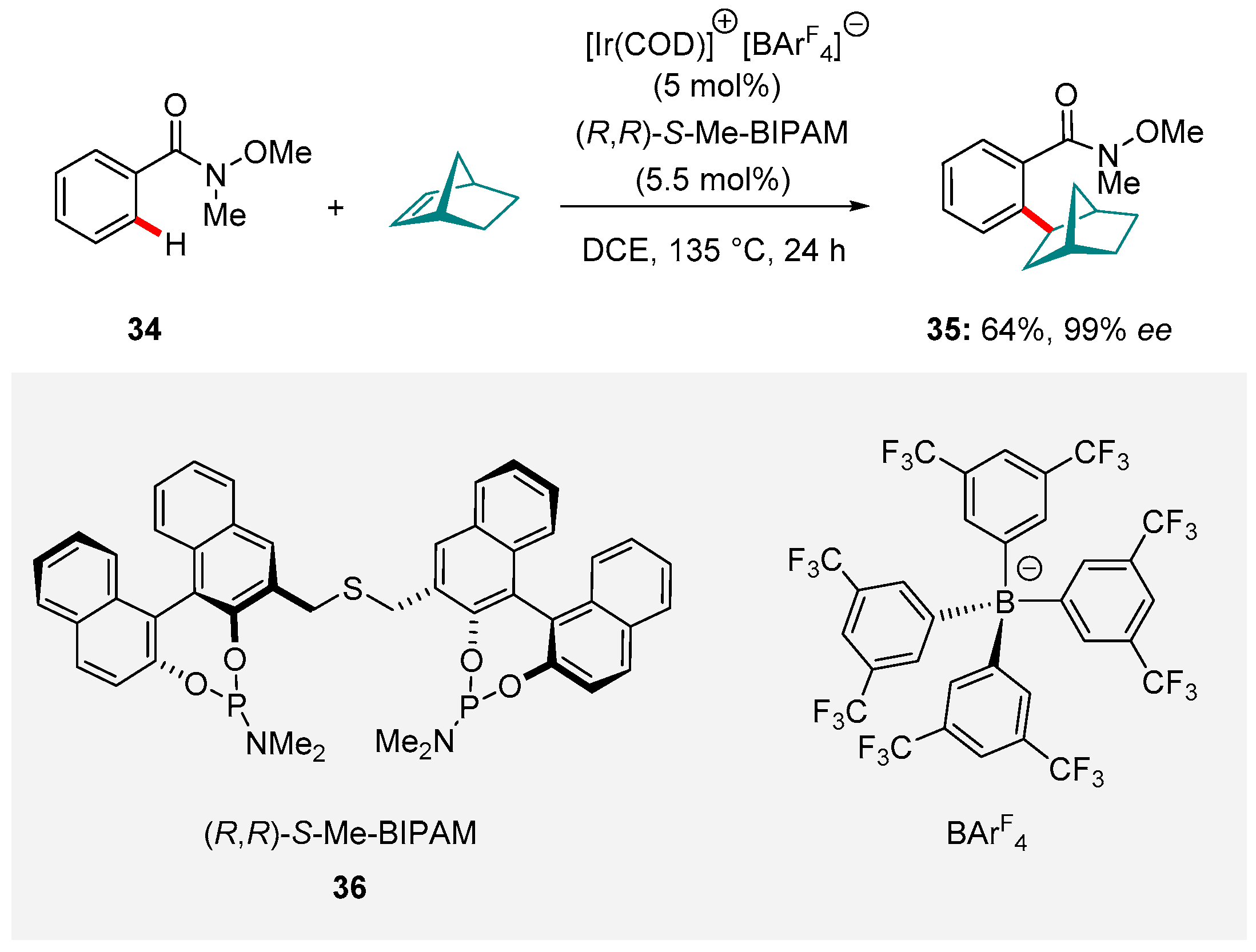
Enantioselective C-H functionalization marries the benefits of directly substituting a C-H bond with with establishing a new stereocenter in the product. This pursuit continues to inspire a growing body of research [121]. In 2015, Yamamoto and Shirai described an Ir-catalyzed protocol for the asymmetric intermolecular hydroarylation of arenes directed by oxygen based directing groups [122]. Although the scope included only a single example exploiting a Weinreb amide directing group (the transformation of 34 to 35), this entry was representative both in terms of yield and selectivity (Scheme 16). Enantioselectivity was induced using the bidentate bis(phosphoramidite) ligand (R,R)-S-Me-BIPAM ligand (36) and the N-O bond was preserved in the product.
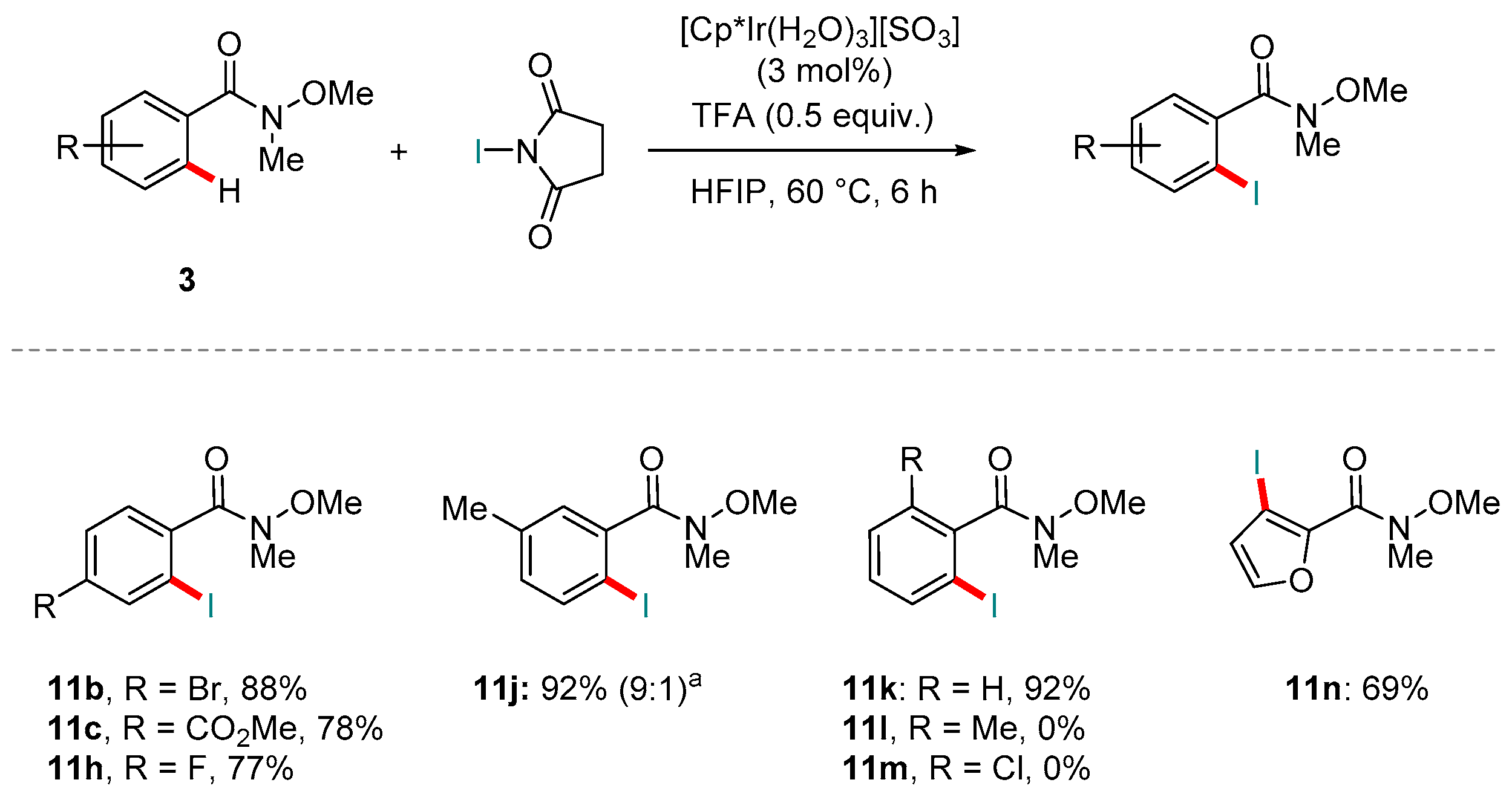
Most recently, the group of Martín-Matute has developed an Ir(III)-catalyzed C-H ortho-iodination of various amides [123]. The scope includes a strong focus on Weinreb amides. Their reaction used [Cp*Ir(H2O)3][SO4] as the catalyst precursor, and N-iodo-succinimide (NIS) as the halogenating reagent in the presence of trifluoroacetic acid. Selected examples are shown below (Scheme 17). It is notable that Weinreb amides returned only the mono-iodinated products 11; a result only tertiary amides gave (primary and secondary amides gave at least small amounts of di-iodination). Whilst the substrate scope included a broad range of functional groups, substituents positioned ortho to the directing group at the outset restricted reactivity, presumably by imposing prohibitive amounts of steric hindrance.
The authors also performed a robustness screen [124,125] to identify the reaction’s tolerance towards various functional groups. To this end, various small molecules with different functional groups were added to the reaction mixture to see how the reaction would be affected. Tolerance towards several common functional groups, including ketones amides, carboxylic acids and alkyl halides proved excellent. Aldehydes and alkenes returned less satisfactory results, however. Mechanistically, the reaction was proposed to follow a pathway closely related to that of Martín-Matute and co-workers’ recently published Ir-catalyzed C-H ortho-iodination of aryl carboxylic acids [126]. The acid additive is understood to play a dual role: 1) activation of NIS via protonation of its carbonyl group and 2) by encouraging the dissociation of the iodinated product from the Ir center. Indeed, Martín-Matute and co-workers found in their optimization study on amides that lowering the amount of acid additive favored the formation of di-iodinated products for non-tertiary amides.
7. Conclusions
The Weinreb amide is a privileged functional group in organic synthesis that enables otherwise impossible transformations. Recent developments have seen C-H functionalization methodology greatly expand the range of chemical contexts in which its advantages may be exploited. Challenges remain, however. Presently, Weinreb amides are rarely explored as a substrate class in their own right; they are most often presented as specialized examples in studies describing a more general scope, typically amidst other amides. Thus, it is usual that only the simplest examples of Weinreb amides are demonstrated to work under newly developed reaction conditions. This is at least a little unfair since, as some of the examples described above demonstrate, Weinreb amides may return different results to other amides, for example by virtue of the reactivity of their N-O bond. Weinreb amides might, in this sense, be considered hitherto as “sleeper” substrates.
Despite this, it is evident that Weinreb amides can direct a wide range of C-H functionalization reactions catalyzed by transition metals. C-H functionalization, and catalytic methodology in general, is undergoing a shift of emphasis towards the use of less “endangered” [127,128], especially first-row transition elements for catalysis [43]. However, their use with Weinreb amides is rare. For example, to the best of our knowledge, no Weinreb amide-directed C-H functionalization reactions catalyzed by Mn, Fe or Ni have yet been reported.
Finally, it is unfortunate that the preponderance of conditions used for Weinreb amide directed C-H functionalization involve toxic halogenated solvents. Increasing legislative pressure is being brought to bear on this problem, with a particular focus against 1,2-dichloroethane (DCE) [129], which features in many of the reactions discussed above. We look forward to the use of Weinreb amides under greener conditions [130,131,132].
Acknowledgments
We thank the Wenner-Gren Foundation for funding (JK).
Conflicts of Interest
The authors declare that no conflicts of interest exist.
References
- Crabtree, R.H.; Lei, A. Introduction: CH Activation. Chem. Rev. 2017, 117, 8481–8482. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Lan, J.; You, J. Oxidative C-H/C-H Coupling Reactions between Two (Hetero)arenes. Chem. Rev. 2017, 117, 8787–8863. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Kim, Y.; Chang, S. Transition Metal-Catalyzed C–H Amination: Scope, Mechanism, and Applications. Chem. Rev. 2017, 117, 9247–9301. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Yamada, S.; Kaneda, T.; Itami, K. C–H Functionalization of Azines. Chem. Rev. 2017, 117, 9302–9332. [Google Scholar] [CrossRef] [PubMed]
- Sandtorv, A.H. Transition Metal-Catalyzed C-H Activation of Indoles. ACS Catal. 2015, 357, 2403–2435. [Google Scholar] [CrossRef]
- Simmons, E.M.; Hartwig, J.F. On the Interpretation of Deuterium Kinetic Isotope Effects in C-H Bond Functionalizations by Transition-Metal Complexes. Angew. Chem. Int. Ed. Engl. 2012, 51, 3066–3072. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Li, Y.; Bai, R.; Lan, Y. Mechanism of Rhodium-Catalyzed C–H Functionalization: Advances in Theoretical Investigation. Acc. Chem. Res. 2017, 50, 2799–2808. [Google Scholar] [CrossRef] [PubMed]
- Davies, D.L.; Macgregor, S.A.; McMullin, C.L. Computational Studies of Carboxylate-Assisted C–H Activation and Functionalization at Group 8–10 Transition Metal Centers. Chem. Rev. 2017, 117, 8649–8709. [Google Scholar] [CrossRef] [PubMed]
- Balcells, D.; Clot, E.; Eisenstein, O. C–H Bond Activation in Transition Metal Species from a Computational Perspective. Chem. Rev. 2010, 110, 749–823. [Google Scholar] [CrossRef] [PubMed]
- Gensch, T.; Hopkinson, M.N.; Glorius, F.; Wencel-Delord, J. Mild metal-catalyzed C-H activation: Examples and concepts. Chem. Soc. Rev. 2016, 45, 2900–2936. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, J.F.; Larsen, M.A. Undirected, Homogeneous C–H Bond Functionalization: Challenges and Opportunities. ACS Cent. Sci. 2016, 2, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Brückl, T.; Baxter, R.D.; Ishihara, Y.; Baran, P.S. Innate and Guided C–H Functionalization Logic. Acc. Chem. Res. 2012, 45, 826–839. [Google Scholar] [CrossRef] [PubMed]
- Ping, L.; Chung, D.S.; Bouffard, J.; Lee, S.G. Transition metal-catalyzed site- and regio-divergent C-H bond functionalization. Chem. Soc. Rev. 2017, 46, 4299–4328. [Google Scholar] [CrossRef] [PubMed]
- Saint-Denis, T.G.; Zhu, R.-Y.; Chen, G.; Wu, Q.-F.; Yu, J.-Q. Enantioselective C(sp3)‒H bond activation by chiral transition metal catalysts. Science 2018, 359, eaao4798. [Google Scholar] [CrossRef] [PubMed]
- Sambiagio, C.; Schönbauer, D.; Blieck, R.; Dao-Huy, T.; Pototschnig, G.; Schaaf, P.; Wiesinger, T.; Zia, M.F.; Wencel-Delord, J.; Besset, T.; et al. A comprehensive overview of directing groups applied in metal-catalysed C–H functionalisation chemistry. Chem. Soc. Rev. 2018, 47, 6603–6743. [Google Scholar] [CrossRef] [PubMed]
- Lyons, T.W.; Sanford, M.S. Palladium-Catalyzed Ligand-Directed C−H Functionalization Reactions. Chem. Rev. 2010, 110, 1147–1169. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Agasti, S.; Maiti, D. Palladium catalysed meta-C–H functionalization reactions. Org. Biomol. Chem. 2016, 14, 5440–5453. [Google Scholar] [CrossRef] [PubMed]
