3. Experimental Section
Nomenclature: In this section, we have applied the systematic nomenclature of the International Union of Pure and Applied Chemistry (IUPAC) for naming synthesized compounds and assigning NMR data. This is different from the other sections, where the common purine nomenclature is used, e.g., the 7-position in the purine nomenclature translates into the 5-position in the IUPAC nomenclature.
Material: Aziridine cofactor
4 [
7], 4-chloro-5-iodo-7
H-pyrrolo[2,3-
d]pyrimidine (
6) [
20], 5-
O-(
t-butyldimethylsilyl)-2,3-
O-isopropylidene-α-
d-ribofuranosyl chloride (
7) [
17],
N-(2-propynyl)-2,2,2-trifluoroacetamide (
11) [
21] and aziridine [
22,
23] were prepared according to literature procedures. (Caution: aziridine is hazardous and should be handled with care).
N-Hydroxysuccinimidyl biotin (NHS biotin),
tert-butyldimethylsilyl chloride (TBS-Cl),
N-ethyldiisopropylamine (EDIA), tetrakis(triphenylphosphine)palladium, triethylamine and trifluoroacetic acid were purchased from Fluka. 4-(Dimethylamino)pyridine (DMAP), methanesulfonyl chloride (MsCl), platinum(IV)oxide, tetrabutylammonium fluoride and tris[2-(2-methoxyethoxy)ethyl]amine (TDA-1) were purchased from Merck. Copper(I) iodide was from Riedel-deHaën. All reagents were of pro analysis (p.a.) grade. Dry solvents were either purchased or dried using common laboratory techniques [
24]. The DNA cytosine-C5 MTase M.HhaI was kindly provided by Dr. S. Klimasauskas (Vilnius, Lithuania). Proteinase K was obtained from QIAGEN; DNase (Grade II) was from Roche Applied Science; phosphodiesterases from
Crotalus adamanteus and from calf spleen were from Sigma; and alkaline phosphatase was from Boehringer. The restriction endonucleases R.HaeII, R.HhaI and R.XmnI were purchased from New England Biolabs; R.TaqI was purchased from MBI Fermentas. Streptavidin was obtained from Gerbu. ODN
I (5′-TGTCAGCGCATGA-3′) and
II (5′-TCATGC
MeGCTGACA-3′; C
Me =
C5-methyl-2′-deoxy–cytidine) were synthesized using a DNA/RNA synthesizer Model 392 from Applied Biosystems and purified by reverse-phase HPLC. pUC19 plasmid DNA was purchased from MBI Fermentas.
General Procedures: All air- or water-sensitive chemical reactions were carried out in dried glassware under an argon atmosphere. Silica gel 60 F254 glass plates (Merck, Darmstadt, Germany) were used for TLC. Flash chromatography was carried out using Merck silica gel 60 (40–63 µm). HPLC was performed using a Waters Breeze System equipped with a binary programmable pump system 1525, a dual wavelength absorbance detector 2487 and a Waters inline degasser. NMR spectra were recorded using a Mercury 300 (75 MHz for 13C), Inova 400 (400 MHz, 100 MHz and 376 MHz for 1H, 13C and 19F, respectively) and a Unity 500 (500 MHz for 1H) (all Varian) in the NMR spectroscopy facility of the institute. CDCl3 (δH = 7.24 and δC = 77.0) or [D6]DMSO (δH = 2.49 and δC = 39.5) were used as solvents. Assignments of 13C signals are based on 1H-, 13C-correlated 2D-NMR and on 13C-DEPT spectra. Electrospray ionization mass spectra (ESI-MS) were obtained using a Finnigan LCQ DECA XP Plus in the mass spectrometry facility of the institute. Measurements were carried out in the positive ion mode. UV absorption measurements were performed in methanol or water using a Varian Cary 3E spectrometer.
