
The HK2 Dependent “Warburg Effect” and Mitochondrial Oxidative Phosphorylation in Cancer: Targets for Effective Therapy with 3-Bromopyruvate

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Knowledge Acquired to Date
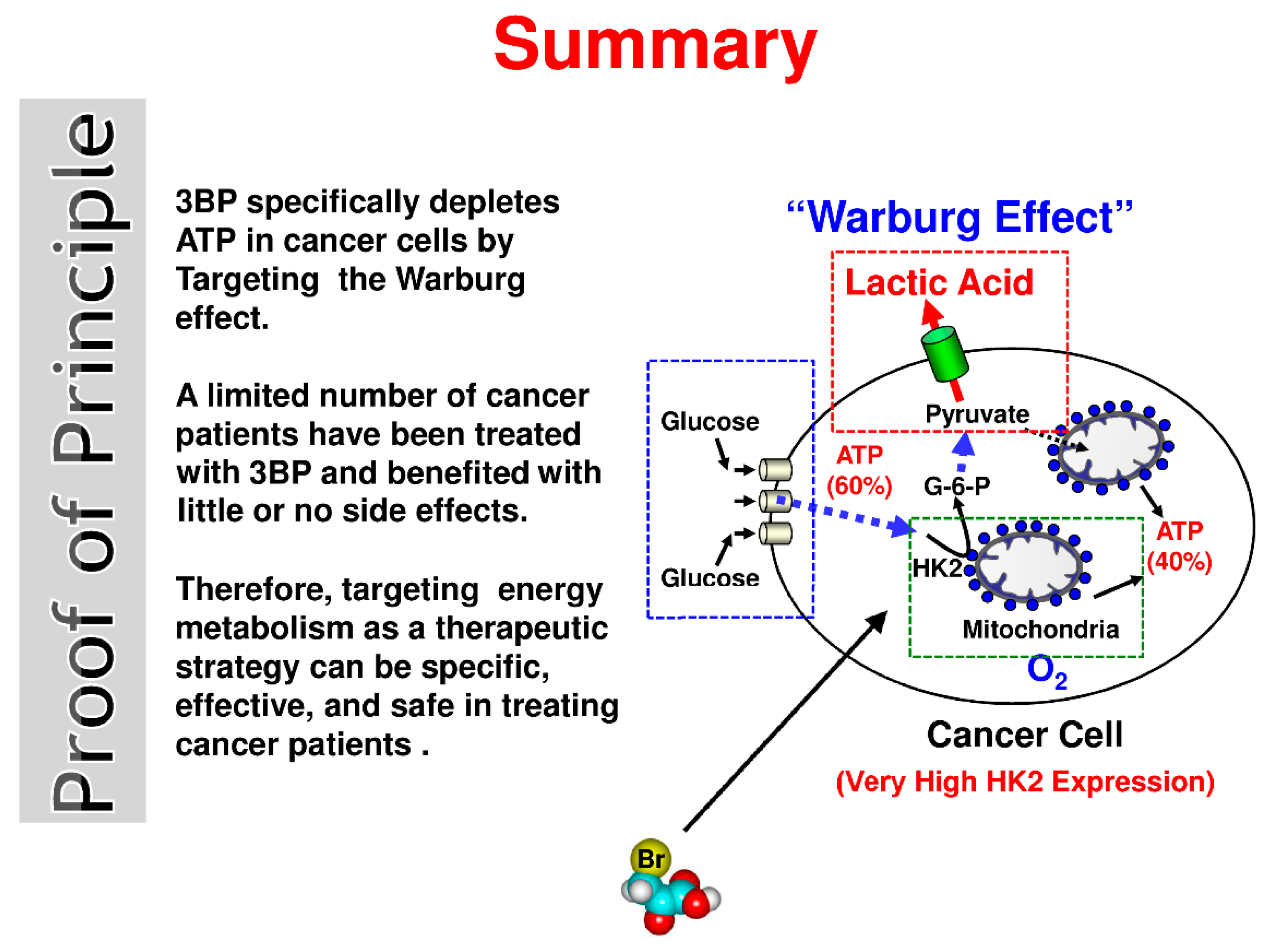
2.1. Energy Production Considerations in Normal Cells and Cancer Cells
2.2. Anti-Energy Metabolism Inhibitors: An Effective Approach for Treating Cancer
2.3. Mechanism of Action of 3BP
2.4. Perspectives
2.5. Proper and Improper Use of 3BP
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, E.P.; Lehninger, A.L. Intracellular structures and the fatty acid oxidase system of rat liver. J. Biol. Chem. 1948, 172, 847–848. [Google Scholar] [PubMed]
- Lehninger, A.L.; Kennedy, E.P. The requirement of the fatty acid oxidase complex of rat liver. J. Biol. Chem. 1948, 173, 753–771. [Google Scholar] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, R.A.; Paggi, M.G.; Pedersen, P.L. Contributions of glycolysis and oxidative phosphorylation to adenosine 5-triphosphate production in AS-30D hepatoma cells. Cancer Res. 1984, 44, 5702–5706. [Google Scholar] [PubMed]
- Icard, P.; Poulain, L.; Lincet, H. Understanding the central role of citrate in the metabolism of cancer cells. Biochim. Biophys. Acta 2012, 1825, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Ruiz, R.; Rigoulet, M.; Devin, A. The Warburg and Crabtree effects: On the origin of cancer cell energy metabolism and of yeast glucose repression. Biochim. Biophys. Acta 2011, 1807, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, P.L.; Greenawalt, J.W.; Chan, T.L.; Morris, H.P. A Comparison of Some Ultrastructural and Biochemical Properties of Mitochondria from Morris Hepatomas 9618A, 7800 and 3924A. Cancer Res. 1970, 30, 2620–2626. [Google Scholar] [PubMed]
- Pedersen, P.L. Tumor mitochondria and the bioenergetics of cancer cells. Prog. Exp. Tumor Res. 1978, 22, 190–274. [Google Scholar] [PubMed]
- Weber, G.; Stubbs, M.; Morris, H.P. Metabolism of hepatomas of different growth rates in situ and during ischemia. Cancer Res. 1971, 31, 2177–2183. [Google Scholar] [PubMed]
- Diaz-Ruiz, R.; Uribe-Carvajal, S.; Devin, A.; Rigoulet, M. Tumor cell energy metabolism and its common features with yeast metabolism. Biochim. Biophys. Acta 2009, 1796, 252–265. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, H.G. Observations on the carbohydrate metabolism of tumors. Biochem. J. 1929, 23, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Broach, J.R. Nutritional control of growth and development in yeast. Genetics 2012, 192, 73–105. [Google Scholar] [CrossRef] [PubMed]
