Spectroscopic and Structural Study of Some Oligosilanylalkyne Complexes of Cobalt, Molybdenum and Nickel


Institut für Anorganische Chemie, Technische Universität Graz, Stremayrgasse 9, A-8010 Graz, Austria
*
Authors to whom correspondence should be addressed.
Molecules 2019, 24(1), 205; https://doi.org/10.3390/molecules24010205
Submission received: 6 December 2018
/
Revised: 23 December 2018
/
Accepted: 27 December 2018
/
Published: 8 January 2019
(This article belongs to the Special Issue Advances in Silicon Chemistry 2018)
Abstract
:Metal induced stabilization of α-carbocations is well known for cobalt- and molybdenum complexed propargyl cations. The same principle also allows access to reactivity enhancement of metal coordinated halo- and hydrosilylalkynes. In a previous study, we have shown that coordination of oligosilanylalkynes to the dicobalthexacarbonyl fragment induces striking reactivity to the oligosilanyl part. The current paper extends this set of oligosilanylalkyne complexes to a number of new dicobalthexacarbonyl complexes but also to 1,2-bis(cyclopentadienyl)tetracarbonyldimolybdenum and (dippe)Ni complexes. NMR-Spectroscopic and crystallographic analysis of the obtained complexes clearly show that the dimetallic cobalt and molybdenum complexes cause rehybridization of the alkyne carbon atoms to sp3, while in the nickel complexes one π-bond of the alkyne is retained. For the dicobalt and dimolybdenum complexes, strongly deshielded 29Si NMR resonances of the attached silicon atoms indicate enhanced reactivity, whereas the 29Si NMR shifts of the respective nickel complexes are similar to that of respective vinylsilanes.
1. Introduction
It is well known that the reactivity of organic molecules can be altered dramatically by coordination to a metal fragment. Alkene coordination to Pd(II) represents a typical example, where electron density from the π-bond is shifted toward the metal. The thus caused polarization allows nucleophilic attack on the olefin, which is otherwise fairly unreactive [1].
Other particularly interesting cases of reactivity enhancement by metal coordination include metal induced stabilization of α-carbocations such as observed for dicobalt- and dimolybdenum complexed propargyl cations. The Nicholas reaction, which is the reaction of a dicobalt-coordinated propargyl cation with a nucleophile, depends on this effect [2,3,4,5]. As also silyl-substituted alkynes can be subjected to complexation with dicobaltoctacarbonyl [6,7,8,9], the question arose whether some kind of sila-Nicholas reaction might be possible. Corriu and co-workers have indeed found that dicobalt complexed methoxy- and halo-silylalkynes undergo facile alkylation, reduction, hydrolysis, and halogenation reactions [10]. In a subsequent study, enhanced reactivity of the Si-H bond of a coordinated hydrosilylalkyne was demonstrated and further investigated [11]. Whether the coordination of alkyne is essential for increased reactivity of hydrosilylalkynes could not be established as it was found that also non-coordinated hydrosilylalkynes would react with alcohols in the presence of 10 mol% of a Co2(CO)6-tolane complex [11].
For dicobalthexacarbonyl stabilized propargyl cations a fair number of isolated and well characterized examples are known [12,13]. For the silicon case, silylium ions are likely to be involved in reactions of the respective complexes, as was demonstrated by Corriu and co-workers, who reacted their dicobalthexacarbonyl coordinated hydrosilylalkyne with Ph3C[BF4] to obtain the respective dicobalthexacarbonyl complexed fluorosilane [11]. Theoretical and experimental aspects of the formation of transition metal-stabilized silylium ions were further studied by Brook and co-workers using again dicobalt- but also dimolybdenum-complexes for the coordination of silylated alkynes [14,15,16].
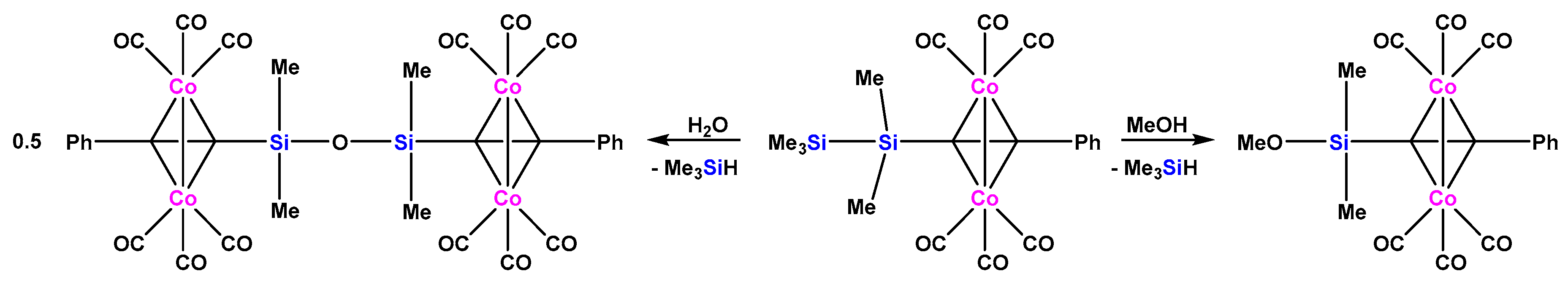
In order to check, whether complexation with dicobaltoctacarbonyl can activate even Si-Si bonds, our group prepared a number of dicobalthexacarbonyl coordinated tris(trimethylsilyl)silylalkyne complexes [17] (see below). These proved to be very reactive against a number of reagents. To provide a simplified system, we thus prepared a pentamethyldisilanylethyne dicobalthexacarbonyl complex, which underwent clean reactions with MeOH and H2O to the respective methoxysilane and disiloxane complexes (Scheme 1) [17].
NMR spectroscopic analysis of the obtained oligosilylalkyne dicobalt complexes showed the respective α-silicon atoms to be substantially deshielded, which is in line with the observed enhanced electrophilicity [17]. The current study aims at extending spectroscopic and structural studies to more elaborate oligosilanyl alkynes and to other transition metals.
2. Results and Discussion
To gain more detailed insight into the spectroscopic and structural properties of metal complexed oligosilanylated alkynes we decided to compare the properties of the already studied examples of cobalt complexes together with a couple of additional alkyne cobalt complexes to a few related complexes of molybdenum and nickel.
2.1. Cobalt Complexes
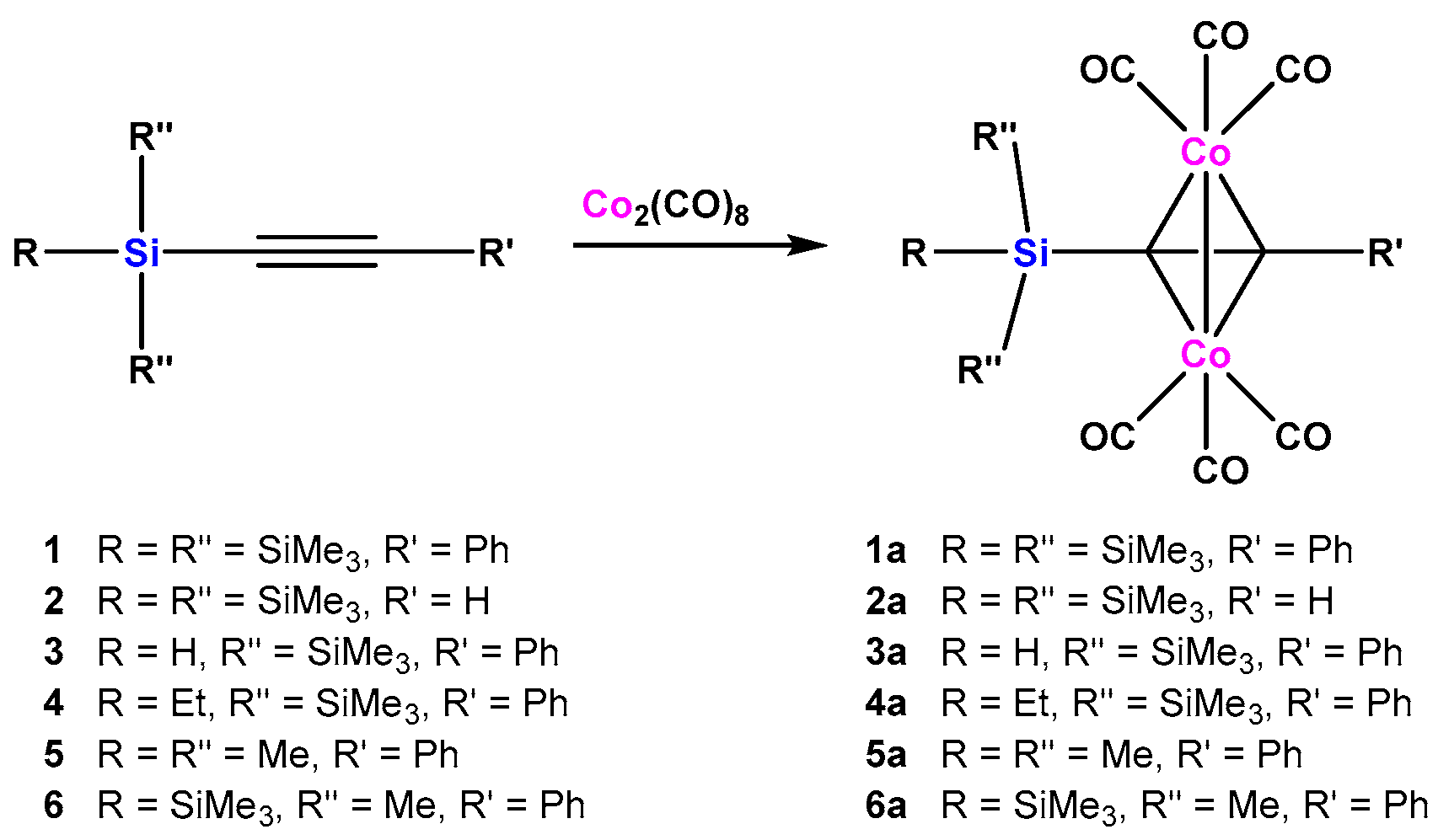
As depicted in Scheme 2 a number of a oligosilanylalkynes (1–6) were reacted previously with Co2(CO)8 to give the expected dicobalthexacarbonyl complexes 1a–6a [17] (Scheme 2). Molecular structures of several of these compounds were studied by single crystal XRD analysis (Table 2). Table 1 features 13C (alkyne C atoms) and 29Si (alkynyl attached Si atoms) NMR chemical shifts of the employed free alkynes together with the respective resonances for the metal coordinated compounds.
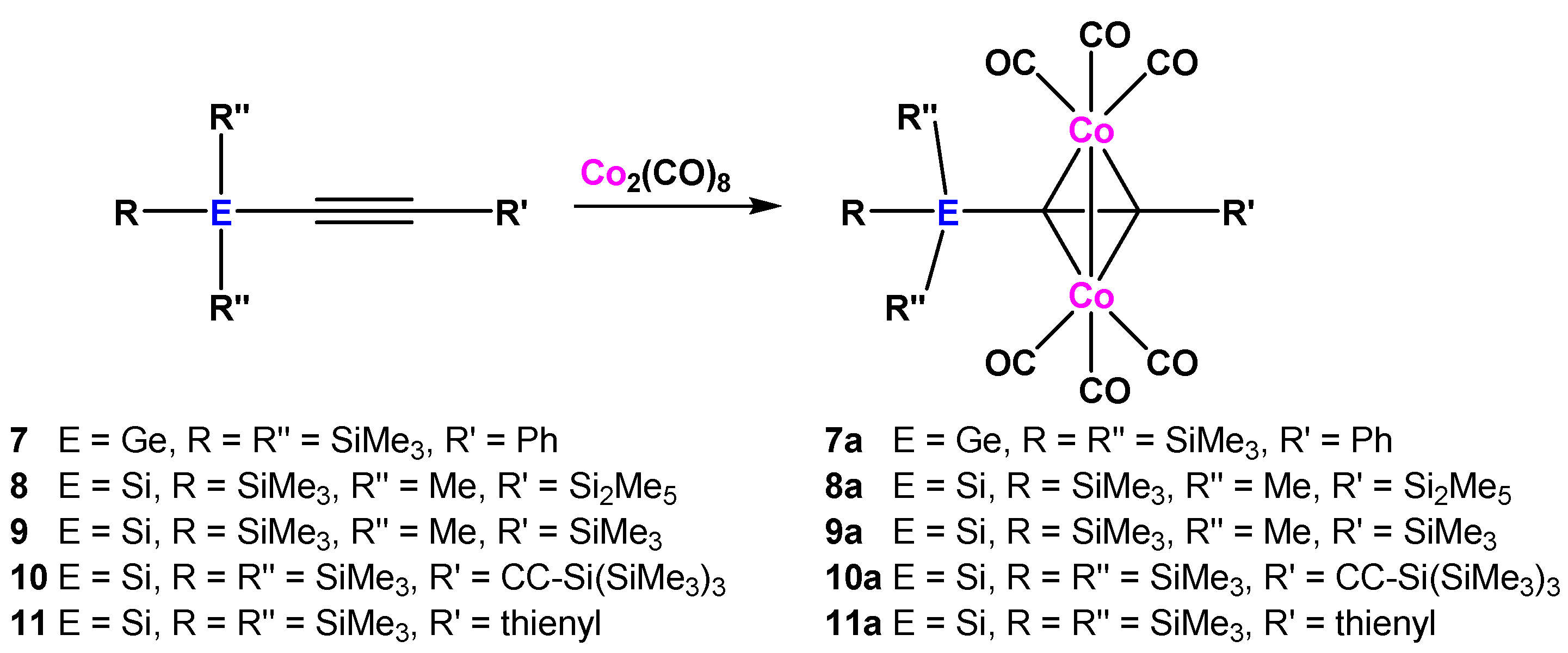
In addition to complexes 1a–6a, we decided to convert also the known alkynes 7 [18], 8 [19], 9 [20], and 10 [18], as well as the novel compound 11 to dicobalthexacarbonyl complexes. Compounds 7 and 11 are similar to tris(trimethylsilyl)silylphenylacetylene (1), but 7 contains a tris(trimethylsilyl)germyl group instead of the tris(trimethylsilyl)silyl unit and 11 features a 2-thienyl substituent instead of the phenyl group. As expected, both alkynes underwent smooth reaction to the respective dicobalthexacarbonyl complexes 7a and 11a (Scheme 3).