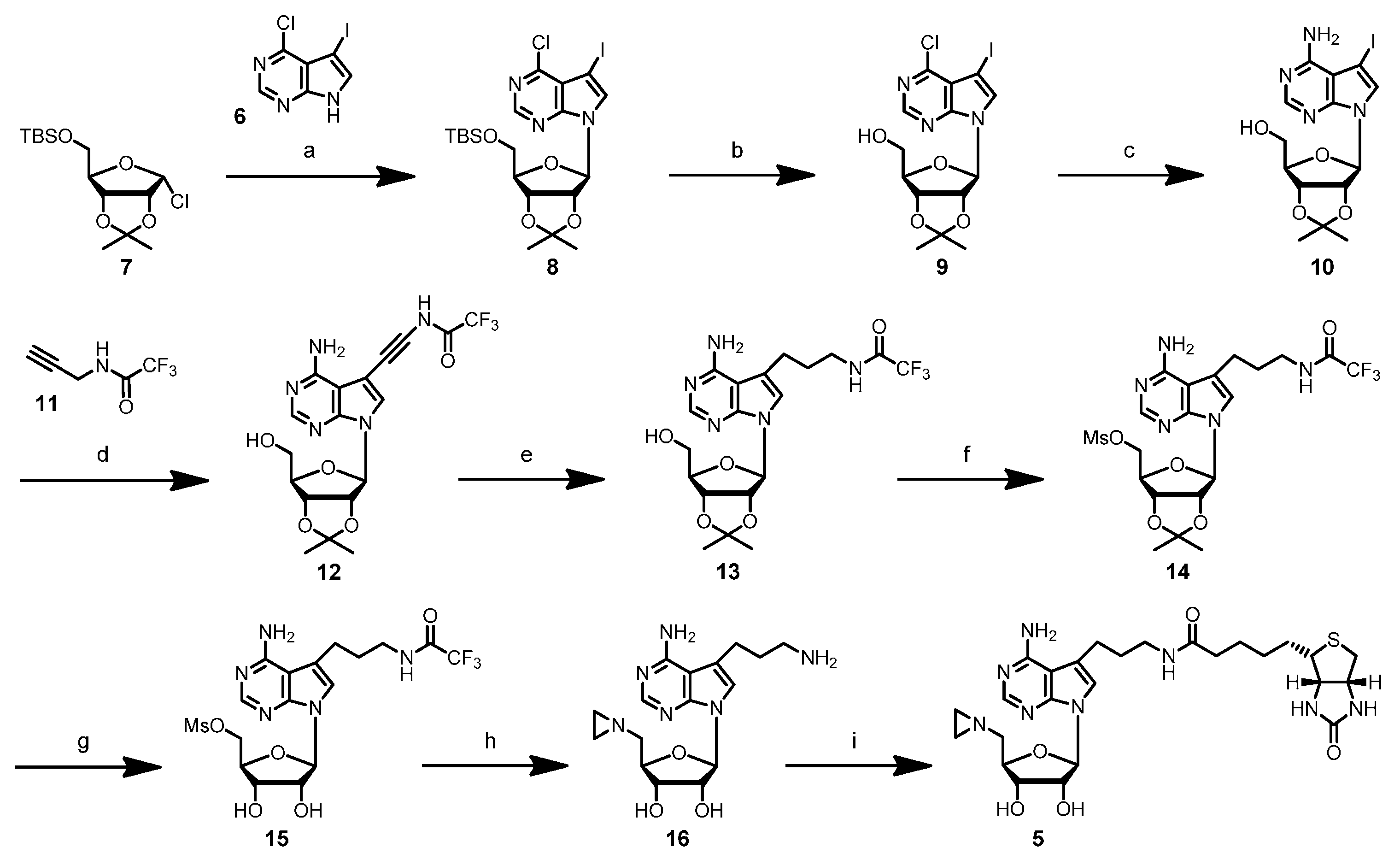
4-Chloro-5-iodo-7-(5′-O-t-butyldimethylsilyl-2′,3′-O-iso-β-d-propylidene-β-d-ribofuranosyl)-7H-pyrrolo[2,3-d]pyrimidine (8): To a suspension of powdered KOH (280 mg, 5.0 mmol) in dry acetonitrile (15 mL) under argon atmosphere was added tris[2-(2-methoxyethoxy)ethyl]amine (TDA-1) (200 µL, 0.6 mmol) and the mixture stirred at room temperature for 30 min. 4-Chloro-5-iodo-7H-pyrrolo[2,3-d]pyrimidine (6) (500 mg, 1.79 mmol) was added, and the mixture was stirred for 30 min at room temperature. A freshly-prepared solution of 5-O-(t-butyldimethylsilyl)-2,3-O-isopropylidene-α-d-ribofuranosyl chloride (7) (3.61 mmol) in THF (8 mL) was added, and the reaction mixture was stirred at room temperature for 2 days. The resulting black suspension was diluted by the addition of ethyl acetate (60 mL) and passed through a paper filter. The solvents were removed under reduced pressure, and the crude product was purified by column chromatography (silica gel, elution with ethyl acetate/hexane 15:85) to give nucleoside 8 (431 mg, 43%) as a light yellow oil (Rf 0.43, ethyl acetate/hexane 20:80). 1H-NMR (500 MHz, CDCl3): δ = 0.12 (s, 3H, SiCH3a), 0.12 (s, 3H, SiCH3b), 0.92 (s, 9H, SiC(CH3)3), 1.38 (s, 3H, isopropylidene-CH3a), 1.65 (s, 3H, isopropylidene-CH3b), 3.81 (dd, 2J = 11.29 Hz, 3J = 3.05 Hz, 1H, H5′a), 3.92 (dd, 2J = 11.60 Hz, 3J = 2.75 Hz, 1H, H5′b), 4.40 (q, 3J = 2.75 Hz, 1H, H4′), 4.90 (dd, 3J = 2.44 Hz, 3J = 6.10 Hz, 1H, H3′), 4.94 (dd, 3J = 3.05 Hz, 3J = 6.10 Hz, 1H, H2′), 6.43 (d, 3J = 3.05 Hz, 1H, H1′), 7.78 (s, 1H, H6), 8.65 (s, 1H, H2); 13C-NMR (100 MHz, CDCl3): δ = −5.38 (SiCH3a), −5.22 (SiCH3b), 18.43 (SiC(CH3)3), 25.38 (isopropylidene-CH3a), 25.98 (SiC(CH3)3), 27.32 (isopropylidene-CH3b), 52.13 (C5), 63.48 (C5′), 80.79 (C4′), 85.38 (C3′), 86.19 (C2′), 90.77 (C1′), 114.02 (isopropylidene-C(CH3)2), 117.24 (C9), 131.89 (C6), 150.25 (C2), 150.86 (C8), 152.40 (C4).