- Van Urk, H.; Voll, W.S.L.; Scheffers, W.A.; van Dijken, J.P. Transient-state analysis of metabolic fluxes in Crabtree-positive and Crabtree-negative yeasts. Appl. Environ. Microbiol. 1990, 56, 282–286. [Google Scholar]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumor growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Romero-Garcia, S.; Moreno-Altamirano, M.M.; Prado-Garcia, H.; Sanchez-Garcia, F.J. Lactate contribution to the tumor microenvironment: Mechanisms, effects on immune cells and therapeutic relevance. Front. Immunol. 2016, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Alfarouk, K.O.; Verduzco, D.; Rauch, C.; Muddathir, A.K.; Adil, H.H.B.; Elhassan, G.O.; Ibrahim, M.E. Glycolysis, tumor metabolism, cancer growth and dissemination. A new pH-based etiopathogenic perspective and therapeutic approach to an old cancer question. Oncoscience 2014, 1, 777–802. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Enriquez, S.; Juárez, O.; Rodríguez-Zavala, J.S.; Moreno-Sánchez, R. Multisite control of the Crabtree effect in ascites hepatoma cells. Eur. J. Biochem. 2001, 8, 2512–2519. [Google Scholar] [CrossRef]
- Marroquin, L.D.; Hynes, J.; Dykens, J.A.; Jamieson, J.D.; Will, Y. Circumventing the crabtree effect: Replacing media glucose with galactose increases susceptibility of HepG2 cells to mitochondrial toxicants. Toxicol. Sci. 2007, 97, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Grüning, N.M.; Lehrach, H.; Ralser, M. Regulatory crosstalk of the metabolic network. Trends Biochem. Sci. 2010, 35, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, Y.Y.; Zhang, Q.W.; Zhao, S.R.; Wu, C.Z.; Cheng, X.; Jiang, C.C.; Jiang, Z.W.; Liu, H. 3-Bromopyruvate induces apoptosis in breast cancer cells by down regulating Mcl-1 through the PI3K/Akt signaling pathway. Anticancer Drugs 2014, 25, 447–455. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Cheng, T. Q’s next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010, 29, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Vithayathil, S.; Kumar, S.; Sung, P.L.; Dobrolecki, L.E.; Putluri, V.; Bhat, V.B.; Bhowmik, S.K.; Gupta, V.; Arora, K.; et al. Fatty acid oxidation-driven Src links mitochondrial energy reprogramming and oncogenic properties in triple-negative breast cancer. Cell Rep. 2016, 14, 2154–2165. [Google Scholar] [CrossRef] [PubMed]
- Bluemlein, K.; Grüning, N.M.; Feichtinger, R.G.; Lehrach, H.; Kofler, B.; Ralser, M. No evidence for a shift in pyruvate kinase PKM1 to PKM2 expression during tumorigenesis. Oncotarget 2011, 2, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Hitosugi, T.; Kang, S.; Vander Heiden, M.G.; Chung, T.W.; Elf, S.; Lythgoe, K.; Dong, S.; Lonial, S.; Wang, X.; Chen, G.Z.; et al. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci. Signal. 2009, 2. [Google Scholar] [CrossRef] [PubMed]