- Leitch, J.A.; Frost, C.G. Ruthenium-catalysed σ-activation for remote meta-selective C–H functionalisation. Chem. Soc. Rev. 2017, 46, 7145–7153. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Maity, S.; Maiti, D. Reaching the south: Metal-catalyzed transformation of the aromatic para-position. Chem. Commun. 2016, 52, 12398–12414. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, G.; Breit, B. Removable Directing Groups in Organic Synthesis and Catalysis. Angew. Chem. Int. Ed. 2011, 50, 2450–2494. [Google Scholar] [CrossRef] [PubMed]
- Yadav, M.R.; Rit, R.K.; Shankar, M.; Sahoo, A.K. Reusable and Removable Directing Groups for C(sp2)−H Bond Functionalization of Arenes. ASIAN J. Org. Chem. 2015, 4, 846–864. [Google Scholar] [CrossRef]
- Zhang, F.; Spring, D.R. Arene C–H functionalisation using a removable/modifiable or a traceless directing group strategy. Chem. Soc. Rev. 2014, 43, 6906–6919. [Google Scholar] [CrossRef] [PubMed]
- Font, M.; Quibell, J.M.; Perry, G.J.P.; Larrosa, I. The use of carboxylic acids as traceless directing groups for regioselective C–H bond functionalisation. Chem. Commun. 2017, 53, 5584–5597. [Google Scholar] [CrossRef] [PubMed]
- Gandeepan, P.; Ackermann, L. Transient Directing Groups for Transformative C–H Activation by Synergistic Metal Catalysis. Chem 2018, 4, 199–222. [Google Scholar] [CrossRef]
- Ihara, H.; Koyanagi, M.; Suginome, M. Anthranilamide: A Simple, Removable ortho-Directing Modifier for Arylboronic Acids Serving also as a Protecting Group in Cross-Coupling Reactions. Org. Lett. 2011, 13, 2662–2665. [Google Scholar] [CrossRef] [PubMed]
- Ihara, H.; Suginome, M. Easily Attachable and Detachable ortho-Directing Agent for Arylboronic Acids in Ruthenium-Catalyzed Aromatic C−H Silylation. J. Am. Chem. Soc. 2009, 131, 7502–7503. [Google Scholar] [CrossRef] [PubMed]
- Ihara, H.; Ueda, A.; Suginome, M. Ruthenium-catalyzed C-H Silylation of Methylboronic Acid Using a Removable α-Directing Modifier on the Boron Atom. Chem. Lett. 2011, 40, 916–918. [Google Scholar] [CrossRef]
- Yamamoto, T.; Ishibashi, A.; Suginome, M. Regioselective Synthesis of o-Benzenediboronic Acids via Ir-Catalyzed o-C–H Borylation Directed by a Pyrazolylaniline-Modified Boronyl Group. Org. Lett. 2017, 19, 886–889. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Poisson, T.; Pannecoucke, X.; Besset, T. The Transient Directing Group Strategy: A New Trend in Transition-Metal-Catalyzed C–H Bond Functionalization. Synthesis 2017, 49, 4808–4826. [Google Scholar] [CrossRef]
- Zhu, R.-Y.; Farmer, M.E.; Chen, Y.-Q.; Yu, J.-Q. A Simple and Versatile Amide Directing Group for C−H Functionalizations. Angew. Chem. Int. Ed. 2016, 55, 10578–10599. [Google Scholar] [CrossRef] [PubMed]
- Nahm, S.; Weinreb, S.M. N-methoxy-n-methylamides as effective acylating agents. Tetrahedron Lett. 1981, 22, 3815–3818. [Google Scholar] [CrossRef]
- Nowak, M. Weinreb Amides. Synlett 2015, 26, 561–562. [Google Scholar] [CrossRef]
- De Sarkar, S.; Liu, W.; Kozhushkov, S.I.; Ackermann, L. Weakly Coordinating Directing Groups for Ruthenium(II)- Catalyzed C—H Activation. Adv. Synth. Catal. 2014, 356, 1461–1479. [Google Scholar] [CrossRef]
- Arockiam, P.B.; Bruneau, C.; Dixneuf, P.H. Ruthenium(II)-catalyzed C-H bond activation and functionalization. Chem. Rev. 2012, 112, 5879–5918. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, S.; Villuendas, P.; Urriolabeitia, E.P. Ru-catalysed C–H functionalisations as a tool for selective organic synthesis. Tetrahedron Lett. 2016, 57, 3413–3432. [Google Scholar] [CrossRef]
- Yang, F.; Ackermann, L. Ruthenium-Catalyzed C–H Oxygenation on Aryl Weinreb Amides. Org. Lett. 2013, 15, 718–720. [Google Scholar] [CrossRef] [PubMed]
- More, N.Y.; Padala, K.; Jeganmohan, M. Ruthenium-Catalyzed C–H Benzoxylation of tert-Benzamides with Aromatic Acids by Weak Coordination. J. Org. Chem. 2017, 82, 12691–12700. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; Kapur, M. Fujiwara-Moritani Reaction of Weinreb Amides using a Ruthenium-Catalyzed C-H Functionalization Reaction. Chem. Asian J. 2015, 10, 1505–1512. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, E.M.; Zhang, H.; Stoltz, B.M. Oxidative Heck-Type Reactions (Fujiwara–Moritani Reactions). In The Mizoroki–Heck Reaction; Oestreich, M., Ed.; Wiley: Chichester, UK, 2009. [Google Scholar]
- Ackermann, L. Carboxylate-Assisted Transition-Metal-Catalyzed C−H Bond Functionalizations: Mechanism and Scope. Chem. Rev. 2011, 111, 1315–1345. [Google Scholar] [CrossRef] [PubMed]
- Mo, J.; Wang, L.; Liu, Y.; Cui, X. Transition-Metal-Catalyzed Direct C–H Functionalization under External-Oxidant-Free Conditions. Synthesis 2015, 47, 439–459. [Google Scholar] [CrossRef]
- Das, R.; Kapur, M. Product Control using Substrate Design: Ruthenium-Catalysed Oxidative C-H Olefinations of Cyclic Weinreb Amides. Chem. Eur. J. 2016, 22, 16986–16990. [Google Scholar] [CrossRef] [PubMed]
- Gandeepan, P.; Müller, T.; Zell, D.; Cera, G.; Warratz, S.; Ackermann, L. 3d Transition Metals for C–H Activation. Chem. Rev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Moselage, M.; Li, J.; Ackermann, L. Cobalt-Catalyzed C–H Activation. ACS Catal. 2016, 6, 498–525. [Google Scholar] [CrossRef]
- Gao, K.; Yoshikai, N. Low-Valent Cobalt Catalysis: New Opportunities for C–H Functionalization. Acc. Chem. Res. 2014, 47, 1208–1219. [Google Scholar] [CrossRef] [PubMed]
- Yoshikai, N. Development of Cobalt-Catalyzed C–H Bond Functionalization Reactions. Bull. Chem. Soc. Jpn. 2014, 87, 843–857. [Google Scholar] [CrossRef]
- Yoshino, T.; Ikemoto, H.; Matsunaga, S.; Kanai, M. Cp*CoIII-Catalyzed C2-Selective Addition of Indoles to Imines. Chem. A Eur. J. 2013, 19, 9142–9146. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, T.; Ikemoto, H.; Matsunaga, S.; Kanai, M. A Cationic High-Valent Cp*CoIII Complex for the Catalytic Generation of Nucleophilic Organometallic Species: Directed C–H Bond Activation. Angew. Chem. Int. Ed. Engl. 2013, 52, 2207–2211. [Google Scholar] [CrossRef] [PubMed]
- Bunno, Y.; Murakami, N.; Suzuki, Y.; Kanai, M.; Yoshino, T.; Matsunaga, S. Cp*CoIII-Catalyzed Dehydrative C–H Allylation of 6-Arylpurines and Aromatic Amides Using Allyl Alcohols in Fluorinated Alcohols. Org. Lett. 2016, 18, 2216–2219. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Yoshino, T.; Matsunaga, S.; Kanai, M. Air-Stable Carbonyl(pentamethylcyclopentadienyl)cobalt Diiodide Complex as a Precursor for Cationic (Pentamethylcyclopentadienyl)cobalt(III) Catalysis: Application for Directed C-2 Selective C-H Amidation of Indoles. Adv. Synth. Catal. 2014, 356, 1491–1495. [Google Scholar] [CrossRef]
- Colomer, I.; Chamberlain, A.E.R.; Haughey, M.B.; Donohoe, T.J. Hexafluoroisopropanol as a highly versatile solvent. Nat. Rev. Chem. 2017, 1, 0088. [Google Scholar] [CrossRef]
- Wencel-Delord, J.; Colobert, F. A remarkable solvent effect of fluorinated alcohols on transition metal catalysed C–H functionalizations. Org. Chem. Front. 2016, 3, 394–400. [Google Scholar] [CrossRef]
- Planas, O.; Chirila, P.G.; Whiteoak, C.J.; Ribas, X. Current Mechanistic Understanding of Cobalt-Catalyzed C–H Functionalization. Adv. Organomet. Chem. 2018, 69, 209–282. [Google Scholar] [CrossRef]
- Gorelsky, S.I.; Lapointe, D.; Fagnou, K. Analysis of the Concerted Metalation-Deprotonation Mechanism in Palladium-Catalyzed Direct Arylation Across a Broad Range of Aromatic Substrates. J. Am. Chem. Soc. 2008, 130, 10848–10849. [Google Scholar] [CrossRef] [PubMed]
- Kawai, K.; Bunno, Y.; Yoshino, T.; Matsunaga, S. Weinreb Amide Directed Versatile C–H Bond Functionalization under (eta(5)-Pentamethylcyclopentadienyl)cobalt(III) Catalysis. Chem. Eur. J. 2018, 24, 10231–10237. [Google Scholar] [CrossRef] [PubMed]
- Kuhl, N.; Schröder, N.; Glorius, F. Rh(III)-Catalyzed Halogenation of Vinylic C–H Bonds: Rapid and General Access to Z-Halo Acrylamides. Org. Lett. 2013, 15, 3860–3863. [Google Scholar] [CrossRef] [PubMed]
- Chirila, P.G.; Adams, J.; Dirjal, A.; Hamilton, A.; Whiteoak, C.J. Cp*Co(III)-Catalyzed Coupling of Benzamides with α,β-Unsaturated Carbonyl Compounds: Preparation of Aliphatic Ketones and Azepinones. Chem. Eur. J. 2018, 24, 3584–3589. [Google Scholar] [CrossRef] [PubMed]