4-Chloro-5-iodo-7-(2′,3′-O-isopropylidene-β-d-ribofuranosyl)-7H-pyrrolo[2,3-d]pyrimidine (9): A solution of nucleoside 8 (348 mg, 0.61 mmol) in THF (10 mL) was cooled to 0 °C. After the addition of tetrabutylammonium fluoride (TBAF) (291 mg, 0.92 mmol), the reaction mixture was stirred at 0 °C for 2 h. The reaction mixture was allowed to warm up to room temperature, and the solvent was removed under reduced pressure. The residue was dissolved in ethyl acetate (10 mL), and the organic layer was washed with water (2 mL) and brine (2 mL). The organic layer was dried over magnesium sulfate, filtered, and the solvent was removed under reduced pressure. Purification by column chromatography (silica gel, elution with ethyl acetate/hexane 40:60) gave nucleoside 9 (192 mg, 69%) as a light yellow foam (Rf 0.23, ethyl acetate/hexane 40:60). 1H-NMR (500 MHz, CDCl3): δ = 1.37 (s, 3H, isopropylidene-CH3a), 1.64 (s, 3H, isopropylidene-CH3b), 3.81 (t, 2J = 3J = 10.68 Hz, 1H, H5′a), 3.95 (dd, 2J = 12.51 Hz, 3J = 1.83 Hz, 1H, H5′b), 4.47–4.49 (m, 1H, H4′), 4.76 (d, 3J = 10.07 Hz, 1H, 5′-OH), 5.09 (dd, 3J = 2.14 Hz, 3J = 6.10 Hz, 1H, H3′), 5.19 (dd, 3J = 4.88 Hz, 3J = 6.10 Hz, 1H, H2′), 5.88 (d, 3J = 4.88 Hz, 1H, H1′), 7.51 (s, 1H, H6), 8.63 (s, 1H, H2); 13C-NMR (75 MHz, CDCl3): δ = 25.30 (isopropylidene-CH3a), 27.54 (isopropylidene-CH3b), 51.94 (C5), 63.09 (C5′), 81.16 (C4′), 83.28 (C3′), 85.76 (C2′), 94.58 (C1′), 114.44 (isopropylidene-C(CH3)2), 118.72 (C9), 134.55 (C6), 149.56 (C2), 150.60 (C8), 153.67 (C4); ESI-MS m/z (relative intensity): 452.3 (9) [M + H]+, 280.5 (100) [4-chloro-5-iodo-7H-pyrrolo[2,3-d]pyrimidine + H]+.