- Christofk, H.R.; Vander Heiden, M.G.; Wu, N.; Asara, J.M.; Cantley, L.C. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature 2008, 452, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Mazurek, S.; Boschek, C.B.; Hugo, F.; Eigenbrodt, E. Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin. Cancer Biol. 2005, 15, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Wellen, K.E.; Mazurek, S.; Bamezai, R.N. Pyruvate kinase M2: Regulatory circuits and potential for therapeutic intervention. Curr. Pharm. Des. 2013, 20, 2595–2606. [Google Scholar] [CrossRef]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, C.; Longatto-Filho, A.; Azevedo-Silva, J.; Casal, M.; Schmitt, F.C.; Baltazar, F. Role of monocarboxylate transporters in human cancers: State of the art. J. Bioenerg. Biomembr. 2012, 44, 127–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doherty, J.R.; Cleveland, J.L. Targeting lactate metabolism for cancer therapeutics. J. Clin. Investig. 2013, 123, 3685–3692. [Google Scholar] [CrossRef] [PubMed]
- Feron, O. Pyruvate into lactate and back: From the Warburg effect to symbiotic energy fuel exchange in cancer cells. Radiother. Oncol. 2009, 92, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Birsoy, K.; Wang, T.; Possemato, R.; Yilmaz, O.H.; Koch, C.E.; Chen, W.W.; Hutchins, A.W.; Gultekin, Y.; Peterson, T.R.; Carette, J.E.; et al. MCT1-mediated transport of a toxic molecule is an effective strategy for targeting glycolytic tumors. Nat. Genet. 2013, 45, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; Flores, R.E.; Poff, A.M.; D’Agostino, D.P. Cancer as a metabolic disease: Implications for novel therapeutics. Carcinogenesis 2014, 35, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Macheda, M.L.; Rogers, S.; Best, J.D. Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J. Cell. Physiol. 2005, 202, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Seino, Y.; Fukumoto, H.; Koh, G.; Yano, H.; Inagaki, N.; Yamada, Y.; Inoue, K.; Manabe, T.; Imura, H. Over-expression of facilitative glucose transporter genes in human cancer. Biochem. Biophys. Res. Commun. 1990, 170, 223–230. [Google Scholar] [CrossRef]
- Bustamante, E.; Pedersen, P.L. High aerobic glycolysis of rat hepatoma cells in culture: Role of mitochondrial hexokinase. Proc. Natl. Acad. Sci. USA 1977, 74, 3735–3739. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, E.; Morris, H.P.; Pedersen, P.L. Energy metabolism of tumor cells: Requirement for a form of hexokinase with a propensity for mitochondrial binding. J. Biol. Chem. 1981, 256, 8699–8704. [Google Scholar] [PubMed]
- Nakashima, R.A.; Paggi, M.G.; Scott, L.J.; Pedersen, P.L. Purification and characterization of a bindable form of mitochondrial bound hexokinase from the highly glycolytic AS-30D rat hepatoma cell line. Cancer Res. 1988, 48, 913–919. [Google Scholar] [PubMed]