Previously, we noted that (Me3Si)3SiC≡CSiMe3 and even (Me3Si)3SiC≡CMe do not react with Co2(CO)8 [17]. Since the respective dicobalthexacarbonyl complex of Me3SiC≡CSiMe3 is known [6,9], we reasoned that the presence of the rather bulky (Me3Si)3Si group requires a small (H) or flat (Ph) substituent on the other side of the ethynyl group in order to allow effective approach of Co2(CO)8 to the C≡C unit. Since the steric properties of the Me3SiSiMe2 substituent are significantly less demanding compared to (Me3Si)3Si, we assumed that the presence of another small silyl group on the alkyne would nevertheless permit coordination to the dicobalthexacarbonyl fragment. For that reason, we prepared Me5Si2C≡CSi2Me5 (8) and Me5Si2C≡CSiMe3 (9) containing either two or one pentamethyldisilanyl substituents. Both compounds smoothly reacted with Co2(CO)8 to give complexes 8a and 9a (Scheme 3).
Substrate 10 is a butadiyne with two terminal tris(trimethylsilyl)silyl groups. Although we were able to achieve complexation of two alkyne units with Co2(CO)8 in 1,4-bis[tris(trimethylsilyl)silylethynyl]benzene and dimethyldi(phenylethynyl)silane [17], compound 10 permits only metalation of one triple bond, leading to formation of complex 10a with tris(trimethylsilyl)silyl and tris(trimethylsilyl)silylethynyl substituents (Scheme 3). Metalation of both triple bonds of 1,4-bis(trimethylsilyl)butyne with 2 equiv. Co2(CO)8 is known [8,21] but again it seems that the steric bulk of the tris(trimethylsilyl)silyl group prevents this process for 10.
With the intention to introduce further metal coordination sites, we decided to study replacing phenyl substituents at the alkyne by thienyl groups. The reaction of the thus obtained 2,4 bis[tris(trimethylsilyl)silylethynyl]thiophene (12) with 2 equiv. Co2(CO)8 can be considered as a variation of the previously reported reaction of 1,4-bis[tris(trimethylsilyl)silylethynyl]benzene [17]. As expected, both alkyne parts of 12 underwent complexation with Co2(CO)6 fragments to give complex 12a (Scheme 4).
2.2. Molybdenum Complexes
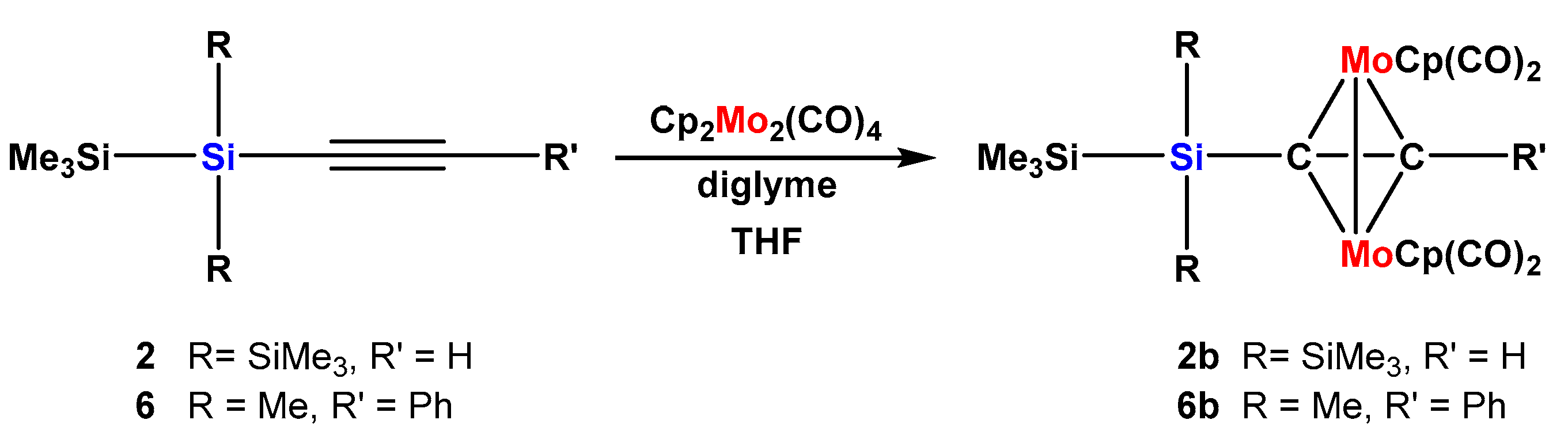
Apart from the dicobalthexacarbonyl unit, also other metal fragments are known to stabilize α-carbocations. The Cp2Mo2(CO)4 unit is one of these and therefore we were interested to utilize some of our silylalkynes for the preparation of dimolybdenum complexes. Thermal dissociation of 2 CO from Cp2Mo2(CO)6 delivers Cp2Mo2(CO)4 [22], which is known to add easily to alkynes [23]. For reasons of comparison, we subjected silylated alkynes 2 and 6 to metalation conditions with Cp2Mo2(CO)4 and obtained complexes 2b and 6b in acceptable yields (Scheme 5).
2.3. Nickel Complexes
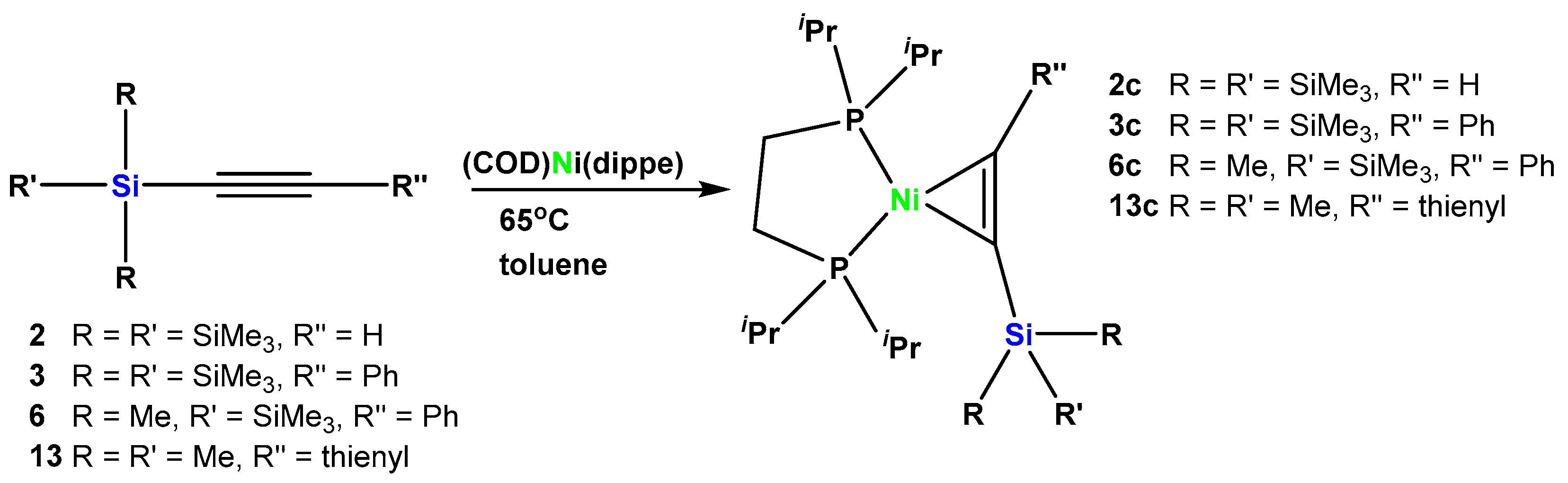
To study also mononuclear complexes of late metals, we decided to prepare Ni(0) complexes of a number of oligosilanylated alkynes. As the nickel fragment of choice (dippe)Ni (dippe = 1,2-di-iso-propylphosphinoethylene) was selected. Formation of the desired complexes (2c, 3c, 6c, and 13c) was achieved by reacting four different silylated alkynes (2, 3, 6, and 13) with (COD)Ni(dippe) [24] in toluene at 65 °C (Scheme 6).
2.4. NMR Spectroscopic Analysis
A substantial number of dicobalthexacarbonyl alkynyl complexes has been studied using NMR spectroscopy, with particular emphasis on 13C NMR properties of the coordinated alkyne carbon atoms [9,35,36]. It is interesting to note that the differences in the chemical shifts of free and coordinated alkyne atoms are surprisingly small. Nevertheless, in accordance also with structural information derived from crystal structure analytical studies, the coordination to the dicobalthexacarbonyl fragment goes along with a change in hybridization of the involved carbon atoms from sp to sp3.
Previously, we have shown that NMR spectroscopic analysis of the complexed oligosilanylalkynes provides a good tool to judge the degree of associated Si-Si bond activation. In particular, the 29Si NMR chemical shift values of the silicon atoms attached to the alkyne unit proved to be significant [17]. Table 1 details 13C and 29Si NMR shifts of the alkyne carbons and the attached silicon atoms of the free alkynes and the respective metal complexes.
The difference in electron donating abilities of phenyl and 2-thienyl units is clearly reflected by the alkynyl 13C NMR resonances of 1-(tris(trimethylsilyl)silyl)-2-(thien-2′-yl)ethyne 11 and 1-(tris(trimethylsilyl)silyl)-2-phenylethyne 1 (Table 1). Compared to the values of 1, the thienyl substituted carbon (δ = 100.4 ppm) of 11 is shifted some 8 ppm down-field, while the respective ß-carbon (δ = 93.5 ppm) experiences some 5 ppm shift in the opposite direction. The 29Si NMR shift of the attached tris(trimethylsilyl)silyl group is more or less identical for 1 and 11. The alkynyl 13C NMR resonances of butadiyne 10 (δ = 79.2 (Si) and 93.4 (C) ppm) are similar to that of tris(trimethylsilyl)silylethyne (2), reflecting not much substituent influence on the not silylated side of the alkyne. The disilylated alkynes 8 and 9 show that the effect on the α-carbon is quite similar (ca. +17.5 ppm) for SiMe3 and Si2Me5 but that Si2Me5 exhibits a stronger effect on the β-carbon than SiMe3. Again the 29Si NMR shifts of alkynyl substituted SiMe3 (δ = ca. −18.5 ppm) and Si2Me5 (δ = ca. −38 ppm) are not much affected by the other substituent on the alkyne. Assignment and approximate prediction of alkynyl 13C shifts is conveniently accomplished by using an increment system as outlined earlier by us [37].
In accordance with the fact that 1 and 11 feature remarkably different alkyne 13C resonances also their coordination behavior to the Co2(CO)6 unit is different. While both silicon substituted carbon atoms experience an up-field shift (see Table 1), the aryl substituted atom is shifted to lower field for the case of 1 being converted to 1a, whereas the thienyl substituted carbon of complex 11a is shifted about 4 ppm to higher field. Again the central 29Si resonances of the Si(SiMe3)3 groups are practically identical for 1a and 11a (Table 1).
For the disubstituted alkynes 8 (δ = 115.3 ppm) and 9 (δ = 112.9 and 116.5 ppm) the 13C values of the alkyne carbon atoms are similar to what is known for bis(trimethylsilyl)acetylene (BTMSA) (δ = 113 ppm) [9]. Also, the effect of coordination to the Co2(CO)6 fragment is mostly identical leading to 13C resonances at 93.1 ppm for 8a and δ = 93.9/92.4 ppm for 9a in accordance with the 93 ppm observed for the respective BTMSA complex [9]. The 29Si NMR down-field shift associated with the coordination to the Co2(CO)6 fragment amounts to ca 20 ppm for the alkynyl substituted silicon atom of the Si2Me5 units of 8a and 9a (Table 1) from δ = ca. −38 ppm for the free alkyne to ca −18 ppm for the complex.