4-Amino-5-iodo-7-(2′,3′-O-isopropylidene-β-d-ribofuranosyl)-7H-pyrrolo[2,3-d]pyrimidine (10): A solution of nucleoside 9 (981 mg, 2.17 mmol) in methanol (80 mL) was cooled to 0 °C and then saturated with gaseous ammonia. A pre-cooled autoclave was filled with the solution, sealed and heated to 80–85 °C overnight. The autoclave was allowed to cool to room temperature; the solution was removed, and the solvent was evaporated under reduced pressure. The crude product was purified by column chromatography (silica gel, elution with methanol/methylene chloride 5:95) to yield nucleoside 10 (557 mg, 59%) as a light yellow foam (Rf 0.21, methanol/methylene chloride 5:95). 1H-NMR (400 MHz, [D6]DMSO): δ = 1.31 (s, 3H, isopropylidene-CH3a), 1.53 (s, 3H, isopropylidene-CH3b), 3.49–3.59 (m, 2H, H5′), 4.10–4.13 (m, 1H, H4′), 4.90 (dd, 3J = 3.02 Hz, 3J = 6.32 Hz, 1H, H3′), 5.11 (dd, 3J = 3.29 Hz, 3J = 6.31 Hz, 1H, H2′), 5.16 (t, 3J = 5.49 Hz, 1H, 5′-OH), 6.19 (d, 3J = 3.30 Hz, 1H, H1′), 6.76 (s, br, 2H, NH2), 7.69 (s, 1H, H6), 8.12 (s, 1H, H2); 13C-NMR (75 MHz, [D6]DMSO): δ = 25.68 (isopropylidene-CH3a), 27.59 (isopropylidene-CH3b), 52.84 (C5), 62.05 (C5′), 81.43 (C3′), 84.03 (C2′), 85.99 (C4′), 89.28 (C1′), 103.67 (C9), 113.66 (isopropylidene-C(CH3)2), 127.81 (C6), 150.22 (C8), 152.65 (C2), 157.76 (C4); ESI-MS m/z (relative intensity): 433.2 (100) [M + H]+, 261.5 (8) [4-amino-5-iodo-7H-pyrrolo[2,3-d]pyrimidine + H]+.
4-Amino-7-(2′,3′-O-isopropylidene-β-d-ribofuranosyl)-5-[1″-(3″-trifluoroacetamido)prop-1″-ynyl]-7H-pyrrolo[2,3-d]pyrimidine (12): To a solution of nucleoside 10 (295 mg, 0.68 mmol) in dry DMF (10 mL) under argon atmosphere were added copper(I) iodide (39 mg, 0.21 mmol), N-(2-propynyl)-2,2,2-trifluoroacetamide (11) (1.05 g, 6.95 mmol), triethylamine (290 µL, 2.08 mmol) and tetrakis(triphenylphosphine)palladium (118 mg, 0.10 mmol). The yellow reaction mixture was stirred at room temperature overnight. The reaction was quenched by the addition of Dowex® 1 × 8 anion exchange resin (1.3 g, loaded with hydrogen carbonate) and methanol/methylene chloride (1:1 mixture, 15 mL). After stirring at room temperature for 40 min, the mixture was filtered through Celite, and the solvents were removed under reduced pressure. The crude product was purified by column chromatography (silica gel, elution with methanol/methylene chloride 7:93) to give nucleoside 12 (305 mg, 98%) as a light yellow foam (Rf 0.23, methanol/methylene chloride 7:93). 1H-NMR (400 MHz, [D6]DMSO): δ = 1.31 (s, 3H, isopropylidene-CH3a), 1.53 (s, 3H, isopropylidene-CH3b), 3.51–3.57 (m, 2H, H5′), 4.13–4.16 (m, 1H, H4′), 4.32 (d, 3J = 5.22 Hz, 2H, H3″), 4.91 (dd, 3J = 2.75 Hz, 3J = 6.04 Hz, 1H, H3′), 5.12 (dd, 3J = 3.30 Hz, 3J = 6.05 Hz, 1H, H2′), 5.17, (t, 3J = 5.36 Hz, 1H, 5′-OH), 6.19 (d, 3J = 3.30 Hz, 1H, H1′), 7.95 (s, 1H, H6), 8.14 (s, 1H, H2), 10.10 (t, 3J = 5.22 Hz, 1H, 3″-NH); 13C-NMR (100 MHz, [D6]DMSO): δ = 25.13 (isopropylidene-CH3a), 27.03 (isopropylidene-CH3b), 29.88 (C3″), 61.49 (C5′), 75,85 (C2″), 80.91 (C3′), 83.55 (C2′), 85.58 (C4′), 86.89 (C1″), 88.95 (C1′), 94.42 (C5), 102.05 (C9), 112.91 (isopropylidene-C(CH3)2), 115.62 (q, 1J = 286 Hz, CF3), 126.83 (C6), 149.08 (C8), 152.72 (C2), 156.07 (q, 2J = 36.4 Hz, COCF3), 157.30 (C4); 19F-NMR (376 MHz, [D6]DMSO): δ = −69.72 (CF3); ESI-MS m/z (relative intensity): 456.4 (12) [M + H]+, 284.4 (100) [4-amino-5-[1′-(3′-trifluoroacetamido)prop-1-ynyl)-7H-pyrrolo[2,3-d]pyrimidine + H]+.