- Nakashima, R.A.; Mangan, P.S.; Colombini, M.; Pedersen, P.L. Hexokinase receptor complex in hepatoma mitochondria: Evidence from N,N’-dicyclohexylcarbodiimide-labelling studies for the involvement of the pore-forming protein VDAC. Biochemistry 1986, 25, 1015–1021. [Google Scholar] [CrossRef] [PubMed]
- Arora, K.K.; Pedersen, P.L. Functional significance of mitochondrial bound hexokinase in tumor cell metabolism. Evidence for preferential phosphorylation of glucose by intra-mitochondrially generated ATP. J. Biol. Chem. 1988, 263, 17422–17428. [Google Scholar] [PubMed]
- Pedersen, P.L. Voltage dependent anion channels (VDACs): A brief introduction with a focus on the outer mitochondrial compartment’s roles together with hexokinase-2 in the “Warburg effect” in cancer. J. Bioenerg. Biomembr. 2008, 40, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Mathupala, S.P.; Ko, Y.H.; Pedersen, P.L. Hexokinase-2 bound to mitochondria: Cancer’s stygian link to the “Warburg Effect” and a pivotal target for effective therapy. Semin. Cancer Biol. 2009, 19, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Vora, S.; Halper, J.P.; Knowles, D.M. Alterations in the activity and isozymic profile of human phosphofructokinase during malignant transformation in vivo and in vitro: Transformation- and progression-linked discriminants of malignancy. Cancer Res. 1985, 45, 2993–3001. [Google Scholar] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Marín-Hernández, A.; Gallardo-Pérez, J.C.; Ralph, S.J.; Rodríguez-Enríquez, S.; Moreno-Sánchez, R. HIF-1alpha modulates energy metabolism in cancer cells by inducing over-expression of specific glycolytic isoforms. Mini Rev. Med. Chem. 2009, 9, 1084–1101. [Google Scholar] [CrossRef] [PubMed]
- Mathupala, S.P.; Rempel, A.; Pedersen, P.L. Glucose catabolism in cancer cells: Identification and characterization of a marked activation response to hypoxic conditions. J. Biol. Chem. 2001, 276, 43407–43412. [Google Scholar] [CrossRef] [PubMed]
- Robey, R.B.; Hay, N. Mitochondrial hexokinases: Guardians of the mitochondria. Cell Cycle 2005, 4, 654–658. [Google Scholar] [CrossRef] [PubMed]
- Pastorino, J.G.; Hoek, J.B. Regulation of hexokinase binding to VDAC. J. Bioenerg. Biomembr. 2008, 40, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Salk, J.J.; Fox, E.J.; Loeb, L.A. Mutational heterogeneity in human cancers: Origin and consequences. Annu. Rev. Pathol. 2010, 5, 51–75. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.D.; Deslandes, E.; Villedieu, M.; Poulain, L.; Duval, M.; Gauduchon, P.; Schwartz, L.; Icard, P. Effect of 2-deoxy-D-glucose on various malignant cell lines in vitro. Anticancer Res. 2006, 26, 3561–3566. [Google Scholar] [PubMed]