Complexes 2b and 6b are the dimolybdenum analogs of the cobalt complexes 2a and 6a. While for cobalt complex 2a the 13C alkyne resonances were not much different from the free alkyne, the dimolybdenum compex 2b features both of these resonances shifted down-field. The 29Si signal of the central silicon atom at −58.8 ppm (Table 1), however, indicates a marked deshielding, which is consistent with what has been found for propargyl cations and which is stronger than found in the dicobalt complex 2a of the same alkyne (δ = −67.7 ppm). The situation for 6b is quite similar. The 13C alkyne resonances at 93.1 and 108.1 ppm are very close to 2b and also the 29Si chemical shift of the attached silicon atom at −16.8 ppm is again somewhat less shielded than in the corresponding dicobalt complex 6a.
The NMR spectroscopic features of the dicobalt and dimolybdenum complexes of silylated alkynes show that the coordinated ligand resembles an alkane. In contrast to that, coordination to Ni(0) displays a different picture. The 13C NMR spectrum of complex 3c, which features alkyne 1 as ligand exhibits resonances at 118.8 (C-Si) and 157.9 (C-Ph) ppm, which clearly indicates a more pronounced C-C double bond character of the coordinated alkyne. The 29Si NMR shift of the central hypersilyl silicon atom of 3c (−90.0 ppm) is also in line with a vinyl substituted tris(trimethylsilyl)silane. 13C (113.5 and 160.4 ppm) and 29Si (−85.1) NMR shifts of (E)-tBuCH=CHSi(SiMe3)3 are supporting these assignments [38]. Very similar spectroscopic pictures were found for complex 2c, which features coordinated alkyne 2, and for 6c with coordinated alkyne 6.
2.5. Crystallographic Analysis
In addition to the spectroscopic analysis of novel Co, Mo and Ni silylalkyne complexes, we have also studied a few of the new complexes by single crystal XRD analysis. Table 2 and Table S1 combine the obtained date with the previous examples from our earlier study [17].
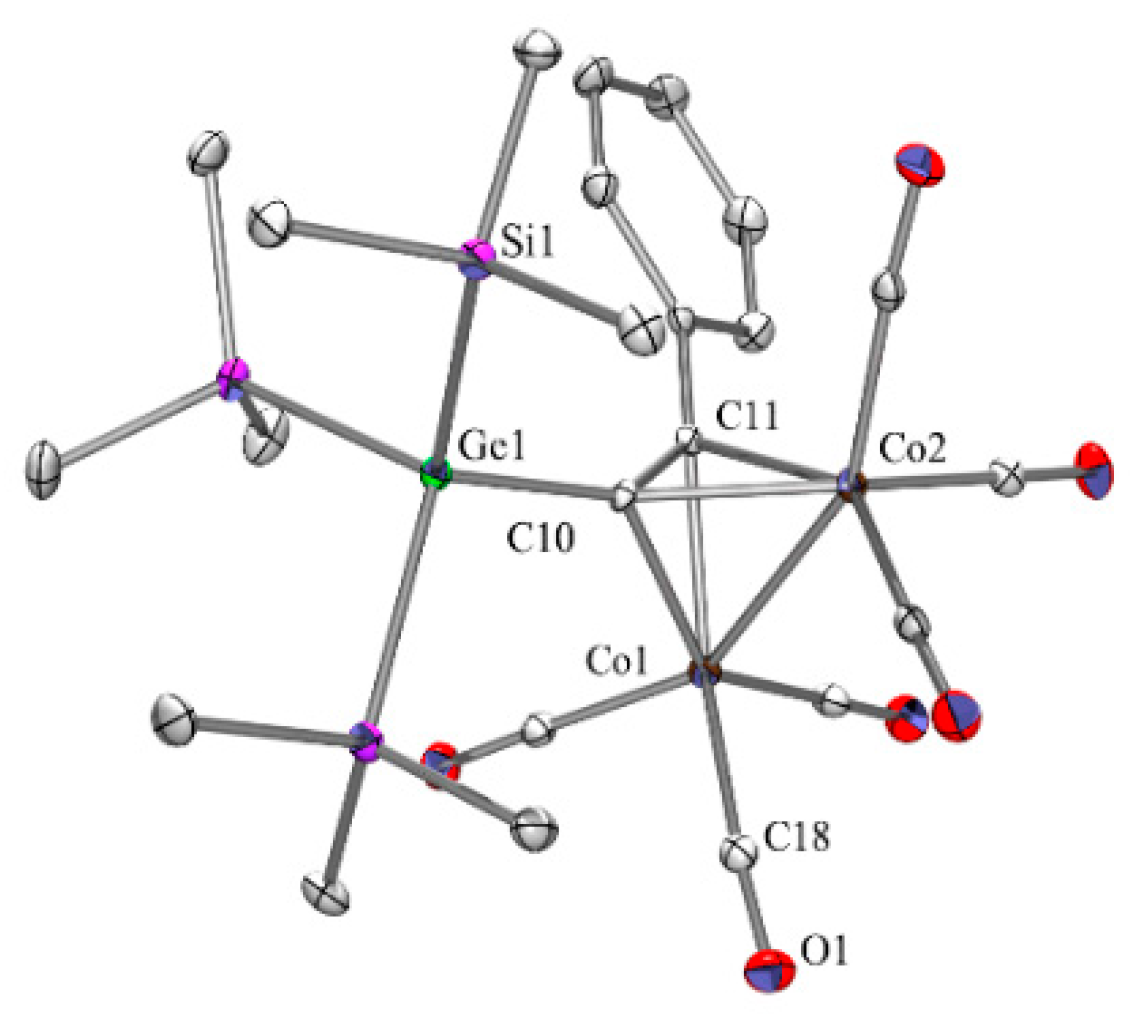
As expected the germyl substituted alkyne complex compound 7a (Figure 1) is isostructural to 1a [17]. Both compounds crystallize in the monoclinic space group P21. Structure parameters of 1a, 7a, and also 11a such as Co-Co, C-C, and Co-C distances are very similar (see Table 2). A C-Ge bond for 7a of 1.966(2) Å is quite typical and the spatial orientations of the phenyl and E(SiMe3)3 (E = Si, Ge) in 1a and 7a are almost identical and very similar to that of 11a.
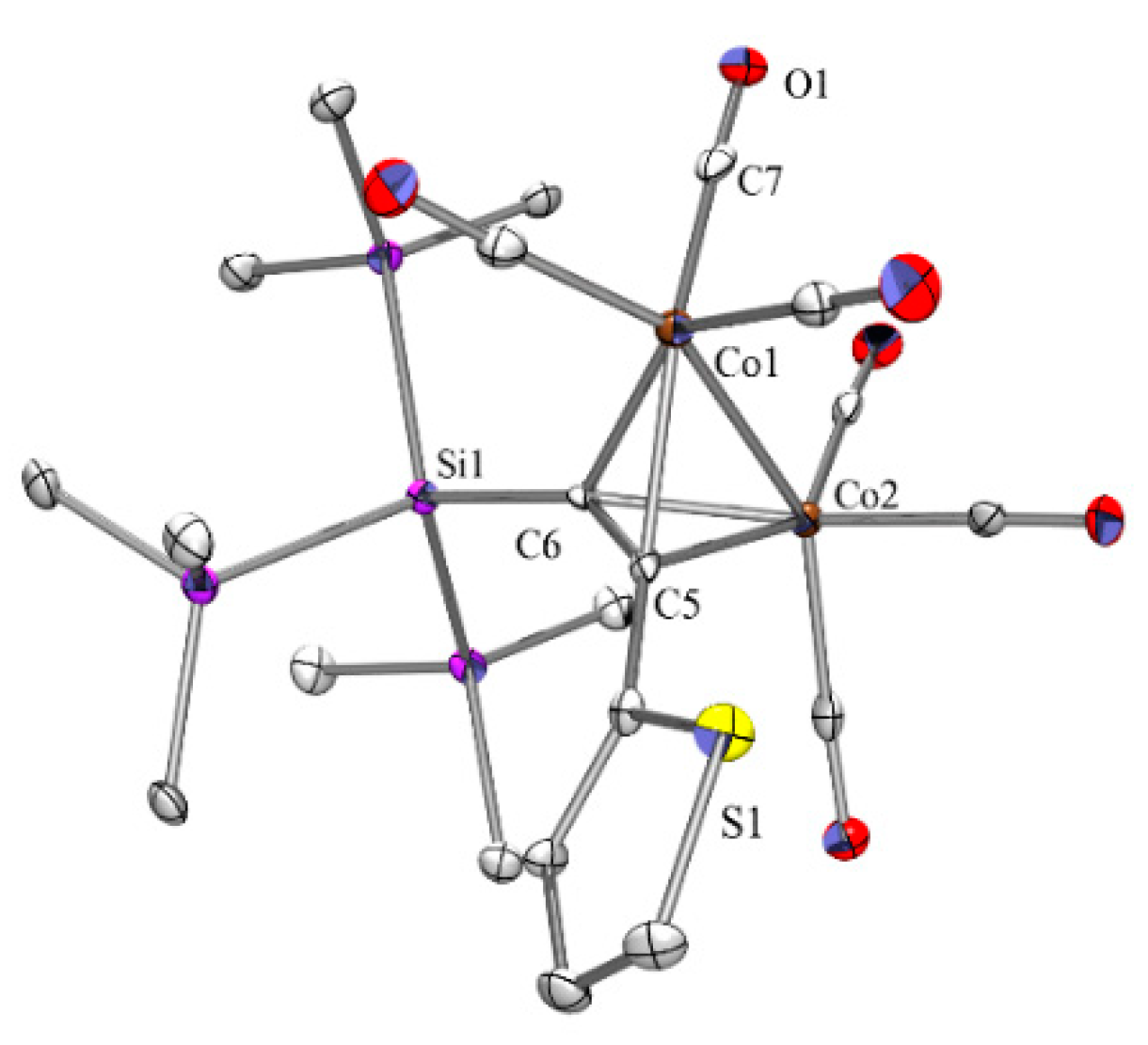
Compound 11a (Figure 2) was found to crystallize in the monoclinic space group P21/n with two crystallographically independent molecules in the asymmetric unit. In one of these molecules, the sulfur atom in the thienyl unit is disordered over two positions. The plane of the thienyl group is aligned with the Si-C-C-C plane of the former alkyne unit.
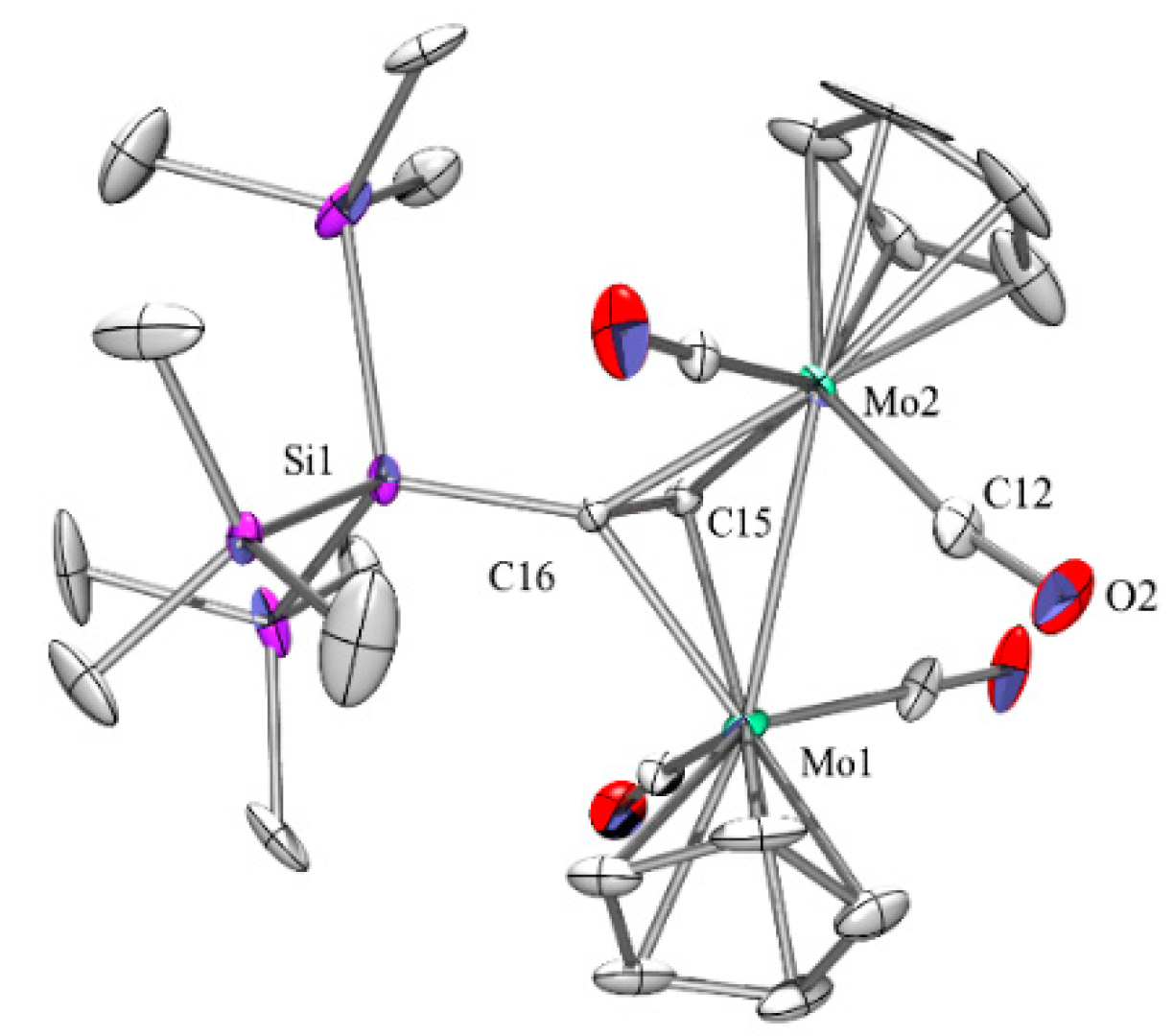
Since molybdenum is a second-row element the Mo-Mo distance in complex 2b (Figure 3) of 2.966(1) Å is much longer than the 2.45–2.47 Å which are typical for the Co-Co distances in the dicobalthexacarbonyl complexes. Nevertheless, the C-C bond in 2b of 1.337(10) Å is quite similar to what is found for dicobalt complexes. Direct comparison with complex 2a, where the C-C distance is 1.310(15) Å reveals that the dimolybdenum complex is able to induce a higher degree of sp3 hybridization consistent with elongated C-C bond. This is also evident by a much-diminished C-C-Si angle of 132.1(7) deg for 2b compared to the 142.7(8) deg found for 2a [17].