4-Amino-7-(2′,3′-O-isopropylidene-β-d-ribofuranosyl)-5-[1″-(3″-trifluoroacetamido)propyl]-7H-pyrrolo[2,3-d]pyrimidine (13): To a solution of nucleoside 12 (305 mg, 0.67 mmol) in methanol (75 mL) was added platinum(IV)oxide (10 mg, 44 µmol), and hydrogen gas was bubbled through the solution at room temperature for 5 h. The catalyst was removed by filtration through Celite and washed with methanol. The solvent was removed under reduced pressure, and the crude product was purified by column chromatography (silica gel, elution with methanol/methylene chloride 5:95). Nucleoside 13 (248 mg, 81%) was obtained as a light yellow solid (Rf 0.30, methanol/methylene chloride 5:95). 1H-NMR (400 MHz, [D6]DMSO): δ = 1.31 (s, 3H, isopropylidene-CH3a), 1.53 (s, 3H, isopropylidene-CH3b), 1.75–1.82 (m, 2H, H2″), 2.75–2.79 (m, 2H, H1″), 3.25–3.30 (m, 2H, H3″), 3.48–3.58 (m, 2H, H5′), 4.06–4.09 (m, 1H, H4′), 4.88 (dd, 3J = 3.02 Hz, 3J = 6.32 Hz, 1H, H3′), 5.11 (dd, 3J = 3.57 Hz, 3J = 6.32 Hz, 1H, H2′), 5.16 (t, 3J = 5.64 Hz, 1H, 5′-OH), 6.14 (d, 3J = 3.84 Hz, 1H, H1′), 6.65 (s, br, 2H, NH2), 7.14 (s, 1H, H6), 8.04 (s, 1H, H2), 9.43 (t, 3J = 5.36 Hz, 3″-NH); 13C-NMR (75 MHz, [D6]DMSO): δ = 23.53 (C2″), 25.70 (isopropylidene-CH3a), 27.62 (isopropylidene-CH3b), 29.76 (C1″), 39.26 (C3″), 62.14 (C5′), 81.53 (C3′), 83.62 (C2′), 85.42 (C4′), 89.07 (C1′), 102.46 (C9), 113.70 (isopropylidene-C(CH3)2), 115.35 (C5), 116.46 (q, 1J = 286.1 Hz, CF3), 120.05 (C6), 150.86 (C8), 151.69 (C2), 156.70 (q, 2J = 35.7 Hz, COCF3), 157.84 (C4); 19F-NMR (376 MHz, [D6]DMSO): δ = −74.74 (CF3); ESI-MS m/z (relative intensity): 460.4 (88) [M + H]+, 288.5 (100) [4-amino-5-[1′-(3′-trifluoroacetamido)propyl]-7H-pyrrolo[2,3-d]pyrimidine + H]+.
4-Amino-7-(2′,3′-O-isopropylidene-5′-O-mesyl-β-d-ribofuranosyl)-5-[1″-(3″-trifluoroacetamido)propyl]-7H-pyrrolo[2,3-d]pyrimidine (14): To a solution of nucleoside 13 (72 mg, 157 µmol) in dry methylene chloride (7 mL) under argon atmosphere were added dimethylaminopyridine (DMAP) (20 mg, 164 µmol) and triethylamine (66 µL, 474 µmol), and the mixture was cooled to 0 °C in an ice bath. Methanesulfonyl chloride (MsCl) (40 µL, 515 µmol) was slowly added, and the reaction mixture was stirred at 0 °C for 2 h. The reaction was quenched by adding an ice-cold, saturated sodium hydrogen carbonate solution (1 mL), and the organic phase was removed. The aqueous layer was extracted with ice-cold chloroform (3 × 2 mL), and the combined organic layers were dried over magnesium sulfate. After filtration, the solvent was removed under reduced pressure, and the crude product was purified by column chromatography (silica gel, methanol/methylene chloride 7:93) to give nucleoside 14 (40 mg, 46%) as a light yellow solid (Rf 0.47, methanol/methylene chloride 10:90). 1H-NMR (400 MHz, [D6]DMSO): δ = 1.33 (s, 3H, isopropylidene-CH3a), 1.54 (s, 3H, isopropylidene-CH3b), 1.77–1.81 (m, 2H, H2″), 2.77 (t, 3J = 7.42 Hz, 2H, H1″), 3.12 (s, 3H, mesyl-CH3), 3.24–3.32 (m, 2H, H3″), 4.31–4.35 (m, 2H, H5′), 4.38–4.42 (m, 1H, H4′), 4.99 (dd, 3J = 2.75 Hz, 3J = 6.32 Hz, 1H, H3′), 5.23 (dd, 3J = 3.02 Hz, 3J = 6.32 Hz, 1H, H2′), 6.23 (d, 3J = 3.02 Hz, 1H, H1′), 6.68 (s, br, 2H, NH2), 7.13 (s, 1H, H6), 8.07 (s, 1H, H2), 9.42 (t, br, 1H, 3″-NH); 19F-NMR (376 MHz, [D6]DMSO): δ = −74.74 (CF3).