- Johansson Seechurn, C.C.C.; Kitching, M.O.; Colacot, T.J.; Snieckus, V. Palladium-Catalyzed Cross-Coupling: A Historical Contextual Perspective to the 2010 Nobel Prize. Angew. Chem. Int. Ed. Engl. 2012, 51, 5062–5085. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.-F.; Neumann, H.; Beller, M. Synthesis of Heterocycles via Palladium-Catalyzed Carbonylations. Chem. Rev. 2013, 113, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Weinstein, A.B.; White, P.B.; Stahl, S.S. Ligand-Promoted Palladium-Catalyzed Aerobic Oxidation Reactions. Chem. Rev. 2018, 118, 2636–2679. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Castillo, P.; Buchwald, S.L. Applications of Palladium-Catalyzed C–N Cross-Coupling Reactions. Chem. Rev. 2016, 116, 12564–12649. [Google Scholar] [CrossRef] [PubMed]
- Sehnal, P.; Taylor, R.J.K.; Fairlamb, I.J.S. Emergence of Palladium(IV) Chemistry in Synthesis and Catalysis. Chem. Rev. 2010, 110, 824–889. [Google Scholar] [CrossRef] [PubMed]
- Timsina, Y.N.; Gupton, B.F.; Ellis, K.C. Palladium-Catalyzed C–H Amination of C(sp2) and C(sp3)–H Bonds: Mechanism and Scope for N-Based Molecule Synthesis. ACS Catal. 2018, 8, 5732–5776. [Google Scholar] [CrossRef]
- Topczewski, J.J.; Sanford, M.S. Carbon–hydrogen (C–H) bond activation at PdIV: A Frontier in C–H functionalization catalysis. Chem. Sci. 2015, 6, 70–76. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Wasa, M.; Chan, K.S.L.; Shao, Q.; Yu, J.-Q. Palladium-Catalyzed Transformations of Alkyl C–H Bonds. Chem. Rev. 2017, 117, 8754–8786. [Google Scholar] [CrossRef] [PubMed]
- Bonney, K.J.; Schoenebeck, F. Experiment and computation: A combined approach to study the reactivity of palladium complexes in oxidation states 0 to iv. Chem. Soc. Rev. 2014, 43, 6609–6638. [Google Scholar] [CrossRef] [PubMed]
- Valente, C.; Çalimsiz, S.; Hoi, K.H.; Mallik, D.; Sayah, M.; Organ, M.G. The Development of Bulky Palladium NHC Complexes for the Most-Challenging Cross-Coupling Reactions. Angew. Chem. Int. Ed. Engl. 2012, 51, 3314–3332. [Google Scholar] [CrossRef] [PubMed]
- Hazari, N.; Melvin, P.R.; Beromi, M.M. Well-defined nickel and palladium precatalysts for cross-coupling. Nat. Rev. Chem. 2017, 1, 0025. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, K.; Lan, Q.; Wang, X.-S. Pd(II)-catalyzed C–H arylation of aryl and benzyl Weinreb amides. Org. Biomol. Chem. 2015, 13, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Mudarra, Á.L.; Martínez de Salinas, S.; Pérez-Temprano, M.H. Beyond the traditional roles of Ag in catalysis: The transmetalating ability of organosilver(i) species in Pd-catalysed reactions. Org. Biomol. Chem. 2018, 17, 1655–1667. [Google Scholar] [CrossRef] [PubMed]
- Yedage, S.L.; Bhanage, B.M. Palladium-Catalyzed Deaminative Phenanthridinone Synthesis from Aniline via C–H Bond Activation. J. Org. Chem. 2016, 81, 4103–4111. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Wan, L.; Zhang, G.; Leow, D.; Spangler, J.; Yu, J.-Q. Pd(II)-Catalyzed C–H Functionalizations Directed by Distal Weakly Coordinating Functional Groups. J. Am. Chem. Soc. 2015, 137, 4391–4397. [Google Scholar] [CrossRef] [PubMed]
- Engle, K.M.; Wang, D.-H.; Yu, J.-Q. Ligand-Accelerated C−H Activation Reactions: Evidence for a Switch of Mechanism. J. Am. Chem. Soc. 2010, 132, 14137–14151. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; Kapur, M. Transition-Metal-Catalyzed Site-Selective C−H Halogenation Reactions. Asian J. Org. Chem. 2018, 7, 1524–1541. [Google Scholar] [CrossRef]
- Das, R.; Kapur, M. Palladium-Catalyzed, ortho-Selective C–H Halogenation of Benzyl Nitriles, Aryl Weinreb Amides, and Anilides. J. Org. Chem. 2017, 82, 1114–1126. [Google Scholar] [CrossRef] [PubMed]
- Powers, D.C.; Lee, E.; Ariafard, A.; Sanford, M.S.; Yates, B.F.; Canty, A.J.; Ritter, T. Connecting Binuclear Pd(III) and Mononuclear Pd(IV) Chemistry by Pd–Pd Bond Cleavage. J. Am. Chem. Soc. 2012, 134, 12002–12009. [Google Scholar] [CrossRef] [PubMed]
- Powers, D.C.; Benitez, D.; Tkatchouk, E.; Goddard, W.A.; Ritter, T. Bimetallic Reductive Elimination from Dinuclear Pd(III) Complexes. J. Am. Chem. Soc. 2010, 132, 14092–14103. [Google Scholar] [CrossRef] [PubMed]
- Powers, D.C.; Ritter, T. Bimetallic Pd(III) complexes in palladium-catalysed carbon–heteroatom bond formation. Nat. Chem. 2009, 1, 419. [Google Scholar] [CrossRef]
- Powers, D.C.; Geibel, M.A.L.; Klein, J.E.M.N.; Ritter, T. Bimetallic Palladium Catalysis: Direct Observation of Pd(III)−Pd(III) Intermediates. J. Am. Chem. Soc. 2009, 131, 17050–17051. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Chekshin, N.; Shen, P.-X.; Yu, J.-Q. Ligand-Enabled, Palladium-Catalyzed β-C(sp3)–H Arylation of Weinreb Amides. ACS Catal. 2018, 8, 9292–9297. [Google Scholar] [CrossRef]
- Willcox, D.; Chappell, B.G.N.; Hogg, K.F.; Calleja, J.; Smalley, A.P.; Gaunt, M.J. A general catalytic β-C–H carbonylation of aliphatic amines to β-lactams. Science 2016, 354, 851–857. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.-Y.; Li, Z.-Q.; Park, H.S.; Senanayake, C.H.; Yu, J.-Q. Ligand-Enabled γ-C(sp3)–H Activation of Ketones. J. Am. Chem. Soc. 2018, 140, 3564–3568. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.-Y.; He, J.; Wang, X.-C.; Yu, J.-Q. Ligand-Promoted Alkylation of C(sp3)–H and C(sp2)–H Bonds. J. Am. Chem. Soc. 2014, 136, 13194–13197. [Google Scholar] [CrossRef] [PubMed]
- Baudoin, O. Ring Construction by Palladium(0)-Catalyzed C(sp3)–H Activation. Acc. Chem. Res. 2017, 50, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- Baudoin, O. Transition metal-catalyzed arylation of unactivated C(sp3)–H bonds. Chem. Soc. Rev. 2011, 40, 4902–4911. [Google Scholar] [CrossRef] [PubMed]
- Chatani, N.; Rej, S. Rh-Catalyzed Removable Directing Group Assisted sp2 or sp3-C-H Bond Functionalization. Angew. Chem. Int. Ed. 2018. [Google Scholar] [CrossRef]
- Colby, D.A.; Bergman, R.G.; Ellman, J.A. Rhodium-Catalyzed C−C Bond Formation via Heteroatom-Directed C−H Bond Activation. Chem. Rev. 2010, 110, 624–655. [Google Scholar] [CrossRef] [PubMed]
- Vásquez-Céspedes, S.; Wang, X.; Glorius, F. Plausible Rh(V) Intermediates in Catalytic C–H Activation Reactions. ACS Catal. 2018, 8, 242–257. [Google Scholar] [CrossRef]
- Davies, D.L.; Ellul, C.E.; Macgregor, S.A.; McMullin, C.L.; Singh, K. Experimental and DFT Studies Explain Solvent Control of C–H Activation and Product Selectivity in the Rh(III)-Catalyzed Formation of Neutral and Cationic Heterocycles. J. Am. Chem. Soc. 2015, 137, 9659–9669. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Qiu, Z. DFT Studies on the Mechanism of the Rhodium(III)-Catalyzed C–H Activation of N-Phenoxyacetamide. J. Org. Chem. 2015, 80, 10686–10693. [Google Scholar] [CrossRef] [PubMed]
- Davis, T.A.; Hyster, T.K.; Rovis, T. Rhodium(III)-catalyzed intramolecular hydroarylation, amidoarylation, and Heck-type reaction: Three distinct pathways determined by an amide directing group. Angew. Chem. Int. Ed. 2013, 52, 14181–14185. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, C.; Li, Y.; Yin, F.; Wang, X.-S. Rhodium-Catalyzed C-H Olefination of Aryl Weinreb Amides. Adv. Synth. Catal. 2013, 355, 1724–1728. [Google Scholar] [CrossRef]
- Wang, S.-M.; Li, C.; Leng, J.; Bukhari, S.N.A.; Qin, H.-L. Rhodium(iii)-catalyzed Oxidative Coupling of N-Methoxybenzamides and Ethenesulfonyl fluoride: A C–H Bond Activation Strategy for the Preparation of 2-Aryl ethenesulfonyl fluorides and Sulfonyl fluoride Substituted γ-Lactams. Org. Chem. Front. 2018, 5, 1411–1415. [Google Scholar] [CrossRef]
- Wang, C.-S.; Dixneuf, P.H.; Soulé, J.-F. Photoredox Catalysis for Building C–C Bonds from C(sp2)–H Bonds. Chem. Rev. 2018, 118, 7532–7585. [Google Scholar] [CrossRef] [PubMed]
- Shaw, M.H.; Twilton, J.; MacMillan, D.W.C. Photoredox Catalysis in Organic Chemistry. J. Org. Chem. 2016, 81, 6898–6926. [Google Scholar] [CrossRef] [PubMed]
- Romero, N.A.; Nicewicz, D.A. Organic Photoredox Catalysis. Chem. Rev. 2016, 116, 10075–10166. [Google Scholar] [CrossRef] [PubMed]