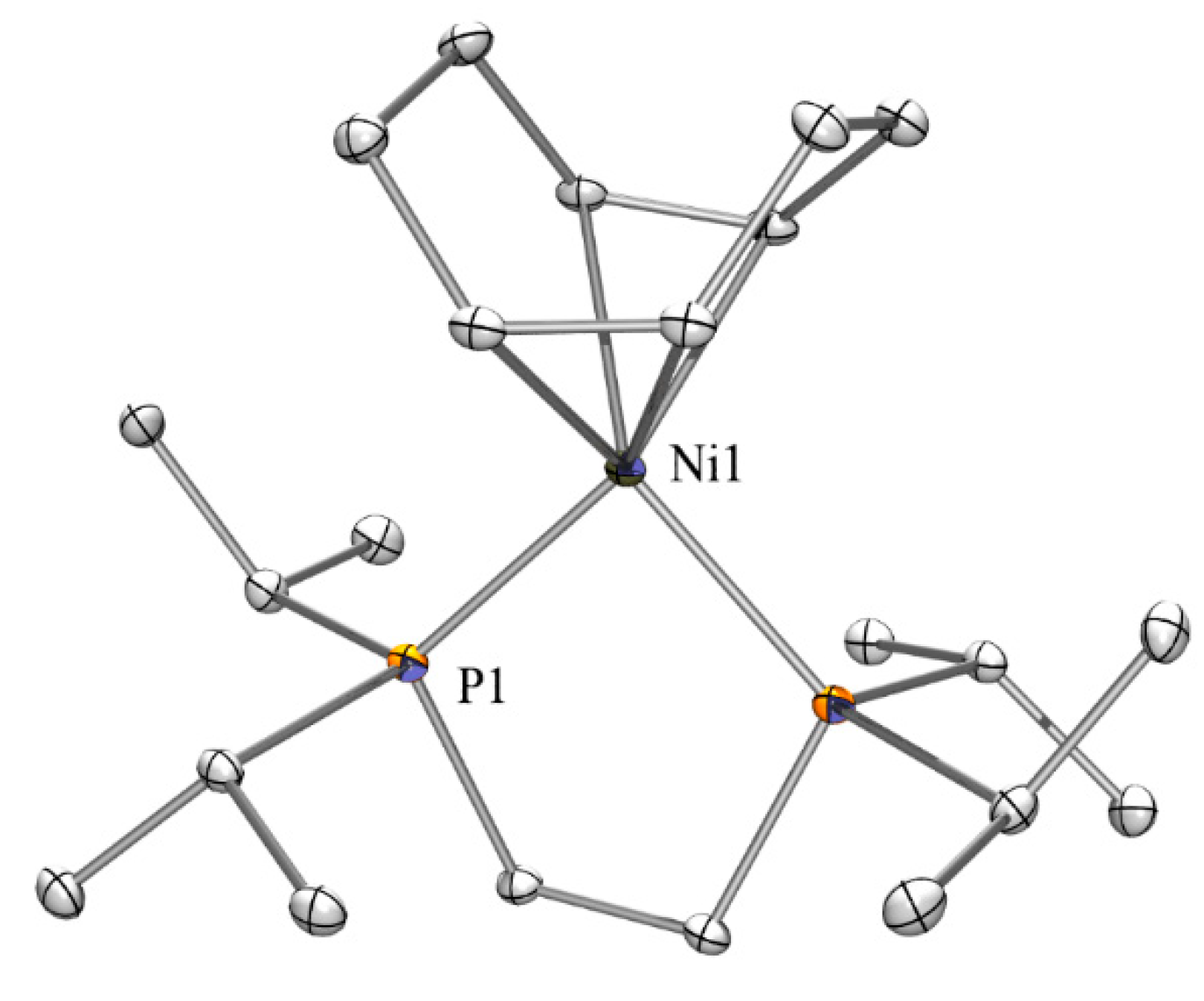
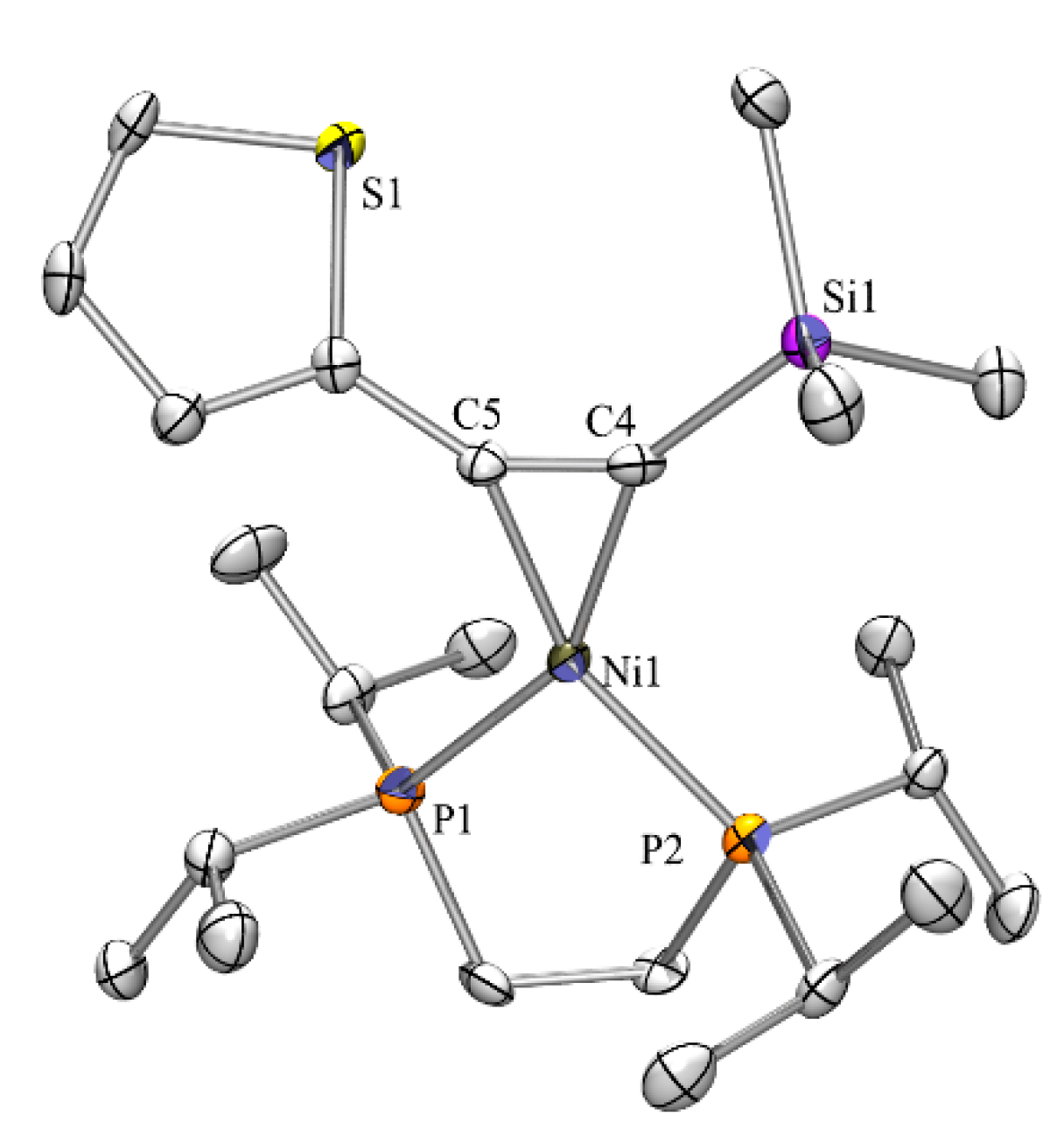
In the course of the nickel complex synthesis we obtained crystals of our starting material (COD)Ni(dippe) [24] and determined its structure by single crystal XRD analysis (Figure 4). The complex crystallizes in the monoclinic space group C2/c and is quite similar to a number of other diphosphine nickel COD complexes [39,40,41,42,43]. Compared to similar complexes the P-Ni distances of 2.161(1) Å and the C-Ni distances between 2.094(2) and 2.107 Å are relatively short. Nevertheless, distances in the analogous dppe complex [44] are almost identical.
Complex 13c (Figure 5) which crystallizes in the monoclinic space group P21/c is the only nickel complex of this study that could be crystallographically analyzed. Its structural features are very close to that of the bis(trimethylsilyl)acetylene nickel complex with the dippe ligand [27] with Ni-P distances of 2.152(1) and 2.162(2) Å and a C-C distance of the coordinated alkyne of 1.296(8) Å.
3. Experimental Section
All reactions involving air-sensitive compounds were carried out under an atmosphere of dry nitrogen or argon using either Schlenk techniques or a glove box. Solvents were dried using a column solvent purification system [45]. If not, otherwise stated chemicals were obtained from different suppliers and were used without further purification.
1H (300 MHz), 13C (75.4 MHz), 29Si (59.3 MHz), and 31P (124.4 MHz) NMR spectra were recorded on a Varian Unity INOVA 300 spectrometer. Samples for 29Si spectra were either dissolved in deuterated solvents or in cases of reaction samples measured with a D2O capillary in order to provide an external lock frequency signal. To compensate for the low isotopic abundance of 29Si the INEPT pulse sequence was used for the amplification of the signal [46,47]. If not noted otherwise the used solvent was C6D6 and all samples were measured at rt. Elementary analysis was carried using a Heraeus VARIO ELEMENTAR EL apparatus.
For X-ray structure analyses, the crystals were mounted onto the tip of glass fibers, and data collection was performed with a BRUKER-AXS SMART APEX CCD diffractometer using graphite-monochromated Mo Kα radiation (0.71073 Å). The data were reduced to F2o and corrected for absorption effects with SAINT [48] and SADABS [49,50], respectively. Structures were solved by direct methods and refined by full-matrix least-squares method (SHELXL97) [51]. All non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms were placed in calculated positions to correspond to standard bond lengths and angles. Crystallographic data (excluding structure factors) for the structures of compounds 7a, 11a, 2b, 13c and (COD)Ni(dippe) reported in this paper have been deposited with the Cambridge Crystallographic Data Center as supplementary publication no. CCDC-1881863 (7a), 1881859 (11a), 1881860 (2b), 1881862 (13c), and 1881861 (COD)Ni(dippe). Copies of data can be obtained free of charge at: http://www.ccdc.cam.ac.uk/products/csd/request/. Figures of solid-state molecular structures were generated using Ortep-3 as implemented in WINGX [52] and rendered using POV-Ray 3.6 [53].