4-Amino-7-(5′-O-mesyl-β-d-ribofuranosyl)-5-[1″-(3″-trifluoracetamido)propyl)-7H-pyrrolo[2,3-d]pyrimidine (15): A solution of nucleoside 14 (40 mg, 74 µmol) in aqueous trifluoroacetic acid (TFA) (70%, 2 mL) was stirred at room temperature for 45 min. The solvent was removed under reduced pressure and the remaining solvent co-evaporated with ethanol and methylene chloride. The product 15 (Rf 0.22, methanol/methylene chloride 10:90) was directly used in the following reaction.
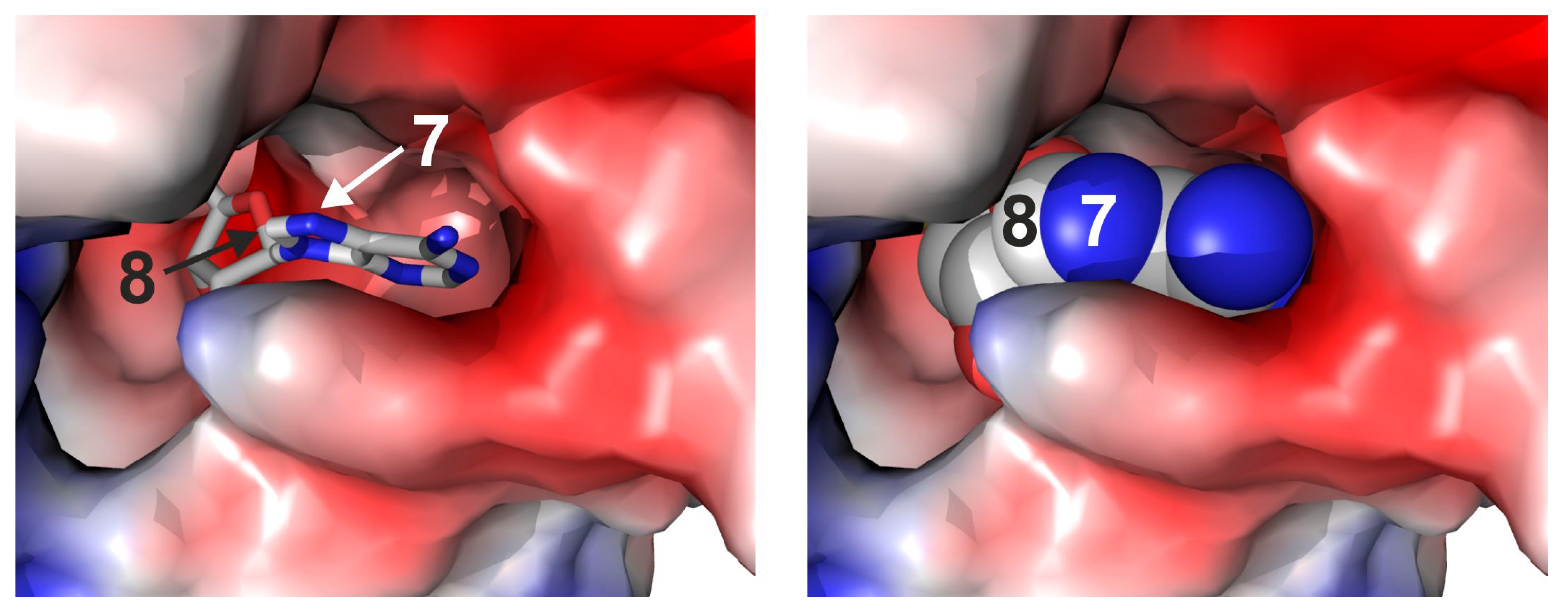
4-Amino-7-(5′-N-aziridinyl-5′-deoxy-β-d-ribofuranosyl)-5-[1″-(3″-aminopropyl)-7H-pyrrolo[2,3-d]pyrimidine (
16): Crude nucleoside
15 from the previous step was dissolved in a mixture of dry aziridine (1 mL, 16.3 mmol) and EDIA (300 µL, 1.8 mmol) under argon atmosphere, and the reaction mixture was stirred at room temperature for 4 days. The reaction progress was monitored by analytical reverse-phase HPLC (Prontosil-ODS, 5 µm, 120 Å, 250 × 4.6 mm, Bischoff, Leonberg, Germany). Compounds were eluted with acetonitrile (7% for 5 min, followed by linear gradients to 31.5% in 10 min, to 35% in 15 min and to 70% in 5 min) in triethylammonium acetate buffer (0.1 M, pH 7.0) and a flow rate of 1 mL/min. The product