- Lu, Y.; Zhang, X.; Zhang, H.; Lan, J.; Huang, G.; Varin, E.; Lincet, H.; Poulain, L.; Icard, P. Citrate induces apoptotic cell death: A promising way to treat gastric carcinoma? Anticancer Res. 2011, 31, 797–805. [Google Scholar] [PubMed]
- Icard, P.; Zhang, X.D.; Varin, E.; Allouche, S.; Coquerel, A.; Paciencia, M.; Joyeux, L.; Gauduchon, P.; Lincet, H.; Poulain, L. Why Anti-Energetic Agents Such as Citrate or 3-Bromopyruvate should be Tested as Anti-Cancer Agents: Experimental In Vitro and In Vivo Studies. In Mesotheliomas—Synonyms and Definition, Epidemiology, Etiology, Pathogenesis, Cyto-Histopathological Features, Clinic, Diagnosis, Treatment, Prognosis; Zubritsky, A., Ed.; InTech: Rijeka, Croatia, 2012; Volume 14, pp. 226–248. [Google Scholar]
- Zhang, X.; Varin, E.; Allouche, S.; Lu, Y.; Poulain, L.; Icard, P. Effect of citrate on malignant pleural mesothelioma cells: A synergistic effect with cisplatin. Anticancer Res. 2009, 29, 1249–1254. [Google Scholar] [PubMed]
- Ko, Y.H.; Smith, B.L.; Wang, Y.; Pomper, M.G.; Rini, D.A.; Torbenson, M.S.; Hullihen, J.; Pedersen, P.L. Advanced cancers: Eradication in all cases using 3-bromopyruvate therapy to deplete ATP. Biochem. Biophys. Res. Commun. 2004, 324, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Sudhakar, A. History of cancer, ancient and modern treatments. J. Cancer Sci. Ther. 2009, 1, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ihrlund, L.S.; Hernlund, E.; Khan, O.; Shoshan, M.C. 3-Bromopyruvate as inhibitor of tumour cell energy metabolism and chemopotentiator of platinum drugs. Mol. Oncol. 2008, 2, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Xu, J.; Yuan, W.; Wu, B.; Wang, H.; Liu, G.; Wang, X.; Du, J.; Cai, S. The Reversal Effects of 3-Bromopyruvate on Multidrug Resistance in Vitro and In Vivo Derived from Human Breast MCF-7/ADR Cells. PLoS ONE 2014, 9, e112132. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.H.; McFadden, B.A. Alkylation of isocitrate lyase from Escherichia coli by 3-bromopyruvate. Arch. Biochem. Biophys. 1990, 278, 373–380. [Google Scholar] [CrossRef]
- Krátký, M.; Vinšová, J. Advances in mycobacterial isocitrate lyase targeting and inhibitors. Curr. Med. Chem. 2012, 19, 6126–6137. [Google Scholar] [CrossRef] [PubMed]
- Dell’Antone, P. Enhancing 3-bromopyruvate toxicity in tumor cells by inducing hyperglycemia. Pharmacologia 2013, 4, 571–575. [Google Scholar]
- Dell’Antone, P. Energy metabolism in cancer cells: How to explain the Warburg and Crabtree effects? Med. Hypotheses 2012, 79, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Sadowska-Bartosz, I.; Soszyński, M.; Ułaszewski, S.; Ko, Y.H.; Bartosz, G. Transport of 3-bromopyruvate across the human erythrocyte membrane. Cell Mol. Biol. Lett. 2014, 19, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.H.; Pedersen, P.L.; Geschwind, J.F. Glucose catabolism in the rabbit VX2 tumor model for liver cancer: Characterization and targeting hexokinase. Cancer Lett. 2001, 173, 83–91. [Google Scholar] [CrossRef]