Co2(CO)8 was freshly sublimed before use. Cp2Mo2(CO)4 was obtained from Cp2Mo2(CO)6 by thermal treatment [22]. (COD)Ni(dippe) [24], tris(trimethylsilyl)silylphenylacetylene (1) [37], tris(trimethylsilyl)silylethyne (2) [54], trimethylsilylphenylacetylene (6) [55], tris(trimethylsilyl)germylphenylacetylene (7) [18], bis(pentamethyldisilanyl)ethyne (8) [19], 1-trimethylsilyl-2-pentamethyldisilanylethyne (9) [20], 1,4-bis[tris(trimethylsilyl)silyl]butadiyne (10) [18], tris(trimethylsilyl)germyl potassium [56], and 1,2-dichlorotetramethyldisilane [57,58] have been prepared according to literature procedures.
3.1. Synthesis of Oligosilanylalkynes
1,2-Bis(pentamethydisilanyl)ethyne (8) A solution of nBuLi (15.98 mmol, 2M in hexane) in Et2O (10 mL) and THF (10 mL) is cooled to −70 °C and trichloroethylene (5.33 mmol) in Et2O (10 mL) is added dropwise. The reaction mixture is allowed to warm up to r.t. and stirring is continued for 3 h. A white precipitate occurred. The reaction is cooled again to −70 °C and chlorpentamethyldisilane in Et2O (10 mL) is added dropwise. The stirring is continued for another 20 h at r.t. and then poured to an ice-cold mixture of 0.5 M H2SO4 und Et2O. The phases were separated, the organic layer washed with brine and then dried over Na2SO4. After filtration, the solvent was removed and subjected to Kugelrohr distillation (0.1 mbar, 50 °C) to separate the product from the also formed pentamethyldisilanylethyne (29Si: −18.8, −34.4.). Compound 8 was obtained as a colorless oil (0.404 g, 26%). NMR (δ in ppm): 1H: 0.19 (s, 12H), 0.14 (s, 18H). 13C: 115.3, −2.5, −2.8. 29Si: −19.4, −38.2.
1-(Tris(trimethylsilyl)silyl)-2-(thien-2′-yl)ethyne (11) A solution 2-bromothiophene (1960 mg, 12 mmol) in freshly distilled diisopropylamine (10 mL) was cooled to 0 °C, CuI (60 mg, 0.32 mmol), PPh3 (180 mg, 0.69 mmol), and Pd(OAc)2 (60 mg, 0.27 mmol) were added and the stirring continued for 30 min. After the addition of tris(trimethylsilyl)silylethyne (2) (4.2 mL, 30 mmol) the mixture was stirred for further 12 h at r.t. The solvent was removed and the residue treated with Et2O (50 mL) and 2M H2SO4. The layers were separated, the organic layer washed with saturated NaHCO3 solution and then dried with Na2SO4. The solvent was removed and the product objected to a flash chromatography on silica gel (eluents: pentane). Oily colorless 11 was obtained (1260 mg, 58%). NMR (δ in ppm, CDCl3): 1H: 7.16 (dd, J = 1 and 7.9 Hz, 1H), 7.15 (dd, J = 1.3 and 6.6 Hz, 1H), 6.93 (dd, J = 3.4 and 5.1 Hz, 1H), 0.27 (s, 27H, SiMe3). 13C: 131.3, 126.7, 126.1, 124.9, 100.4, 93.5, 0.4. 29Si: −11.4, −100.2.
2,5-Bis[tris(trimethylsilyl)silylethynyl)thiophene (12) The reaction was done analogously to that for the preparation of 11 using 2,5-dibromothiophene (1230 mg, 5.08 mmol), freshly distilled diisopropylamine (10 mL), CuI (30 mg, 0.16 mmol), PPh3 (90 mg, 0.35 mmol), Pd(OAc)2 (30 mg, 0.14 mmol), and tris(trimethylsilyl)silylethyne (2) (2000 mg, 7.2 mmol). The mixture was stirred for further 12 h at 60 °C. Oily colorless 12 was obtained (2480 mg, 55%). NMR (δ in ppm, CDCl3): 1H: 7.29 (s, 2H), 0.68 (s, 27H, SiMe3). 13C: 131.4, 130.3, 129.6, 111.5, 0.4. 29Si: −11.3, −100.2.
3.2. Oligosilanylalkyne Dicobalthexacarbonyl Complexes
1-Phenyl-2-tris(trimethylsilyl)germylethyne-Co2(CO)6 (7a). A solution of tris(trimethylsilyl)germylphenylacetylene (7) (150 mg, 0.381 mmol) in pentane (2 mL) was added dropwise to Co2(CO)8 (130 mg, 0.381 mmol) dissolved in pentane (3 mL). The dark red solution was stirred for 1.5 h and then the solvent removed. Black crystalline 7a (248 mg, 93%) was obtained. NMR (δ in ppm): 1H: 7.54 (m, 2H), 7.05 (m, 2H), 6.95 (m, 1H), 0.31 (s, 27H, SiMe3). 13C: 201.2, 201.1, 139.4, 130.0, 128.8, 128.0, 109.7, 80.9, 2.7. 29Si: −4.4. Anal. Calcd. For C23H32Co2GeO6Si3 (679.23): C 40.67, H 4.75. Found: C 40.49, H 4.66.
1,2-Bis(pentamethyldisilanyl)ethyne-Co2(CO)6 (8a). The reaction was done analogously to that for the preparation of 7a using Co2(CO)8 (83 mg, 0.244 mmol) and bis(pentamethyldisilanyl)ethyne (8) (70 mg, 0.244 mmol). A dark red oil of 8a (113 mg, 81%) was obtained. NMR (δ in ppm): 1H: 0.37 (s, 12H), 0.15 (s, 18H). 13C: 201.8, 196.0, 93.1, 0.3, −1.6. 29Si: −15.9, −18.3.
1-Trimethylsilyl-2-pentamethyldisilanylethyne-Co2(CO)6 (9a). The reaction was done analogously to that for the preparation of 7a using Co2(CO)8 (244 mg, 0.563 mmol) and 1-trimethylsilyl-2-pentamethyldisilanylethyne (9) (150 mg, 0.563 mmol). Black crystalline 9a (324 mg, 95%) was obtained. NMR (δ in ppm): 1H: 0.33 (s, 6H), 0.26 (s, 9H), 0.12 (s, 9H). 13C: 201.2, 93.9, 92.4, 1.3, −0.2, −1.9. 29Si: 0.3, −15.8, −18.6.
1,4-Bis[tris(trimethylsilyl)silyl]buta-1,3-diyne-Co2(CO)6 (10a). Reaction was done according to 7a using Co2(CO)8 (63 mg, 0.184 mmol) and 10 (100 mg, 0.184 mmol). After cooling the solution to −70 °C black crystalline 10a (136 mg, 89%) was obtained. NMR (δ in ppm): 1H: 0.88 (s, 27H), 0.79 (s, 27H). 13C: 201.1; 105.1; 103.9; 88.6; 72.6; 2.1; 0.5. 29Si: −10.6; −10.9; −65.9; −99.2.
1-(2′-Thienyl)-2-tris(trimethylsilyl)silylethyne-Co2(CO)6 (11a). A solution of trimethylsilyl-2′-thienylacetylene (11) (200 mg, 0.564 mmol) in pentane (2 mL) was added dropwise to Co2(CO)8 (193 mg, 0.564 mmol) dissolved in pentane (3 mL). The dark red solution was stirred for 3 h and then cooled to −70 °C. Black crystalline 11a (296 mg, 82%) was obtained. NMR (δ in ppm): 1H: 7.08 (dd, J = 3.6 and 1.3 Hz), 6.68 (dd, J = 5.2 and 1.2 Hz), 6.54 (dd, J = 5.2 and 3.6 Hz), 0.32 (s, 27H). 13C: 200.5, 200.4, 200.3, 143.2, 128.0, 127.8, 126.3, 96.6, 76.9, 2.3. 29Si: −11.9, −67.1.
2,5-Bis[tris(trimethylsilyl)silylethynyl]thiophene-Co2(CO)6 (12a). The reaction was done analogously to that for the preparation of 7a using Co2(CO)8 (164 mg, 0.480 mmol) and 2,5-bis[tris(trimethylsilyl)silylethynyl)thiophene (12) (150 mg, 0.240 mmol). After cooling the solution to −70 °C black crystalline 12a (218 mg, 76%) was obtained. NMR (δ in ppm): 1H: 6.23 (s, 2H), 0.28 (s, 54H). 13C: 201.3, 130.7, 127.9, 112.2, 111.8, 2.3. 29Si: −11.9, −66.9.
3.3. Oligosilanylalkyne Dimolybdenyum-1,2-dicyclopentadienyltetraacarbonyl Complexes
Tris(trimethylsilyl)silylethyne-Cp2(CO)4Mo2 (2b). Cp2Mo2(CO)6 (719 mg, 1.47 mmol) in diglyme (10 mL) was kept under reflux for 3h. After cooling to rt, tris(trimethylsilyl)silylethyne (2) (200 mg, 0.733 mmol) in THF (3 mL) was added. After 14 d the solvent was removed, the residue treated with pentane and the insoluble remaining removed by centrifugation. Pentane was removed and 2b (270 mg, 52%) was obtained as a red oily residue obtained which crystallized after several days. NMR (δ in ppm): 1H: 6.46 (s, 1H), 4.98 (s, 10H, Cp), 0.26 (s, 27H, SiMe3). 13C: 241.2 (CO), 233.0 (CO), 101.9, 96.6, 92.4 (Cp), 3.4. 29Si: −11.8, −58.8.
1-Phenylethynylpentamethyldisilan-Cp2(CO)4Mo2 (6b). The reaction was done analogously to that for the preparation of 2b using Cp2Mo2(CO)6 (843 mg, 1.72 mmol) and pentamethyldisilanylphenylacetylene (6) (200 mg, 0.860 mmol). Recrystallization with pentane gave red crystalline 6b (320 mg, 56%). NMR (δ in ppm): 1H: 7.55 (m, 2H), 7.25 (m, 2H), 7.02 (m, 1H), 5.03 (s, 10H, Cp), 0.55 (s, 6H, SiMe2), 0.28 (s, 9H, −SiMe3). 13C: 236.8, 231.8, 231.5, 147.2, 130.0, 128.2, 126.5, 116.0, 92.1, 1.4, 0.0. 29Si: −7.1, −16.8.
3.4. Oligosilanylalkyne Nickel-Ethylenebis(diisopropylphosphine) Complexes
Di-iso-propylphosphinoethylene[tris(trimethylsilyl)silylethynyl]nickel (2c). (COD)Ni(dippe) (56 mg, 0.130 mmol) and tris(trimethylsilyl)silylethyne (2) (36 mg, 0.130 mmol) were dissolved in toluene (3 mL) and stirred for 4 d at 65 °C. The progress of the reaction was checked by NMR. After removing the solvent dark green crystalline 2c (59 mg, 85%) was obtained. NMR (δ in ppm): 1H: 8.12 (dd, JH-P = 11.2 Hz, JH-P = 31.8 Hz, 1H), 2.01 (m, 2H), 1.90 (m, 2H), 1.29–1.15 (m, 10H), 1.02–0.81 (m, 18H), 0.39 (d, 27H, JH-P = 3 Hz, SiMe3). 13C: 145.3 (dd, JC-P = 8 Hz, JC-P = 34 Hz), 124.3 (dd, JC-P = 7 Hz, JC-P = 42 Hz), 25.7, 19.6 (d, JC-P = 7 Hz), 18.9, 1.6 (SiMe3). 29Si: −14.0 (d, JSi-P = 3 Hz, SiMe3), −89.4 (dd, JSi-P = 2 Hz, JSi-P = 15 Hz, Siq). 31P: 81.6 (d, JP-P = 48.5 Hz), 76.0 (d, JP-P =48.5 Hz).
Di-iso-propylphosphinoethylene-[1-phenyl-2-tris(trimethylsilyl)silylethynyl]nickel (3c). (COD)Ni(dippe) (130 mg, 0.303 mmol) and tris(trimethylsilyl)silylphenylacetylene (1) (106 mg, 0.303 mmol) were dissolved in toluene (4 mL) and stirred for 7 d at 65 °C. The progress of the reaction was checked by NMR. After removing the solvent dark green crystalline 3c (166 mg, 82%) was obtained. NMR (δ in ppm): 1H: 7.23 (m, 1H), 7.15 (m, 2H), 6.94 (m, 2H), 2.10 (m, 2H), 1.62 (m, 2H), 1.21 (m, 4H), 0.98 (m, 24H), 0.34 (s, 27H). 13C: 157.9 (dd, JC-P = 5 Hz, JC-P = 32 Hz), 146.4 (dd, JC-P = 4 Hz, JC-P = 14 Hz), 128.4, 127.9, 126.4 (d, JC-P = 2 Hz), 123.8 (d, JC-P = 2 Hz), 118.6 (dd, JC-P = 6 Hz, JC-P = 42 Hz), 26.1 (dd, JC-P = 5 Hz, JC-P =13 Hz), 24.9 (dd, JC-P = 4 Hz, JC-P = 16 Hz), 22.0 (d, JC-P = 11 Hz), 19.8 (d, JC-P = 8 Hz), 18.7 (d, JC-P = 16 Hz), 2.2. 29Si: −14.1 (d, JSi-P = 3 Hz), −90.0 (t, JSi-P = 14 Hz).31P: 78.3 (d, JP-P = 49.1 Hz), 77.2 (d, JP-P = 49.1 Hz).