16 eluted with a retention time of 10.2 min (UV detection at 280 nm and 300 nm). Volatile compounds were removed under reduced pressure, and the residue was dissolved in triethylammonium hydrogen carbonate buffer (4 mL, 0.1 M, pH 8.6) to cleave off the trifluoroacetamido group completely. The crude product was purified by preparative reverse-phase HPLC (Prontosil-ODS, 5 µm, 120 Å, 250 × 8 mm, Bischoff). Compounds were eluted with acetonitrile (7% for 5 min, followed by linear gradients to 21% in 15 min and to 70% in 5 min) in triethylammonium hydrogen carbonate buffer (0.01 M, pH 8.6) at a flow rate of 3 mL/min. Fractions containing the product
16 (retention time 16.8 min, UV detection at 280 nm and 310 nm) were combined and stored at −80 °C. The amount of product
16 (10 mg, 39% from
14) in the combined fractions was determined by UV spectroscopy using the published extinction coefficient ε
279 = 8500 L·mol
−1·cm
−1 of 4-amino-7-(2′-deoxy-β-
d-erythro-pentofuranosyl)-5-[1″-(5″-trifluoroacetamido)pentyl]-7
H-pyrrolo[2,3-
d]pyrimidine [
18].
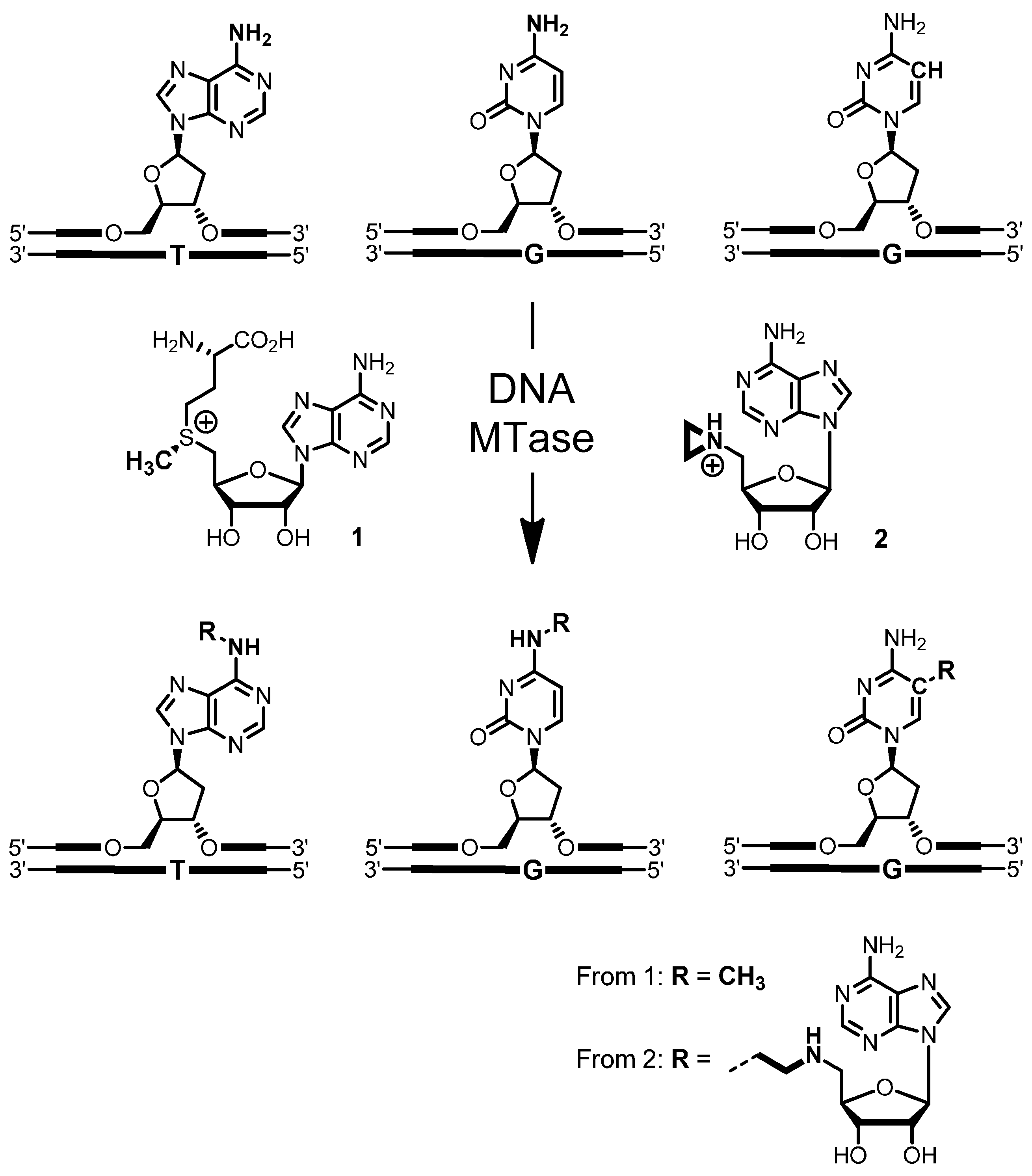
4-Amino-(5′-N-aziridinyl-5′-deoxy-β-d-ribofuranosyl)-5-[1″-(N-biotinyl-3″-aminopropyl)]-7H-pyrrolo[2,3-d]pyrimidine (5): To a solution of nucleoside 16 (10 mg, 29 µmol) in triethylammonium hydrogen carbonate buffer (12 mL, 0.01 M, pH 8.6) containing acetonitrile was added N-hydroxysuccinimidyl biotin (NHS biotin) (10.2 mg, 30 µmol) in DMSO (500 µL). The reaction was stirred at room temperature for 2 h. The progress of the reaction was monitored by analytical reverse-phase HPLC (Prontosil-ODS, 5 µm, 120 Å, 250 × 4.6 mm, Bischoff). Compounds