- Lang, X.; Zhao, J.; Chen, X. Cooperative photoredox catalysis. Chem. Soc. Rev. 2016, 45, 3026–3038. [Google Scholar] [CrossRef] [PubMed]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W.C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef] [PubMed]
- Fabry, D.C.; Rueping, M. Merging Visible Light Photoredox Catalysis with Metal Catalyzed C–H Activations: On the Role of Oxygen and Superoxide Ions as Oxidants. Acc. Chem. Res. 2016, 49, 1969–1979. [Google Scholar] [CrossRef] [PubMed]
- Fabry, D.C.; Zoller, J.; Raja, S.; Rueping, M. Combining rhodium and photoredox catalysis for C-H functionalizations of arenes: Oxidative Heck reactions with visible light. Angew. Chem. Int. Ed. 2014, 53, 10228–10231. [Google Scholar] [CrossRef] [PubMed]
- Fyfe, J.W.B.; Watson, A.J.B. Recent Developments in Organoboron Chemistry: Old Dogs, New Tricks. Chem 2017, 3, 31–55. [Google Scholar] [CrossRef]
- Xu, L.; Zhang, S.; Li, P. Boron-selective reactions as powerful tools for modular synthesis of diverse complex molecules. Chem. Soc. Rev. 2015, 44, 8848–8858. [Google Scholar] [CrossRef] [PubMed]
- Lennox, A.J.J.; Lloyd-Jones, G.C. Selection of boron reagents for Suzuki–Miyaura coupling. Chem. Soc. Rev. 2014, 43, 412–443. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, J.W.; Blair, D.J.; Burke, M.D. Towards the generalized iterative synthesis of small molecules. Nat. Rev. Chem. 2018, 2, 0115. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Grillo, A.S.; Burke, M.D. From Synthesis to Function via Iterative Assembly of N-Methyliminodiacetic Acid Boronate Building Blocks. Acc. Chem. Res. 2015, 48, 2297–2307. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ballmer, S.G.; Gillis, E.P.; Fujii, S.; Schmidt, M.J.; Palazzolo, A.M.E.; Lehmann, J.W.; Morehouse, G.F.; Burke, M.D. Synthesis of many different types of organic small molecules using one automated process. Science 2015, 347, 1221–1226. [Google Scholar] [CrossRef] [PubMed]
- Woerly, E.M.; Roy, J.; Burke, M.D. Synthesis of most polyene natural product motifs using just 12 building blocks and one coupling reaction. Nat. Chem. 2014, 6, 484. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, J.F. Borylation and Silylation of C–H Bonds: A Platform for Diverse C–H Bond Functionalizations. Acc. Chem. Res. 2011, 45, 864–873. [Google Scholar] [CrossRef] [PubMed]
- Mkhalid, I.A.; Barnard, J.H.; Marder, T.B.; Murphy, J.M.; Hartwig, J.F. C-H activation for the construction of C-B bonds. Chem. Rev. 2010, 110, 890–931. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.R.; Bisht, R.; Haldar, C.; Pandey, G.; Dannatt, J.E.; Ghaffari, B.; Maleczka, R.E.; Chattopadhyay, B. Achieving High Ortho Selectivity in Aniline C–H Borylations by Modifying Boron Substituents. ACS Catal. 2018, 8, 6216–6223. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-L.; Kanai, M.; Kuninobu, Y. Iridium/Bipyridine-Catalyzed ortho-Selective C–H Borylation of Phenol and Aniline Derivatives. Org. Lett. 2017, 19, 5944–5947. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, B.; Dannatt, J.E.; Andujar-De Sanctis, I.L.; Gore, K.A.; Maleczka, R.E.; Singleton, D.A.; Smith, M.R. Ir-Catalyzed ortho-Borylation of Phenols Directed by Substrate–Ligand Electrostatic Interactions: A Combined Experimental/in Silico Strategy for Optimizing Weak Interactions. J. Am. Chem. Soc. 2017, 139, 7864–7871. [Google Scholar] [CrossRef] [PubMed]
- Hale, L.V.A.; Emmerson, D.G.; Ling, E.F.; Roering, A.J.; Ringgold, M.A.; Clark, T.B. An ortho-directed C–H borylation/Suzuki coupling sequence in the formation of biphenylbenzylic amines. Org. Chem. Front. 2015, 2, 661–664. [Google Scholar] [CrossRef]
- Crawford, K.M.; Ramseyer, T.R.; Daley, C.J.A.; Clark, T.B. Phosphine-Directed C—H Borylation Reactions: Facile and Selective Access to Ambiphilic Phosphine Boronate Esters. Angew. Chem. 2014, 53, 7589–7593. [Google Scholar] [CrossRef] [PubMed]
- Kawamorita, S.; Ohmiya, H.; Hara, K.; Fukuoka, A.; Sawamura, M. Directed Ortho Borylation of Functionalized Arenes Catalyzed by a Silica-Supported Compact Phosphine−Iridium System. J. Am. Chem. Soc. 2009, 131, 5058–5059. [Google Scholar] [CrossRef] [PubMed]
- Hoque, M.E.; Bisht, R.; Haldar, C.; Chattopadhyay, B. Noncovalent Interactions in Ir-Catalyzed C–H Activation: L-Shaped Ligand for Para-Selective Borylation of Aromatic Esters. J. Am. Chem. Soc. 2017, 139, 7745–7748. [Google Scholar] [CrossRef] [PubMed]
- Tajuddin, H.; Harrisson, P.; Bitterlich, B.; Collings, J.C.; Sim, N.; Batsanov, A.S.; Cheung, M.S.; Kawamorita, S.; Maxwell, A.C.; Shukla, L.; et al. Iridium-catalyzed C–H borylation of quinolines and unsymmetrical 1,2-disubstituted benzenes: Insights into steric and electronic effects on selectivity. Chem. Sci. 2012, 3, 3505–3515. [Google Scholar] [CrossRef]
- Larsen, M.A.; Hartwig, J.F. Iridium-Catalyzed C–H Borylation of Heteroarenes: Scope, Regioselectivity, Application to Late-Stage Functionalization, and Mechanism. J. Am. Chem. Soc. 2014, 136, 4287–4299. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, J.F. Regioselectivity of the borylation of alkanes and arenes. Chem. Soc. Rev. 2011, 40, 1992–2002. [Google Scholar] [CrossRef] [PubMed]
- Ghaffari, B.; Preshlock, S.M.; Plattner, D.L.; Staples, R.J.; Maligres, P.E.; Krska, S.W.; Maleczka, R.E.; Smith, M.R. Silyl Phosphorus and Nitrogen Donor Chelates for Homogeneous Ortho Borylation Catalysis. J. Am. Chem. Soc. 2014, 136, 14345–14348. [Google Scholar] [CrossRef] [PubMed]
- Newton, C.G.; Wang, S.-G.; Oliveira, C.C.; Cramer, N. Catalytic Enantioselective Transformations Involving C–H Bond Cleavage by Transition-Metal Complexes. Chem. Rev. 2017, 117, 8908–8976. [Google Scholar] [CrossRef] [PubMed]
- Shirai, T.; Yamamoto, Y. Cationic Iridium/S-Me-BIPAM-Catalyzed Direct Asymmetric Intermolecular Hydroarylation of Bicycloalkenes. Angew. Chem. Int. Ed. 2015, 54, 9894–9897. [Google Scholar] [CrossRef] [PubMed]
- Erbing, E. Development of New Efficient Iridium-Catalyzed Methods for the Construction of Carbon-Heteroatom Bonds. Ph.D. Thesis, Stockholm University, Stockholm, Sweden, 2018. [Google Scholar]
- Collins, K.D.; Glorius, F. A robustness screen for the rapid assessment of chemical reactions. Nat. Chem. 2013, 5, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Gensch, T.; Teders, M.; Glorius, F. Approach to Comparing the Functional Group Tolerance of Reactions. J. Org. Chem. 2017, 82, 9154–9159. [Google Scholar] [CrossRef] [PubMed]
- Erbing, E.; Sanz-Marco, A.; Vázquez-Romero, A.; Malmberg, J.; Johansson, M.J.; Gómez-Bengoa, E.; Martín-Matute, B. Base- and Additive-Free Ir-Catalyzed ortho-Iodination of Benzoic Acids: Scope and Mechanistic Investigations. ACS Catal. 2018, 8, 920–925. [Google Scholar] [CrossRef]
- Egorova, K.S.; Ananikov, V.P. Which Metals are Green for Catalysis? Comparison of the Toxicities of Ni, Cu, Fe, Pd, Pt, Rh, and Au Salts. Angew. Chem. Int. Ed. 2016, 55, 12150–12162. [Google Scholar] [CrossRef] [PubMed]
- Hayler, J.D.; Leahy, D.K.; Simmons, E.M. A Pharmaceutical Industry Perspective on Sustainable Metal Catalysis. Organometallics 2019, 38, 36–46. [Google Scholar] [CrossRef]
- Sherwood, J. European Restrictions on 1,2-Dichloroethane: C-H Activation Research and Development Should Be Liberated and not Limited. Angew. Chem. Int. Ed. 2018, 57, 14286–14290. [Google Scholar] [CrossRef] [PubMed]
- Clarke, C.J.; Tu, W.-C.; Levers, O.; Bröhl, A.; Hallett, J.P. Green and Sustainable Solvents in Chemical Processes. Chem. Rev. 2018, 118, 747–800. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K.L.; Murray, J.; Sneddon, H.F.; Wheelhouse, K.M.P.; Watson, A.J.B. Connecting the Dots: Method Development Using Sustainable Solvents. Chem 2017, 3, 365–368. [Google Scholar] [CrossRef]
- Welton, T. Solvents and sustainable chemistry. Proc. R. Soc. A Math. Phys. Eng. Sci. 2015, 471. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The ‘textbook’ application of Weinreb amides: generation of mono-addition products resulting from nucleophilic attack on their carbonyl groups.
Figure 1.
The ‘textbook’ application of Weinreb amides: generation of mono-addition products resulting from nucleophilic attack on their carbonyl groups.