Di-iso-propylphosphinoethylene(1-phenyl-2-pentamethyldisilanylethynyl)nickel (6c). (COD)Ni(dippe) (82 mg, 0.191 mmol) and pentamethyldisilanylphenylacetylene (6) (44 mg, 0.191 mmol) were dissolved in toluene (3 mL) and stirred for 18 h at 65 °C. The progress of the reaction was checked by NMR. After removing the solvent dark green oily 6c (92 mg, 87%) was obtained. NMR (δ in ppm): 1H: 7.44 (d, 2H), 7.21 (t, 2H), 7.02 (t, 1H), 1.99 (sept, 2H), 1.80 (m, 2H), 1.24-1.16 (m, 10H), 0.97-0.79 (m, 18H), 0.55 (s, 6H, SiMe2), 0.19 (s, 9H, SiMe3). 13C: 159.6 (dd, JC-P = 5 Hz, JC-P = 34 Hz), 144.8 (dd, JC-P = 5 Hz, JC-P = 12.1 Hz), 132.2 (dd, JC-P = 4 Hz, JC-P = 32 Hz), 128.8, 126.7 (d, JC-P = 3 Hz), 124.2 (d, JC-P = 1 Hz), 26.2 (dd, JC-P =5 Hz, JC-P = 14 Hz), 25.5 (d, JC-P = 5 Hz, JC-P = 16 Hz), 22.0 (t, JC-P = 20 Hz), 21.8 (t, JC-P = 18 Hz), 20.8 (d, JC-P = 9 Hz), 19.8 (d, JC-P = 8 Hz), 19.0 (d, JC-P = 21 Hz), −0.2 (d, JC-P = 2 Hz, SiMe2), −1.2 (SiMe3). 29Si: −19.6 (d, JSi-P = 4 Hz, SiMe3), −31.4 (dd, JSi-P = 4 Hz, JSi-P = 14 Hz, SiMe2). 31P: 78.3 (d, JP-P = 49.0 Hz), 77.3 (d, JP-P = 49.0 Hz).
Di-iso-propylphosphinoethylene(1-(2′-thienyl)-2-tris(trimethylsilyl)silylethynyl)nickel (13c). Reaction was done according to 2c using (COD)Ni(dippe) (0.172 mmol) and 13 (61 mg, 0.172 mmol). Orange crystals of 13c (72 mg, 75%) were obtained. NMR (δ in ppm): 1H: 7.01 (dd, J = 3.5 and 1.3 Hz), 6.76 (dd, J = 5.2 and 1.2 Hz), 6.40 (dd, J = 5.2 and 3.5 Hz), 2.14 (m, 2H), 1.63 (m, 2H), 1.14 (m, 4H), 1.01 (m, 24H), 0.17 (s, 9H). 13C: 154.2 (dd, JC-P = 5 Hz, JC-P = 30 Hz), 148.3 (dd, JC-P = 4 Hz, JC-P = 14 Hz), 143.2 (dd, JC-P = 5 Hz, JC-P = 39 Hz), 129.3, 126.6, 126.1, 25.6 (dd, JC-P = 5 Hz, JC-P =13 Hz), 24.9 (dd, JC-P = 4 Hz, JC-P = 12 Hz), 22.5 (d, JC-P = 12 Hz), 19.7 (d, JC-P = 8 Hz), 19.2 (d, JC-P = 16 Hz), 5.2. 29Si: −19.1 (d, JSi-P = 12 Hz).31P: 79.1 (d, JP-P = 51 Hz), 77.7 (d, JP-P = 51 Hz).
4. Conclusions
Reactivity enhancement of metal coordinated halo- and hydrosilylalkynes is a well-documented fact [10,11,14,15,16]. In a previous study, we have shown that oligosilanylalkyne coordination to the dicobalthexacarbonyl fragment results in the formation of highly reactive compounds [17]. This reactivity was explained as activation of Si-Si bonds in accordance to what was observed previously for Si-H bonds. 29Si NMR spectroscopic analysis confirmed down-field shift of the alkynyl substituted silicon atom resonances, which is in agreement with increased electrophilic properties. The current study extends the range of oligosilanylalkyne complexes to a number of new dicobalthexacarbonyl complexes but also to 1,2-bis(cyclopentadienyl)tetracarbonyldimolybdenum and (dippe)Ni complexes. Similar to what was observed before for metal stabilized propargyl cations, also for the oligosilanylalkyne complexes the respective dimolybdenum complexes were found to cause even stronger 29Si NMR deshielding than was found for the dicobalt complexes. While the dimetallic complexes cause rehybridization of the alkyne carbon atoms to sp3 (alkane type), in the respective nickel complexes one of the π-bonds of the alkynes was found to be retained. The NMR spectroscopic properties of the nickel complexes are completely different from those of the dicobalt and dimolybdenum complexes. In the latter, strongly deshielded 29Si NMR chemical shifts of the attached silicon atoms indicate enhanced reactivity, whereas the 29Si NMR resonances of the respective nickel complexes are similar to that of respective vinylsilanes. Thus, the strong deshielding of the alkyne attached silicon atoms was not observed for the nickel complexes and therefore no pronounced Si-Si bond activation can be expected.
Supplementary Materials
The following are available online at https://www.mdpi.com/1420-3049/24/1/205/s1, Table S1: Crystallographic data for compounds 2b, 7a, 11a, 13c, and dippeNiCOD.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. M.Z., C.M., and J.B. conceived the experiments. M.Z. and to a minor extent J.B. did all the synthetic and characterization work. C.M. and J.B. provided supervision, financial support via projects and wrote the paper.
Funding
This research was funded by the Austrian Fonds zur Förderung der wissenschaftlichen Forschung (FWF) via projects P-30955 (J.B.) and P-19338 (C.M.).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hartwig, J. Organotransition Metal Chemistry: From Bonding to Catalysis; University Science Books: Sausalito, CA, USA, 2009; ISBN 1-891389-53-X. [Google Scholar]
- Nicholas, K.M. A Forty Year Odyssey in Metallo–Organic Chemistry. J. Org. Chem. 2015, 80, 6943–6950. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.J.J. Stereoselective Propargylations with Transition-Metal-Stabilized Propargyl Cations. Eur. J. Org. Chem. 2001, 2001, 2021–2033. [Google Scholar] [CrossRef]
- Teobald, B.J. The Nicholas reaction: The use of dicobalt hexacarbonyl-stabilised propargylic cations in synthesis. Tetrahedron 2002, 58, 4133–4170. [Google Scholar] [CrossRef]
- Nicholas, K.M. Chemistry and synthetic utility of cobalt-complexed propargyl cations. Acc. Chem. Res. 1987, 20, 207–214. [Google Scholar] [CrossRef]
- Krüerke, U.; Hübel, W. Über Organometall-Komplexe, VIII. Reaktionen von Kobaltcarbonyl-Verbindungen mit Alkinen. Chem. Ber. 1961, 94, 2829–2856. [Google Scholar] [CrossRef]
- Seyferth, D.; White, D.L. Silicon-, germanium- and tin-substituted acetylenes and their dicobalt hexacarbonyl complexes. J. Organomet. Chem. 1971, 32, 317–322. [Google Scholar] [CrossRef]
- Pannell, K.H.; Crawford, G.M. Organometalloidal Derivatives of the Transition Metals I. J. Coord. Chem. 1973, 2, 251–256. [Google Scholar] [CrossRef]
- Galow, P.; Sebald, A.; Wrackmeyer, B. Darstellung und NMR-spektroskopie organometallisch substituierter dikobalthexacarbonyl-alkin-komplexe. J. Organomet. Chem. 1983, 259, 253–268. [Google Scholar] [CrossRef]
- Corriu, R.J.P.; Moreau, J.J.E.; Praet, H. Cobalt carbonyl complexes of functional ethynylsilanes. Reactivity at the silicon atom. Organometallics 1989, 8, 2779–2786. [Google Scholar] [CrossRef]
- Corriu, R.J.P.; Moreau, J.J.E.; Praet, H. (Ethynylhydrosilane)cobalt carbonyl complexes. Reactivity of the silicon-hydrogen bond. Organometallics 1990, 9, 2086–2091. [Google Scholar] [CrossRef]
- Connor, R.E.; Nicholas, K.M. Isolation, characterization, and stability of α-[(ethynyl)dicobalt hexacarbonyl] carbonium ions. J. Organomet. Chem. 1977, 125, C45–C48. [Google Scholar] [CrossRef]
- Melikyan, G.G.; Bright, S.; Monroe, T.; Hardcastle, K.I.; Ciurash, J. Overcoming a Longstanding Challenge: X-Ray Structure of a [Co2(CO)6]-Complexed Propargyl Cation. Angew. Chem. Int. Ed. 1998, 37, 161–164. [Google Scholar] [CrossRef]
- Ruffolo, R.; Decken, A.; Girard, L.; Gupta, H.K.; Brook, M.A.; McGlinchey, M.J. Toward Metal-Stabilized Silylium Cations: An EHMO Study of [(HC≡C-SiH2)Co2(CO)6]+ and X-ray Crystal Structures of (Me3SiC≡CSiPh2H)Mo2(CO)4Cp2 and [(Me3SiC≡CSiMe2)Co2(CO)6]2O. Organometallics 1994, 13, 4328–4335. [Google Scholar] [CrossRef]
- Ruffolo, R.; Kainz, S.; Gupta, H.; Brook, M.; Mcglinchey, M. A synthetic and structural study on metal cluster complexes of allyl—alkynyl—silanes: Does protonation lead to metal-stabilized silyl cations? J. Organomet. Chem. 1997, 547, 217–226. [Google Scholar] [CrossRef]
- Stradiotto, M.; Brook, M.A.; McGlinchey, M.J. Can metal clusters assist silicon migrations? An NMR spectroscopic and X-ray crystallographic study. Inorg. Chem. Commun. 1998, 1, 105–108. [Google Scholar] [CrossRef]
- Zirngast, M.; Marschner, C.; Baumgartner, J. Cobalt-Assisted Silicon−Silicon Bond Activation. Organometallics 2006, 25, 4897–4908. [Google Scholar] [CrossRef]
- Zirngast, M.; Marschner, C.; Baumgartner, J. Group 4 Metallocene Complexes of Tris(trimethylsilyl)silylacetylene and Related Alkynes. Organometallics 2008, 27, 2570–2583. [Google Scholar] [CrossRef]
- West, R.; Quass, L.C. Preparation of ethynylsilanes from polychloroethylenes. J. Organomet. Chem. 1969, 18, 55–67. [Google Scholar] [CrossRef]
- Kerst, C.; Rogers, C.W.; Ruffolo, R.; Leigh, W.J. Direct Detection and Characterization of a Transient 1-Silaallene Derivative in Solution. J. Am. Chem. Soc. 1997, 119, 466–471. [Google Scholar] [CrossRef]
- Bruce, M.I.; Low, P.J.; Werth, A.; Skelton, B.W.; White, A.H. Some transition-metal complexes derived from silylated 1,3-diynes. J. Chem. Soc. Dalton Trans. 1996, 1551–1566. [Google Scholar] [CrossRef]
- Ginley, D.S.; Bock, C.R.; Wrighton, M.S. Photogeneration of dinuclear metal carbonyls containing a metal-metal triple bond. Inorg. Chim. Acta 1977, 23, 85–94. [Google Scholar] [CrossRef] [Green Version]
- Curtis, M.D. Reactions of the metal-metal triple bond in Cp2Mo2(CO)4 and related complexes. Polyhedron 1987, 6, 759–782. [Google Scholar] [CrossRef]
- Bonrath, W.; Pörschke, K.R.; Wilke, G.; Angermund, K.; Krüger, C. Ein- und zweikernige Nickel(0)-Komplexe von Butadiin. Angew. Chem. 1988, 100, 853–855. [Google Scholar] [CrossRef]
- Rosenthal, U.; Schulz, W.; Goerls, H. [(C6H5)3P]2Ni(Me3SiC≡CSiMe3). Preparation, properties, and structure of the first stable nickel(0) complex with bis(trimethylsilyl)acetylene. Z. Anorg. Chem. 1987, 550, 169–176. [Google Scholar] [CrossRef]
- Bartik, T.; Happ, B.; Iglewsky, M.; Bandmann, H.; Boese, R.; Heimbach, P.; Hoffmann, T.; Wenschuh, E. Synthesis and characterization of bis(phosphine)nickel(0) complexes containing nonsymmetrically substituted acetylenes. Organometallics 1992, 11, 1235–1241. [Google Scholar] [CrossRef]