- Kaplan, R.S.; Pratt, R.D.; Pedersen, P.L. Purification and characterization of the reconstitutively active phosphate transporter from rat liver mitochondria. J. Biol. Chem. 1986, 261, 12767–12773. [Google Scholar] [PubMed]
- Cardaci, S.; Desideri, E.; Ciriolo, M.R. Targeting aerobic glycolysis: 3-bromopyruvate as a promising anticancer drug. J. Bioenerg. Biomembr. 2012, 44, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Ramos, D.; Pellín-Carcelén, A.; Agustí, J.; Murgui, A.; Jordá, E.; Pellín, A.; Monteagudo, C. Deregulation of glyceraldehyde-3-phosphate dehydrogenase expression during tumor progression of human cutaneous melanoma. Anticancer Res. 2015, 35, 439–444. [Google Scholar] [PubMed]
- Wang, D.; Moothart, D.R.; Lowy, D.R.; Qian, X. The expression of glyceraldehyde-3-phosphate dehydrogenase associated cell cycle (GACC) genes correlates with cancer stage and poor survival in patients with solid tumors. PLoS ONE 2013, 8, e61262. [Google Scholar] [CrossRef] [PubMed]
- Dyląg, M.; Lis, P.; Niedźwiecka, K.; Ko, Y.H.; Pedersen, P.L.; Goffeau, A.; Ułaszewski, S. 3-Bromopyruvate: A novel antifungal agent against the human pathogen Cryptococcus neoformans. Biochem. Biophys. Res. Commun. 2013, 434, 322–327. [Google Scholar]
- Lis, P.; Jurkiewicz, P.; Cal-Bąkowska, M.; Ko, Y.H.; Pedersen, P.L.; Goffeau, A.; Ułaszewski, S. Screening the yeast genome for energetic metabolism pathways involved in a phenotypic response to the anti-cancer agent 3-bromopyruvate. Oncotarget 2016, 9, 10153–10173. [Google Scholar]
- Sun, Y.; Liu, Z.; Zou, X.; Lan, Y.; Sun, X.; Wang, X.; Zhao, S.; Jiang, C.; Liu, H. Mechanisms underlying 3-bromopyruvate- induced cell death in colon cancer. J. Bioenerg. Biomembr. 2015, 47, 319–329. [Google Scholar] [CrossRef] [PubMed]
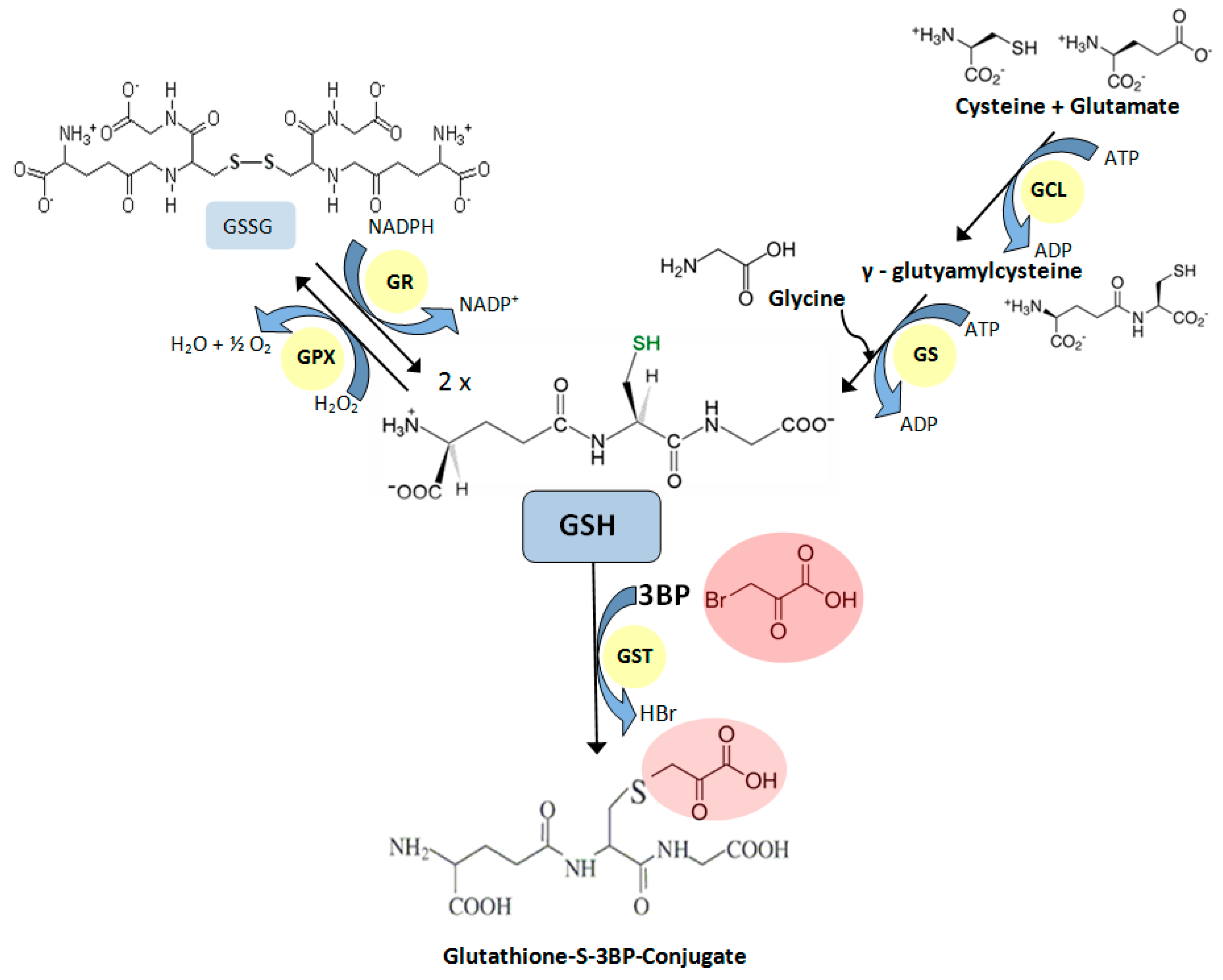
- Niedźwiecka, K.; Dyląg, M.; Augustyniak, D.; Majkowska-Skrobek, G.; Cal-Bąkowska, M.; Ko, Y.H.; Pedersen, P.L.; Goffeau, A.; Ułaszewski, S. Glutathione may have implications in the design of 3-bromopyruvate treatment protocols for both fungal and algal infections as well as multiple myeloma. Oncotarget 2016, 7, 65614–65626. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.H.; Verhoeven, H.A.; Lee, M.J.; Corbin, D.J.; Vogl, T.J.; Pedersen, P.L. A translational study “case report” on the small molecule “energy blocker” 3-bromopyruvate (3BP) as a potent anticancer agent: From bench side to bedside. J. Bioenerg. Biomembr. 