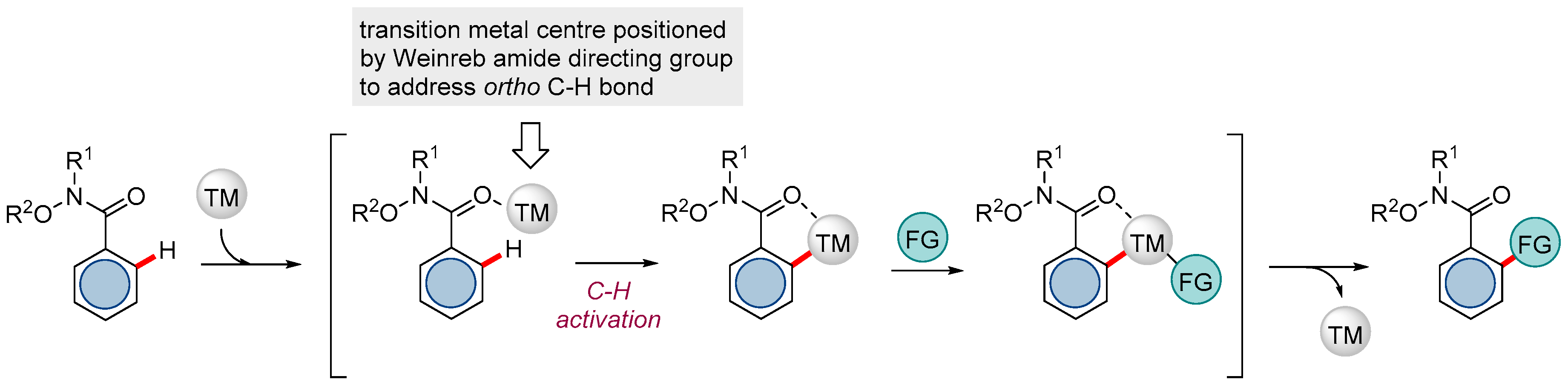
Figure 2.
A generic representation of a Weinreb amide-directed catalytic C-H functionalization (TM = transition metal).
Figure 2.
A generic representation of a Weinreb amide-directed catalytic C-H functionalization (TM = transition metal).