- Edelbach, B.L.; Lachicotte, R.J.; Jones, W.D. Catalytic Carbon-Carbon and Carbon-Silicon Bond Activation and Functionalization by Nickel Complexes. Organometallics 1999, 18, 4660–4668. [Google Scholar] [CrossRef]
- Edelbach, B.L.; Lachicotte, R.J.; Jones, W.D. Catalytic Carbon−Carbon Bond Activation and Functionalization by Nickel Complexes. Organometallics 1999, 18, 4040–4049. [Google Scholar] [CrossRef]
- Tillack, A.; Pulst, S.; Baumann, W.; Baudisch, H.; Kortus, K.; Rosenthal, U. Hydrosilylation of symmetric disubstituted alkynes and butadiynes with L2Ni(0)-butadiyne complexes [L = Ph3P, (o-Tol-O)3P] as catalysts. J. Organomet. Chem. 1997, 532, 117–123. [Google Scholar] [CrossRef]
- Rosenthal, U.; Pulst, S.; Arndt, P.; Baumann, W.; Tillack, A.; Kempe, R. Influence of substituents in new Ni(0) butadiyne complexes. Z. Naturforsch. B 1995, 50, 377–384. [Google Scholar] [CrossRef]
- Rosenthal, U.; Pulst, S.; Arndt, P.; Baumann, W.; Tillack, A.; Kempe, R. Influence of ligands in new Ni(0) complexes of disubstituted butadiynes. Z. Naturforsch. B 1995, 50, 368–376. [Google Scholar] [CrossRef]
- Rosenthal, U.; Pulst, S.; Arndt, P.; Ohff, A.; Tillack, A.; Baumann, W.; Kempe, R.; Burlakov, V.V. Heterobimetallic σ,π-Acetylide-Bridged Complexes from Disubstituted 1,3-Butadiynes. Organometallics 1995, 14, 2961–2968. [Google Scholar] [CrossRef]
- Rosenthal, U.; Oehme, G.; Burlakov, V.V.; Petrovskii, P.V.; Shur, V.B.; Vol’pin, M.E. Carbon-13 and proton NMR studies of selected transition metal alkyne complexes. J. Organomet. Chem. 1990, 391, 119–122. [Google Scholar] [CrossRef]
- Chan, W.Y.; Berenbaum, A.; Clendenning, S.B.; Lough, A.J.; Manners, I. Toward Highly Metalized Polymers: Synthesis and Characterization of Silicon-Bridged [1]Ferrocenophanes with Pendent Cluster Substituents. Organometallics 2003, 22, 3796–3808. [Google Scholar] [CrossRef]
- Aime, S.; Milone, L.; Rossetti, R.; Stanghellini, P.L. 1H and 13C NMR studies of acetylenic complexes of Co2(CO)8. Inorg. Chim. Acta 1977, 22, 135–139. [Google Scholar] [CrossRef]
- Happ, B.; Bartik, T.; Zucchi, C.; Rossi, M.C.; Ghelfi, F.; Palyi, G.; Varadi, G.; Szalontai, G.; Horvath, I.T.; Guastini, C. On the Reactivity of Acetylenes Coordinated to Cobalt. 9. Effects of Substitution and Coordination on the 13C-NMR Chemical Shifts of the sp Carbons of (μ2-R1C2R2)Co2(CO)6 Complexes. Molecular Structure of μ2-2-PhC2SiPh3)Co2(CO)6. Organometallics 1995, 14, 809–819. [Google Scholar] [CrossRef]
- Mechtler, C.; Zirngast, M.; Baumgartner, J.; Marschner, C. Synthesis and Reactions of Alkynyl Oligosilanes. Eur. J. Inorg. Chem. 2004, 2004, 3254–3261. [Google Scholar] [CrossRef]
- Markov, J.; Baumgartner, J.; Marschner, C.; Oehme, H.; Gross, T. Vinyloligosilyl anions—A new class of compounds. In Organosilicon Chemistry VI; Auner, N., Weis, J., Eds.; Wiley-VCH: Hoboken, NJ, USA, 2005; pp. 309–313. [Google Scholar]
- Kempe, R.; Sieler, J.; Walther, D. Crystal structure of bis(ethyldiphenylphosphane)cycloocta-1,5-dienenickel(0), C36H42NiP2. Z. Kristallogr. 2010, 211, 565–566. [Google Scholar] [CrossRef]
- Yin, G.; Kalvet, I.; Englert, U.; Schoenebeck, F. Fundamental Studies and Development of Nickel-Catalyzed Trifluoromethylthiolation of Aryl Chlorides: Active Catalytic Species and Key Roles of Ligand and Traceless MeCN Additive Revealed. J. Am. Chem. Soc. 2015, 137, 4164–4172. [Google Scholar] [CrossRef]
- Maciejewski, H.; Sydor, A.; Marciniec, B.; Kubicki, M.; Hitchcock, P.B. Intermediates in nickel(0)–phosphine complex catalyzed dehydrogenative silylation of olefins. Inorg. Chim. Acta 2006, 359, 2989–2997. [Google Scholar] [CrossRef]
- Liu, N.; Li, X.; Xu, X.; Wang, Z.; Sun, H. Synthesis, structure and DFT study of dinuclear iron, cobalt and nickel complexes with cyclopentadienyl-metal moieties. Dalton Trans. 2011, 40, 6886–6892. [Google Scholar] [CrossRef]
- Freund, R.R.A.; Görls, H.; Langer, J. Nickelalactones with an allyl subunit—The effect of penta-coordination on structures and stability. Dalton Trans. 2014, 43, 13988–14000. [Google Scholar] [CrossRef] [PubMed]
- Skelton, B.W.; Clavell, K.J.; Nielsen, D.J. (Cycloocta-1,5-diene)-((ethane-1,2-diyl)-bis(diphenylphosphane))-nickel. CSD Commun. 2017. Refcode: RECHIT. [Google Scholar]
- Pangborn, A.B.; Giardello, M.A.; Grubbs, R.H.; Rosen, R.K.; Timmers, F.J. Safe and Convenient Procedure for Solvent Purification. Organometallics 1996, 15, 1518–1520. [Google Scholar] [CrossRef]
- Morris, G.A.; Freeman, R. Enhancement of Nuclear Magnetic Resonance Signals by Polarization Transfer. J. Am. Chem. Soc. 1979, 101, 760–762. [Google Scholar] [CrossRef]
- Helmer, B.J.; West, R. Enhancement of 29Si NMR Signals by Proton Polarization Transfer. Organometallics 1982, 1, 877–879. [Google Scholar] [CrossRef]
- Bruker. SAINTPLUS: Software Reference Manual, Version 6.45; Bruker-AXS: Madison, WI, USA, 1997–2003.
- Blessing, R.H. An empirical correction for absorption anisotropy. Acta Cryst. A 1995, 51, 33–38. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. SADABS. Version 2.10; Bruker AXS Inc.: Madison, WI, USA, 2003. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An Update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- POVRAY 3.6; Persistence of Vision Pty. Ltd.: Williamstown, Australia, 2004; Available online: http://www.povray.org/download/ (accessed on 9 July 2008).
- Fischer, R.; Frank, D.; Gaderbauer, W.; Kayser, C.; Mechtler, C.; Baumgartner, J.; Marschner, C. α,ω-Oligosilyl Dianions and Their Application in the Synthesis of Homo- and Heterocyclosilanes. Organometallics 2003, 22, 3723–3731. [Google Scholar] [CrossRef]
- Benkeser, R.A.; Hickner, R.A. The Stereochemistry of the Addition of Silicochloroform to Acetylenes. J. Am. Chem. Soc. 1958, 80, 5298–5300. [Google Scholar] [CrossRef]
- Fischer, J.; Baumgartner, J.; Marschner, C. Silylgermylpotassium Compounds. Organometallics 2005, 24, 1263–1268. [Google Scholar] [CrossRef]
- Ishikawa, M.; Kumada, M.; Sakurai, H. Preparation of some polysilicon halides by aluminum halide catalyzed interchange of methyl and halogen on silicon. J. Organomet. Chem. 1970, 23, 63–69. [Google Scholar] [CrossRef]
- Marschner, C.; Baumgartner, J. 4.4.5 Product Subclass 5: Disilanes and Oligosilanes. In Science of Synthesis: Houben-Weyl Methods of Molecular Transformations; Oestreich, M., Ed.; Thieme: Stuttgart, Germany, 2013. [Google Scholar]
Sample Availability: Not available. |
Scheme 1.
Reactions of a pentamethyldisilanylethyne dicobalthexacarbonyl complex with methanol and water [17].
Scheme 1.
Reactions of a pentamethyldisilanylethyne dicobalthexacarbonyl complex with methanol and water [17].

Scheme 2.
Formation of dicobalthexacarbonyl complexes of oligosilanyl substituted alkynes [17].
Scheme 2.
Formation of dicobalthexacarbonyl complexes of oligosilanyl substituted alkynes [17].

Scheme 3.
Formation of dicobalthexacarbonyl complexes of additional oligosilanyl substituted alkynes.
Scheme 3.
Formation of dicobalthexacarbonyl complexes of additional oligosilanyl substituted alkynes.

Scheme 4.
Formation of the bis(dicobalthexacarbonyl) complex of 2,4 bis[tris(trimethylsilyl)silylethynyl]thiophene (12a).
Scheme 4.
Formation of the bis(dicobalthexacarbonyl) complex of 2,4 bis[tris(trimethylsilyl)silylethynyl]thiophene (12a).

Scheme 5.
Formation of dimolybdenum oligosilanylalkyne complexes.

Scheme 6.
Formation of nickel complexes of oligosilanyl substituted alkynes.

Figure 1.
Crystal structure of 7a. Thermal ellipsoids are represented at the 30% level and hydrogen atoms have been omitted for clarity (bond lengths in Å, angles in deg). Co(1)-C(18) 1.822(3), Co(1)-C(11) 1.973(2), Co(1)-C(10) 2.020(2), Co(1)-Co(2) 2.4582(6), Co(2)-C(11) 1.983(2), Co(2)-C(10) 2.009(2), O(1)-C(18) 1.129(3), Ge(1)-C(10) 1.966(2), Ge(1)-Si(1) 2.3929(9), Ge(1)-Si(3) 2.4128(10), Si(1)-C(1) 1.879(3), C(10)-C(11) 1.337(3), C(19)-Co(1)-C(18) 98.83(12), C(11)-Co(1)-C(10) 39.11(10), C(11)-Co(1)-Co(2) 51.75(7), C(10)-Co(1)-Co(2) 52.21(7), C(11)-Co(2)-C(10) 39.13(10), C(10)-Ge(1)-Si(1) 109.47(7), Si(1)-Ge(1)-Si(2) 108.78(3), C(11)-C(10)-Ge(1) 143.46(19), C(11)-C(10)-Co(2) 69.35(14), Ge(1)-C(10)-Co(2) 136.68(12), Co(2)-C(10)-Co(1) 75.19(8).
Figure 1.
Crystal structure of 7a. Thermal ellipsoids are represented at the 30% level and hydrogen atoms have been omitted for clarity (bond lengths in Å, angles in deg). Co(1)-C(18) 1.822(3), Co(1)-C(11) 1.973(2), Co(1)-C(10) 2.020(2), Co(1)-Co(2) 2.4582(6), Co(2)-C(11) 1.983(2), Co(2)-C(10) 2.009(2), O(1)-C(18) 1.129(3), Ge(1)-C(10) 1.966(2), Ge(1)-Si(1) 2.3929(9), Ge(1)-Si(3) 2.4128(10), Si(1)-C(1) 1.879(3), C(10)-C(11) 1.337(3), C(19)-Co(1)-C(18) 98.83(12), C(11)-Co(1)-C(10) 39.11(10), C(11)-Co(1)-Co(2) 51.75(7), C(10)-Co(1)-Co(2) 52.21(7), C(11)-Co(2)-C(10) 39.13(10), C(10)-Ge(1)-Si(1) 109.47(7), Si(1)-Ge(1)-Si(2) 108.78(3), C(11)-C(10)-Ge(1) 143.46(19), C(11)-C(10)-Co(2) 69.35(14), Ge(1)-C(10)-Co(2) 136.68(12), Co(2)-C(10)-Co(1) 75.19(8).

Figure 2.
Crystal structure of 11a. Thermal ellipsoids are represented at the 30% level and hydrogen atoms have been omitted for clarity (bond lengths in Å, angles in deg). S(1)-C(1) 1.715(5), Co(1)-C(7) 1.811(6), Co(1)-C(5) 1.949(4), Co(1)-C(6) 2.024(4), Co(1)-Co(2) 2.4555(10), O(1)-C(7) 1.144(6), Si(1)-C(6) 1.883(5), Si(1)-Si(2) 2.3626(18), C(5)-C(6) 1.344(6), C(4)-S(1)-C(1) 91.9(2), C(8)-Co(1)-C(7) 99.5(2), C(7)-Co(1)-C(9) 105.0(2), Si(2)-Si(1)-Si(3) 108.33(7), Si(2)-Si(1)-Si(4) 106.75(7), Si(3)-Si(1)-Si(4) 109.88(7), C(6)-C(5)-Co(2), C(5)-C(6)-Si(1) 143.8(4), C(5)-C(6)-Co(2).
Figure 2.