2012, 44, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Ahn, K.J.; Kim, J.A.; Kim, H.M.; Lee, J.D.; Lee, J.M.; Kim, S.J.; Park, J.H. Role of reactive oxygen species-mediated mitochondrial dysregulation in 3-bromopyruvate induced cell death in hepatoma cells: ROS-mediated cell death by 3-BrPA. J. Bioenerg. Biomembr. 2008, 40, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, Y.; Zhang, P.; Chao, Z.; Xia, F.; Jiang, C.; Zhang, X.; Jiang, Z.; Liu, H. Hexokinase II inhibitor, 3-BrPA induced autophagy by stimulating ROS formation in human breast cancer cells. Genes Cancer 2014, 5, 100–112. [Google Scholar] [PubMed]
- Lis, P.; Zarzycki, M.; Ko, Y.H.; Casal, M.; Pedersen, P.L.; Goffeau, A.; Ułaszewski, S. Transport and cytotoxicity of the anticancer drug 3-bromopyruvate in the yeast Saccharomyces cerevisiae. J. Bioenerg. Biomembr. 2012, 44, 155–161. [Google Scholar] [CrossRef] [PubMed]
- El Sayed, S.M.; El-Magd, R.M.; Shishido, Y.; Yorita, K.; Chung, S.P.; Tran, D.H.; Sakai, T.; Watanabe, H.; Kagami, S.; Fukui, K. D-Amino acid oxidase-induced oxidative stress, 3-bromopyruvate and citrate inhibit angiogenesis, exhibiting potent anticancer effects. J. Bioenerg. Biomembr. 2012, 44, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Sadowska-Bartosz, I.; Bartosz, G. The effect of 3-bromopyruvic acid on human erythrocyte antioxidant defense system. Cell Biol. Int. 2013, 37, 1285–1290. [Google Scholar] [CrossRef] [PubMed]
- Sadowska-Bartosz, I.; Szewczyk, R.; Jaremko, L.; Jaremko, M.; Bartosz, G. Anticancer agent 3-bromopyruvic acid forms a conjugate with glutathione. Pharmacol. Rep. 2016, 68, 502–505. [Google Scholar] [CrossRef] [PubMed]
- Reliene, R.; Schiestl, R.H. Glutathione depletion by buthionine sulfoximine induces DNA deletions in mice. Carcinogenesis 2006, 27, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Meister, A. Glutathione metabolism and its selective modification. J. Biol. Chem. 1988, 263, 17205–17208. [Google Scholar] [PubMed]
- Wolchok, J.D.; Williams, L.; Pinto, J.T.; Fleisher, M.; Krown, S.E.; Hwu, W.J.; Livingston, P.O.; Chang, C.; Chapman, P.B. Phase I trial of high dose paracetamol and carmustine in patients with metastatic melanoma. Melanoma Res. 2003, 13, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Hinson, J.A.; Roberts, D.W.; James, L.P. Mechanisms of acetaminophen-induced liver necrosis. Handb. Exp. Pharmacol. 2010, 196, 369–405. [Google Scholar]
- Dyląg, M.; Lis, P.; Ko, J.H.; Pedersen, P.L.; Goffeau, A.; Ułaszewski, S. Use of the Composition of 3-Bromopyruvate as a Second Application of a Medicament for the Treatment of Fungal Infections, PAT.219802. 31 July 2015. Available online: http://regserv.uprp.pl/register/application;jsessionid=84F17C141B9A6E93D3C97D4FA2EB3188?lng=en&tab=no_data&number=P.399978 (accessed on 6 September 2016).