Scheme 1.
Ru(II)-catalyzed C-H oxidation of arenes directed by a Weinreb amide group. (a) Representative scope of the reaction with respect to arene substituents; (b) Reduction of the Weinreb amide to reveal aldehyde functionality.
Scheme 1.
Ru(II)-catalyzed C-H oxidation of arenes directed by a Weinreb amide group. (a) Representative scope of the reaction with respect to arene substituents; (b) Reduction of the Weinreb amide to reveal aldehyde functionality.

Scheme 2.
Ru(II)-catalyzed oxidative C-H ortho-alkenylation of Weinreb amides; selected products and proposed mechanism.
Scheme 2.
Ru(II)-catalyzed oxidative C-H ortho-alkenylation of Weinreb amides; selected products and proposed mechanism.

Scheme 3.
Ru-catalyzed C-H olefination using directed by cyclic Weinreb amides without N-O bond cleavage. Putative intermediates 9a–c are suggested to arise from the insertion of Ru into the substrate N-O bond.
Scheme 3.
Ru-catalyzed C-H olefination using directed by cyclic Weinreb amides without N-O bond cleavage. Putative intermediates 9a–c are suggested to arise from the insertion of Ru into the substrate N-O bond.

Scheme 4.
An initial example of the Weinreb amide-directed Co(III)-catalyzed ortho-C-H allylation.

Scheme 5.
Selected examples of C-H transformations enabled by a Co(III)-catalyzed system.

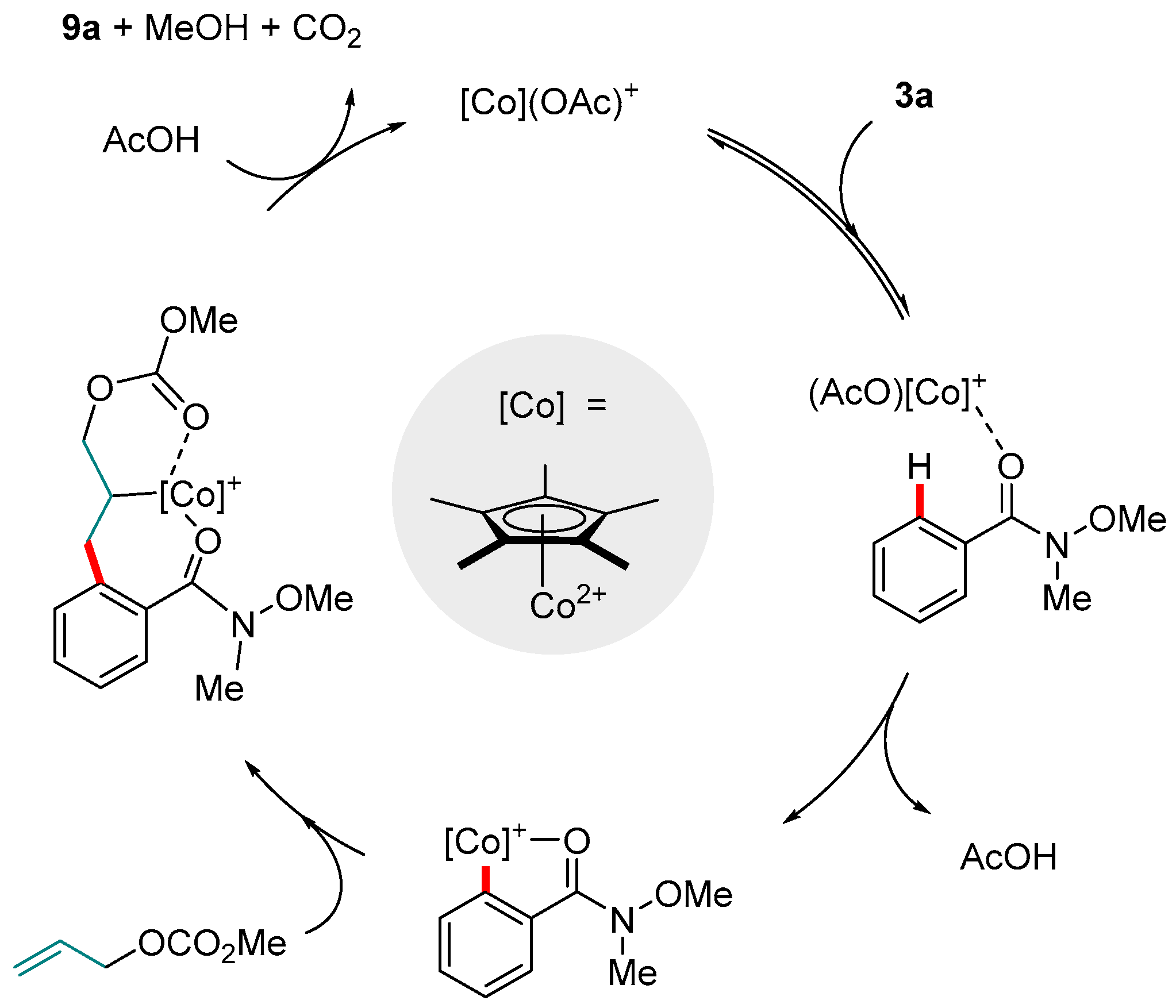
Figure 3.
A mechanism proposed by Yoshino and Matsunaga for the Co(III)-catalyzed C-H allylation of Weinreb amides.
Figure 3.
A mechanism proposed by Yoshino and Matsunaga for the Co(III)-catalyzed C-H allylation of Weinreb amides.

Scheme 6.
Pd-catalyzed C-H arylation of (a) aryl and (b) benzyl amides. Yields in parentheses refer to the amount of di-arylated products detected.
Scheme 6.
Pd-catalyzed C-H arylation of (a) aryl and (b) benzyl amides. Yields in parentheses refer to the amount of di-arylated products detected.

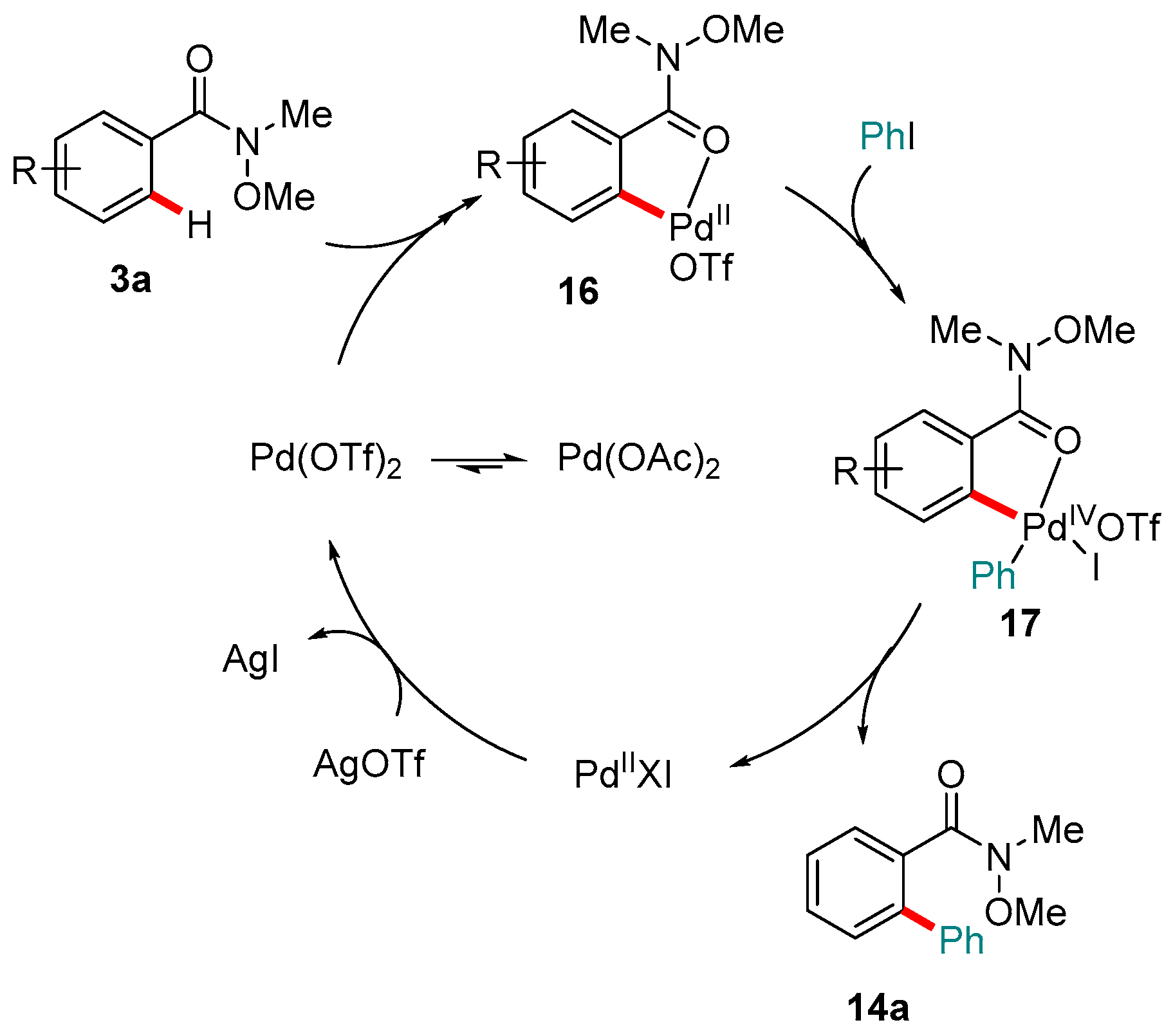
Figure 4.
Mechanism proposed by Wang and co-workers for the C-H ortho-arylation of Weinreb amides proceeding via Pd(IV) intermediates.
Figure 4.
Mechanism proposed by Wang and co-workers for the C-H ortho-arylation of Weinreb amides proceeding via Pd(IV) intermediates.

Scheme 7.
Bhanage’s C-H arylation using aniline as the electrophile source.