Crystal structure of 11a. Thermal ellipsoids are represented at the 30% level and hydrogen atoms have been omitted for clarity (bond lengths in Å, angles in deg). S(1)-C(1) 1.715(5), Co(1)-C(7) 1.811(6), Co(1)-C(5) 1.949(4), Co(1)-C(6) 2.024(4), Co(1)-Co(2) 2.4555(10), O(1)-C(7) 1.144(6), Si(1)-C(6) 1.883(5), Si(1)-Si(2) 2.3626(18), C(5)-C(6) 1.344(6), C(4)-S(1)-C(1) 91.9(2), C(8)-Co(1)-C(7) 99.5(2), C(7)-Co(1)-C(9) 105.0(2), Si(2)-Si(1)-Si(3) 108.33(7), Si(2)-Si(1)-Si(4) 106.75(7), Si(3)-Si(1)-Si(4) 109.88(7), C(6)-C(5)-Co(2), C(5)-C(6)-Si(1) 143.8(4), C(5)-C(6)-Co(2).

Figure 3.
Crystal structure of 2b. Thermal ellipsoids are represented at the 30% level and hydrogen atoms have been omitted for clarity (bond lengths in Å, angles in deg). Mo(1)-C(15) 2.171(9), Mo(1)-C(16) 2.223(8), Mo(1)-Mo(2) 2.9656(14), Mo(2)-C(15) 2.139(8), Mo(2)-C(16) 2.278(8), O(2)-C(12) 1.166(13), Si(1)-C(16) 1.875(8), Si(1)-Si(2) 2.393(3), Si(2)-C(18) 1.824(12), C(15)-C(16) 1.336(11), C(15)-Mo(1)-C(16) 35.4(3), C(15)-Mo(1)-Mo(2) 46.1(2), C(16)-Mo(1)-Mo(2) 49.6(2), C(15)-Mo(2)-C(16) 35.0(3), Si(4)-Si(1)-Si(3) 105.21(15), C(15)-C(16)-Si(1) 132.1(6), C(15)-C(16)-Mo(1) 70.2(5).
Figure 3.
Crystal structure of 2b. Thermal ellipsoids are represented at the 30% level and hydrogen atoms have been omitted for clarity (bond lengths in Å, angles in deg). Mo(1)-C(15) 2.171(9), Mo(1)-C(16) 2.223(8), Mo(1)-Mo(2) 2.9656(14), Mo(2)-C(15) 2.139(8), Mo(2)-C(16) 2.278(8), O(2)-C(12) 1.166(13), Si(1)-C(16) 1.875(8), Si(1)-Si(2) 2.393(3), Si(2)-C(18) 1.824(12), C(15)-C(16) 1.336(11), C(15)-Mo(1)-C(16) 35.4(3), C(15)-Mo(1)-Mo(2) 46.1(2), C(16)-Mo(1)-Mo(2) 49.6(2), C(15)-Mo(2)-C(16) 35.0(3), Si(4)-Si(1)-Si(3) 105.21(15), C(15)-C(16)-Si(1) 132.1(6), C(15)-C(16)-Mo(1) 70.2(5).

Figure 4.
Crystal structure of (COD)Ni(dippe). Thermal ellipsoids are represented at the 30% level and hydrogen atoms have been omitted for clarity (bond lengths in Å, angles in deg). Ni(1)-C(11) 2.094(2), Ni(1)-C(8) 2.107(2), Ni(1)-P(1) 2.1613(7), P(1)-C(1) 1.862(2), P(1)-C(5) 1.865(2), C(2)-C(3) 1.534(3), C(11)-Ni(1)-C(8) 84.89(9), (11)-Ni(1)-P(1) 135.18(7), C(8)-Ni(1)-P(1) 133.48(6).
Figure 4.
Crystal structure of (COD)Ni(dippe). Thermal ellipsoids are represented at the 30% level and hydrogen atoms have been omitted for clarity (bond lengths in Å, angles in deg). Ni(1)-C(11) 2.094(2), Ni(1)-C(8) 2.107(2), Ni(1)-P(1) 2.1613(7), P(1)-C(1) 1.862(2), P(1)-C(5) 1.865(2), C(2)-C(3) 1.534(3), C(11)-Ni(1)-C(8) 84.89(9), (11)-Ni(1)-P(1) 135.18(7), C(8)-Ni(1)-P(1) 133.48(6).

Figure 5.
Crystal structure of 13c. Thermal ellipsoids are represented at the 30% level and hydrogen atoms have been omitted for clarity (bond lengths in Å, angles in deg). (C4)-C(5) 1.296(8), Ni(1)-C(5) 1.872(5), Ni(1)-C(4) 1.917(5), Ni(1)-P(1) 2.1523(16), Ni(1)-P(2) 2.1620(15), P(1)-C(13) 1.849(5), S(1)-C(9) 1.718(5), Si(1)-C(4) 1.834(5), Si(1)-C(1) 1.871(5), C(5)-Ni(1)-C(4) 40.0(2), C(5)-Ni(1)-P(1) 111.80(15), C(4)-Ni(1)-P(1) 151.55(16), C(5)-Ni(1)-P(2) 156.52(15), C(4)-Ni(1)-P(2) 116.57(16), P(1)-Ni(1)-P(2) 91.68(6), Si(1)-C(4)-Ni(1) 148.6(3).
Figure 5.
Crystal structure of 13c. Thermal ellipsoids are represented at the 30% level and hydrogen atoms have been omitted for clarity (bond lengths in Å, angles in deg). (C4)-C(5) 1.296(8), Ni(1)-C(5) 1.872(5), Ni(1)-C(4) 1.917(5), Ni(1)-P(1) 2.1523(16), Ni(1)-P(2) 2.1620(15), P(1)-C(13) 1.849(5), S(1)-C(9) 1.718(5), Si(1)-C(4) 1.834(5), Si(1)-C(1) 1.871(5), C(5)-Ni(1)-C(4) 40.0(2), C(5)-Ni(1)-P(1) 111.80(15), C(4)-Ni(1)-P(1) 151.55(16), C(5)-Ni(1)-P(2) 156.52(15), C(4)-Ni(1)-P(2) 116.57(16), P(1)-Ni(1)-P(2) 91.68(6), Si(1)-C(4)-Ni(1) 148.6(3).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
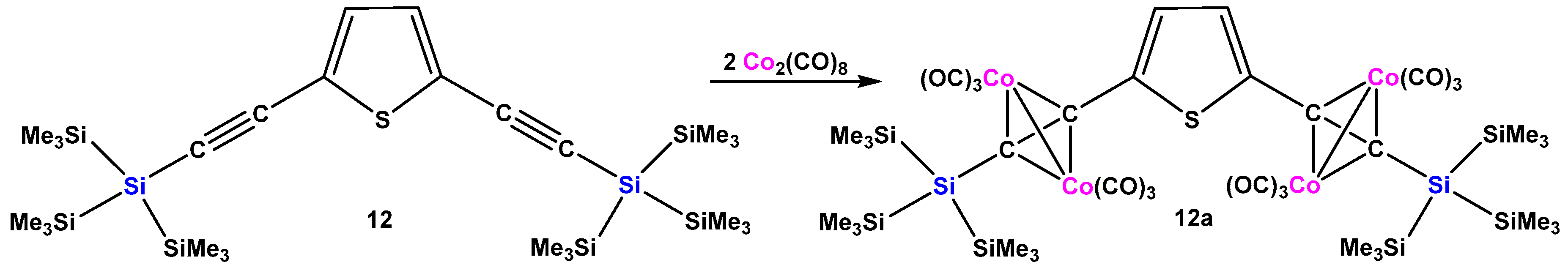{kind=link}
{kind=link}
{kind=link}
Table 1.
29Si and 13C NMR data of the alkynyl substituted Si and the alkynyl C atoms of the free alkynes as well as of the respective Co, Mo and Ni complexes.
Table 1.
29Si and 13C NMR data of the alkynyl substituted Si and the alkynyl C atoms of the free alkynes as well as of the respective Co, Mo and Ni complexes.
| Entry | Alkyne | 29Si | 13C a | Metal | 29Si | 13C a |
|---|---|---|---|---|---|---|
| 1 | (Me3Si)3SiCA≡CBPh (1) b | −100.5 | 88.4/108.6 | Co (1a) b Ni (3c) | −67.1 −90.0 | 75.8/111.4 118.6/157.9 |
| 1a | (Me3Si)3GeCA≡CBPh (7) c | n.a. | 89.6/106.8 | Co (7a) | n.a. | 80.9/109.7 |
| 2 | (Me3Si)3SiCA≡CBH (2) b | −100.7 | 83.4/96.8 | Co (2a) b Mo (2b) Ni (2c) | −67.7 −58.8 −89.4 | 82.9/90.5 96.6/101.9 124.3/145.3 |
| 3 | H(Me3Si)2SiCA≡CBPh (3) b | −90.2 | 86.2/110.2 | Co (3a) b | −53.3 | 71.5/107.9 |
| 4 | Et(Me3Si)2SiCA≡CBPh (4) b | −57.7 | 90.2/109.9 | Co (4a) b | −33.8 | 77.9/109.1 |
| 5 | Me3SiCA≡CBPh (5) b | −18.2 | 94.1/105.8 | Co (5a) b | 0.7 | 79.8/106.0 |
| 6 | Me5Si2CA≡CBPh (6) b | −36.9 | 93.1/108.1 | Co (6a) b Mo (6b) Ni (6c) | −18.0 −16.8 −31.4 | 80.1/107.0 92.1/116.0 132.2/159.6 |
| 7 | (Me3Si)3SiCA≡CBthio (11) | −102.2 | 93.5/100.4 | Co (11c) | −67.1 | 76.6/96.6 |
| 8 | [(Me3Si)3SiC≡C]2 (10) | −100.5 | 79.2/93.4 | Co (10a) | −65.9 | 72.6/88.6 |
| 9 | Me5Si2CA≡CBSiMe3 (9) | −37.6/−18.6 | 112.9/116.5 | Co (9a) | −18.6/0.3 | 93.9/92.4 |
| 10 | Me5Si2C≡CSi2Me5 (8) | −38.2 | 115.3 | Co (8a) | −18.3 | 93.1 |
Table 2.
Structural data of Co and Mo complexes of silyl substituted alkynes.
| Entry | Alkyne | M | M-M Distance (Å) | C-C Distance (Å) | C-Si Distance (Å) | CA-M Distance (Å) | CB-M Distance (Å) |
|---|---|---|---|---|---|---|---|
| 1 | (Me3Si)3SiCA≡CBPh (1) a | Co (1a) | 2.4573(7) | 1.349(5) | 1.873(4) | 2.032(4)/2.022(4) | 1.965(4)/1.987(3) |
| 2 | (Me3Si)3SiCA≡CBH (2) | Co (2a) Mo (2b) | 2.469(2) 2.966(1) | 1.310(15) 1.337(10) | 1.878(10) 1.875(9) | 2.021(9)/2.012(10) 2.278(7)/2.223(10) | 1.938(10)/1.958(10) 2.139(7)/2.170(10) |
| 5 | Me3SiCA≡CBPh (5) a | Co (5a) | 2.4717(13) | 1.330(10) | 1.858(7) | 1.998(7)/1.991(7) | 1.987(7)/1.966(7) |
| 6 | Me5Si2CA≡CBPh (6) a | Co (6a) | 2.4798(9) | 1.345(4) | 1.855(3) | 1.996(3)/1.993(3) | 1.984(3)/1.970(3) |
| 7 | (Me3Si)3SiCA≡CBthio (11) | Co (11a) | 2.456(1)/ 2.473(1) | 1.343(5)/ 1.331(6) | 1.884(4)/ 1.883(4) | 2.023(5)/2.013(4)/ 2.022(4)/2.004(5) | 1.947(6)/1.983(5)/ 1.947(6)/1.983(5) |
| 8 | (Me3Si)3GeCA≡CBPh (7) | Co (7a) | 2.4582(5) | 1.338(3) | 1.966(2) C-Ge | 2.020(2)/2.009(2) | 1.974(2)/1.982(2) |
a Values taken from [17]. thio = 2-thienyl.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zirngast, M.; Marschner, C.; Baumgartner, J. Spectroscopic and Structural Study of Some Oligosilanylalkyne Complexes of Cobalt, Molybdenum and Nickel. Molecules 2019, 24, 205. https://doi.org/10.3390/molecules24010205
AMA Style
Zirngast M, Marschner C, Baumgartner J. Spectroscopic and Structural Study of Some Oligosilanylalkyne Complexes of Cobalt, Molybdenum and Nickel. Molecules. 2019; 24(1):205. https://doi.org/10.3390/molecules24010205
Chicago/Turabian StyleZirngast, Michaela, Christoph Marschner, and Judith Baumgartner. 2019. "Spectroscopic and Structural Study of Some Oligosilanylalkyne Complexes of Cobalt, Molybdenum and Nickel" Molecules 24, no. 1: 205. https://doi.org/10.3390/molecules24010205