- Geschwind, J.F.; Georgiades, C.S.; Ko, Y.H.; Pedersen, P.L. Recently elucidated energy catabolism pathways provide opportunities for novel treatments in hepatocellular carcinoma. Expert. Rev. Anticancer Ther. 2004, 4, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Majkowska-Skrobek, G.; Augustyniak, D.; Lis, P.; Bartkowiak, A.; Gonchar, M.; Ko, Y.H.; Pedersen, P.L.; Goffeau, A.; Ułaszewski, S. Killing multiple myeloma cells with the small molecule 3-bromopyruvate: Implications for therapy. Anticancer Drugs 2014, 25, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Forman, D.; Ferlay, J. The global and regional burden of cancer. In World Cancer Report 2014; Stewart, B.W., Wild, C.P., Eds.; International Agency for Research on Cancer: Lyon, France, 2014; Volume 1.1, pp. 16–53. [Google Scholar]
- Pedersen, P.L. 3-Bromopyruvate (3BP) a fast acting, promising, powerful, specific, and effective “small molecule” anti-cancer agent taken from labside to bedside: Introduction to a special issue. J. Bioenerg. Biomembr. 2012, 44, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Azevedo-Silva, J.; Queirós, O.; Baltazar, F.; Ułaszewski, S.; Goffeau, A.; Ko, Y.H.; Pedersen, P.L.; Preto, A.; Casal, M. The anticancer agent 3-bromopyruvate: A simple but powerful molecule taken from the lab to the bedside. J. Bioenerg. Biomembr. 2016, 48, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Patrick, G.L. An Introduction to Medicinal Chemistry, 5th ed.; Oxford University Press: Oxford, UK, 2013; pp. 258–264. [Google Scholar]
- Yuan, S.; Wang, F.; Chen, G.; Zhang, H.; Feng, L.; Wang, L.; Colman, H.; Keating, M.J.; Li, X.; Xu, R.H.; et al. Effective elimination of cancer stem cells by a novel drug combination strategy. Stem. Cells 2013, 31, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Liederer, B.M.; Borchardt, R.T. Enzymes involved in the bioconversion of ester-based prodrugs. J. Pharm. Sci. 2006, 95, 1177–1195. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.D. Improved passive oral drug delivery via prodrugs. Adv. Drug Deliv. Rev. 1996, 19, 131–148. [Google Scholar] [CrossRef]
- Xu, R.H.; Pelicano, H.; Zhang, H.; Giles, F.J.; Keating, M.J.; Huang, P. Synergistic effect of targeting mTOR by rapamycin and depleting ATP by inhibition of glycolysis in lymphoma and leukemia cells. Leukemia 2005, 19, 2153–2158. [Google Scholar] [CrossRef] [PubMed]
- Pierre, K.; Pellerin, L. Monocarboxylate transporters in the central nervous system: Distribution, regulation and function. J. Neurochem. 2005, 94, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Gandham, S.K.; Talekar, M.; Singh, A.; Amiji, M.M. Inhibition of hexokinase-2 with targeted liposomal 3-bromopyruvate in an ovarian tumor spheroid model of aerobic glycolysis. Int. J. Nanomed. 2015, 10, 4405–4423. [Google Scholar]
- Pedersen, P.L. Mitochondria in relation to cancer metastasis: Introduction to a mini-review series. J. Bioenerg. Biomembr. 2012, 44, 615–617. [Google Scholar] [CrossRef] [PubMed]
- Park, B.J.; Wannemuehler, K.A.; Marston, B.J.; Govender, N.; Pappas, P.G.; Chiller, T.M. Estimation of the current global burden of cryptococcal meningitis among persons living with HIV/AIDS. AIDS 2009, 23, 525–530. [Google Scholar] [CrossRef] [PubMed]
- De Lima, L.P.O.; Seabra, S.H.; Carneiro, H.; Barbosa, H.S. Effect of 3-Bromopyruvate and Atovaquone on Infection during In Vitro Interaction of Toxoplasma gondii and LLC-MK2 Cells. Antimicrob. Agents Chemother. 2015, 59, 5239–5249. [Google Scholar] [CrossRef] [PubMed]
- PreScience Labs Announced that the FDA Accepts IND Application for Novel Oncology Drug. Available online: http://www.presciencelabs.com/prescience-labs-investors/press.php (accessed on 6 September 2016).
- Feldwisch-Drentrup, H. Candidate cancer drug suspected after death of three patients at an alternative medicine clinic. Science 2016. [Google Scholar] [CrossRef]


© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lis, P.; Dyląg, M.; Niedźwiecka, K.; Ko, Y.H.; Pedersen, P.L.; Goffeau, A.; Ułaszewski, S. The HK2 Dependent “Warburg Effect” and Mitochondrial Oxidative Phosphorylation in Cancer: Targets for Effective Therapy with 3-Bromopyruvate. Molecules 2016, 21, 1730. https://doi.org/10.3390/molecules21121730
Lis P, Dyląg M, Niedźwiecka K, Ko YH, Pedersen PL, Goffeau A, Ułaszewski S. The HK2 Dependent “Warburg Effect” and Mitochondrial Oxidative Phosphorylation in Cancer: Targets for Effective Therapy with 3-Bromopyruvate. Molecules. 2016; 21(12):1730. https://doi.org/10.3390/molecules21121730
Chicago/Turabian StyleLis, Paweł, Mariusz Dyląg, Katarzyna Niedźwiecka, Young H. Ko, Peter L. Pedersen, Andre Goffeau, and Stanisław Ułaszewski. 2016. "The HK2 Dependent “Warburg Effect” and Mitochondrial Oxidative Phosphorylation in Cancer: Targets for Effective Therapy with 3-Bromopyruvate" Molecules 21, no. 12: 1730. https://doi.org/10.3390/molecules21121730