Scheme 8.
Yu and co-workers’ protocol for the oxidative ortho C-H acetoxylation of benzylic Weinreb amides. Yields in parentheses refer to the amount of di-olefinated products detected.
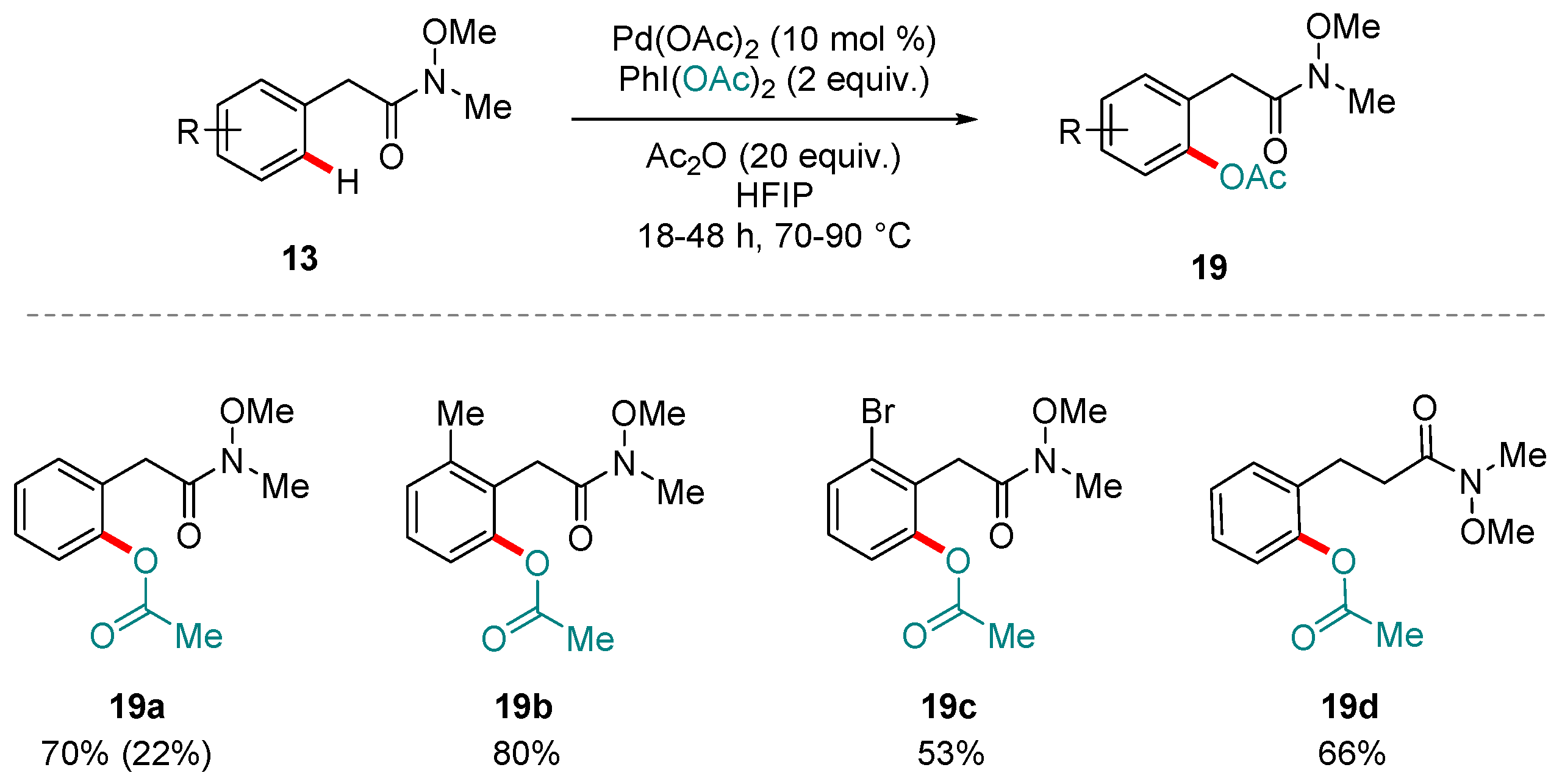
Scheme 8.
Yu and co-workers’ protocol for the oxidative ortho C-H acetoxylation of benzylic Weinreb amides. Yields in parentheses refer to the amount of di-olefinated products detected.

Scheme 9.
Yu and co-workers’ protocol for the oxidative ortho C-H acetoxylation of benzylic Weinreb amides. Yields in parentheses refer to the amount of di-acetoxylated products detected.
Scheme 9.
Yu and co-workers’ protocol for the oxidative ortho C-H acetoxylation of benzylic Weinreb amides. Yields in parentheses refer to the amount of di-acetoxylated products detected.

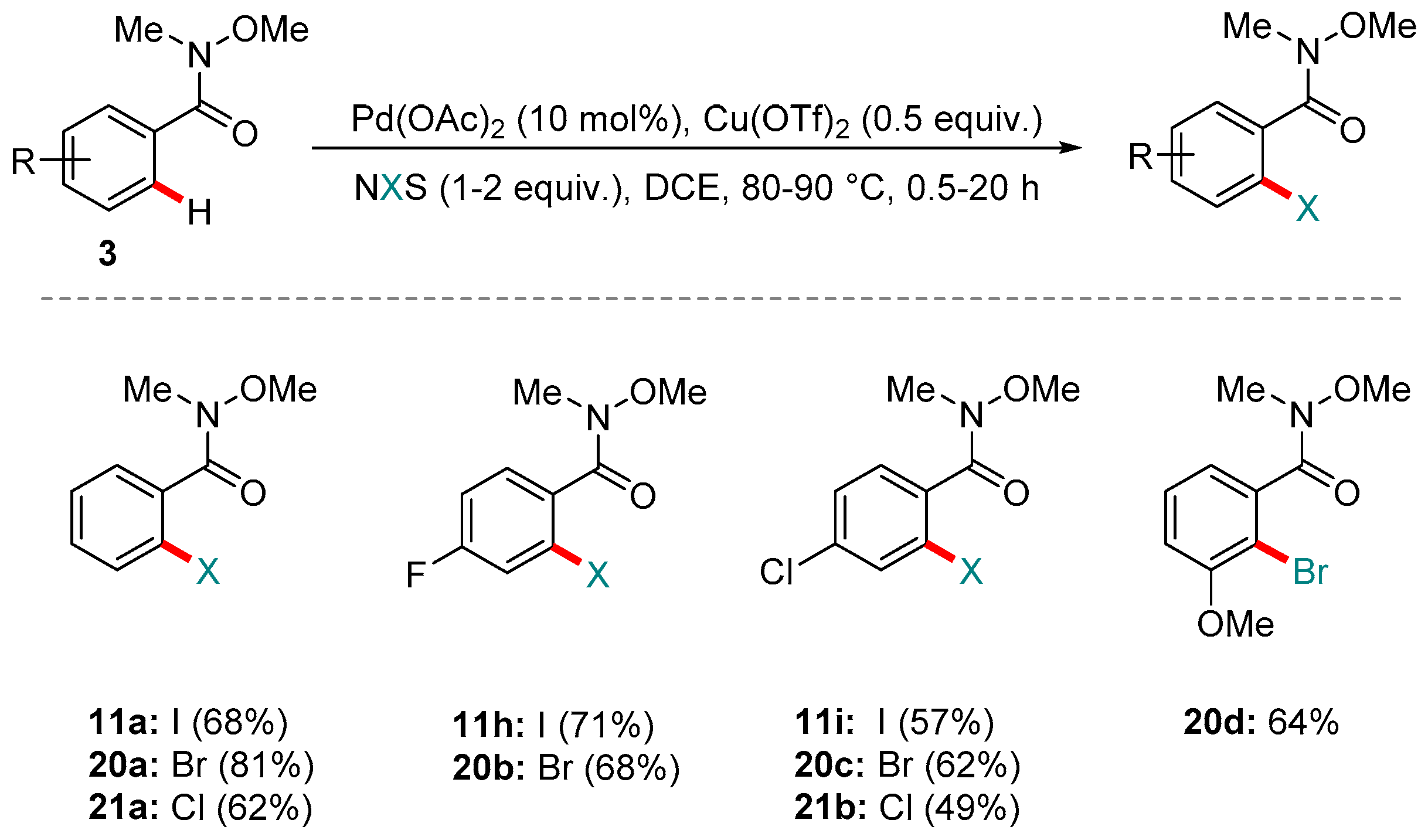
Scheme 10.
Kapur and co-workers’ reaction for the ortho C-H halogenation of aromatic Weinreb amides.
Scheme 10.
Kapur and co-workers’ reaction for the ortho C-H halogenation of aromatic Weinreb amides.

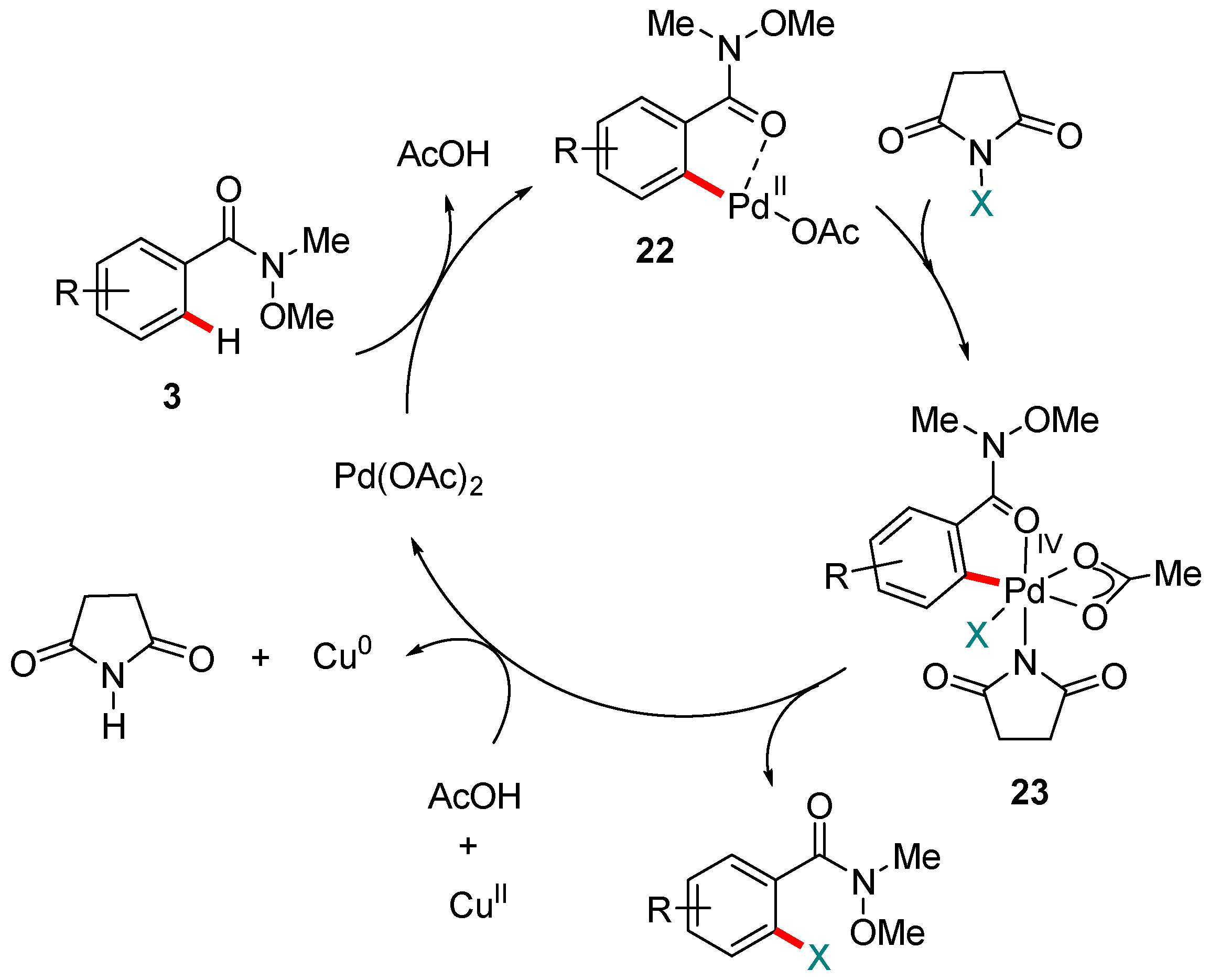
Figure 5.
An adapted version of the mechanism proposed by Kapur and co-workers for the Pd-catalyzed ortho C-H halogenation of aromatic Weinreb amides. X = Cl−, Br− or I−.
Figure 5.
An adapted version of the mechanism proposed by Kapur and co-workers for the Pd-catalyzed ortho C-H halogenation of aromatic Weinreb amides. X = Cl−, Br− or I−.

Scheme 11.
Selected scope from Yu and co-workers’ Pd-catalyzed C(sp3)-H arylation protocol enabled by Weinreb amide directing groups.
Scheme 11.
Selected scope from Yu and co-workers’ Pd-catalyzed C(sp3)-H arylation protocol enabled by Weinreb amide directing groups.

Scheme 12.
A Rh(III)-catalyzed, Weinreb amide-directed alkene hydroarylation.

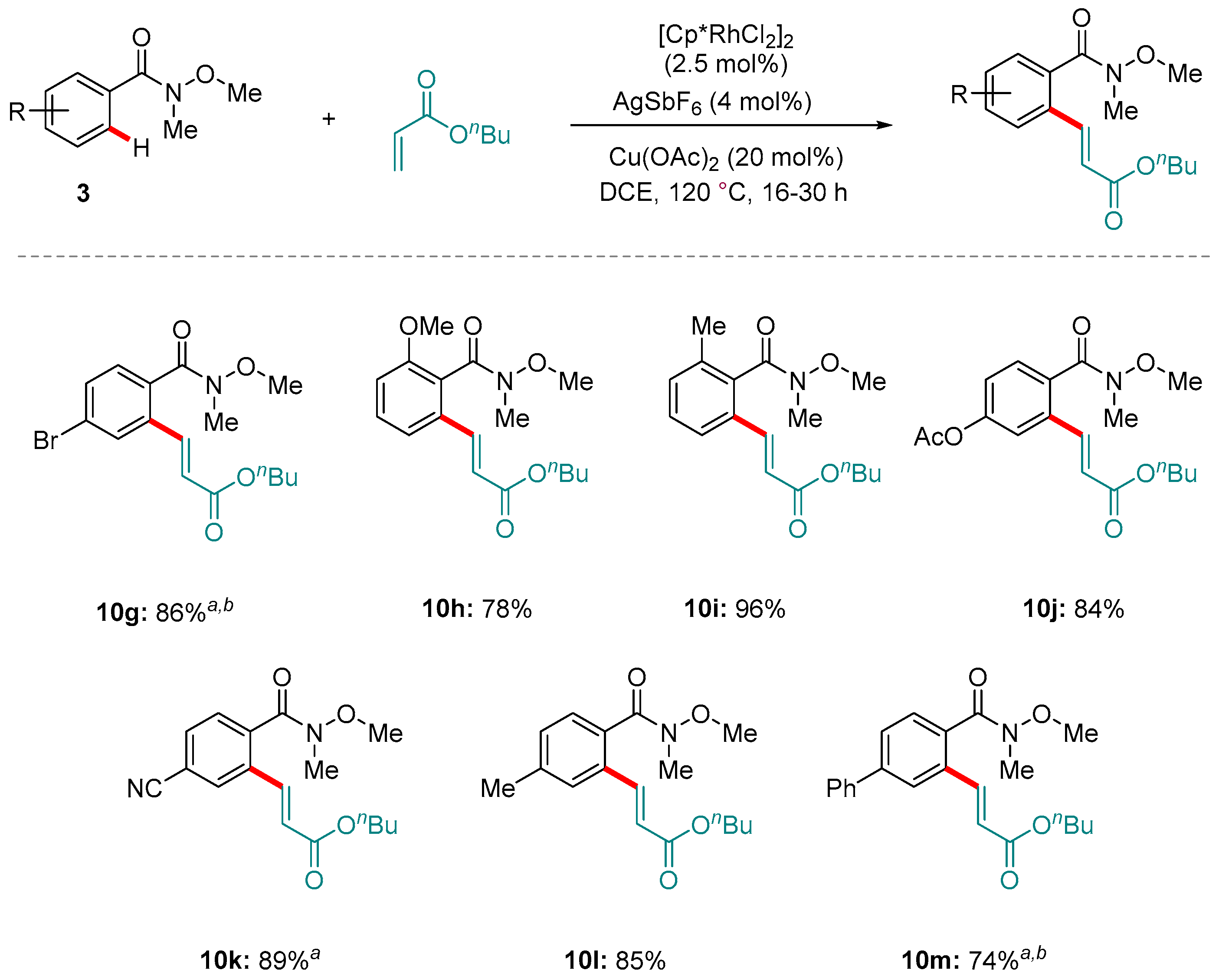
Scheme 13.
Selected examples from Wang and co-workers’ oxidative Rh-catalyzed C-H alkenylation directed by Weinreb amides. a [Cp*RhCl2]2 (2.5 mol%) and AgSbF6 (10 mol%) loadings were used. b A reaction temperature of 130 °C was used.
Scheme 13.
Selected examples from Wang and co-workers’ oxidative Rh-catalyzed C-H alkenylation directed by Weinreb amides. a [Cp*RhCl2]2 (2.5 mol%) and AgSbF6 (10 mol%) loadings were used. b A reaction temperature of 130 °C was used.

Figure 6.
An adapted version of the mechanism proposed by Wang and co-workers for the oxidative C-H alkenylation of aromatic Weinreb amides catalyzed by Rh(III).
Figure 6.
An adapted version of the mechanism proposed by Wang and co-workers for the oxidative C-H alkenylation of aromatic Weinreb amides catalyzed by Rh(III).

Scheme 14.
Rh-catalyzed olefination of aromatic Weinreb amides enabled by Ru photocatalysis.

Figure 7.
Mechanism proposed by Rueping and co-workers for the oxidative C-H olefination of aromatic Weinreb amides catalyzed by Rh(III) in the presence of a Ru photocatalyst.
Figure 7.
Mechanism proposed by Rueping and co-workers for the oxidative C-H olefination of aromatic Weinreb amides catalyzed by Rh(III) in the presence of a Ru photocatalyst.

Scheme 15.
Ir-catalyzed ortho-C-H borylation of an arene using the Weinreb amide as a directing group.
Scheme 15.
Ir-catalyzed ortho-C-H borylation of an arene using the Weinreb amide as a directing group.

Scheme 16.
Ir-catalyzed enantioselective C-H hydroarylation of an olefin, directed by a Weinreb amide.
Scheme 16.
Ir-catalyzed enantioselective C-H hydroarylation of an olefin, directed by a Weinreb amide.

Scheme 17.
Ir-catalyzed C-H iodination of Weinreb amides developed by the Martín-Matute group. a Ratio of isomers is indicated in parenthesis; the major isomer is shown.
Scheme 17.
Ir-catalyzed C-H iodination of Weinreb amides developed by the Martín-Matute group. a Ratio of isomers is indicated in parenthesis; the major isomer is shown.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kalepu, J.; Pilarski, L.T. Weinreb Amides as Directing Groups for Transition Metal-Catalyzed C-H Functionalizations. Molecules 2019, 24, 830. https://doi.org/10.3390/molecules24050830
AMA Style
Kalepu J, Pilarski LT. Weinreb Amides as Directing Groups for Transition Metal-Catalyzed C-H Functionalizations. Molecules. 2019; 24(5):830. https://doi.org/10.3390/molecules24050830
Chicago/Turabian StyleKalepu, Jagadeesh, and Lukasz T. Pilarski. 2019. "Weinreb Amides as Directing Groups for Transition Metal-Catalyzed C-H Functionalizations" Molecules 24, no. 5: 830. https://doi.org/10.3390/molecules24050830