were eluted with acetonitrile (7% for 5 min, followed by linear gradients to 31.5% in 10 min, to 35% in 15 min and to 70% in 5 min) in triethylammonium acetate buffer (0.1 M, pH 7.0) at a flow rate of 1 mL/min. The product 5 eluted with a retention time of 20.8 min (UV detection at 280 nm and 300 nm). The crude product was purified by preparative reverse-phase HPLC (Prontosil-ODS, 5 µm, 120 Å, 250 × 8 mm, Bischoff). Compounds were eluted with acetonitrile (7% for 5 min, followed by linear gradients to 31.5% in 5 min, to 35% in 10 min and to 70% in 5 min) in triethylammonium hydrogen carbonate buffer (0.01 M, pH 8.6) at a flow rate of 3 mL/min. Fractions containing the product 5 (retention time 14.8 min, UV detection at 280 nm and 300 nm) were combined, and the solvent was removed by lyophilization. The aziridine cofactor 5 (5.2 mg, 32%) was obtained as a white solid. 1H-NMR (300 MHz, [D6]DMSO): δ = 1.10–1.15 (m, 2H, aliphat. biotin-H), 1.25–1.35 (m, 4H, aziridine-H), 1.45–1.55 (m, 2H, aliphat. biotin-H), 1.55–1.63 (m, 2H, aliphat. biotin-H), 1.64–1.72 (m, 2H, H2″), 2.04–2.06 (m, 2H, aliphat. biotin-H), 2.28–2.34 (m, 1H, H5′a), 2.46–2.50 (m, 1H, H5′b), 2.54–2.58 (m, 1H, biotin-SCH2a), 2.72–2.77 (m, 2H, H1″), 2.77–2.83 (m, 1H, biotin-SCH2b), 3.05–3.16 (m, 3H, biotin-SCHR, H3″), 3.89–3.94 (m, 1H, H4′), 4.08–4.15 (m, 2H, biotin-SCHRCH, H3′), 4.25–4.34 (m, 2H, biotin-SCH2CH, H2′), 6.05 (d, 3J = 5.69 Hz, 1H, H1′), 6.35 (s, 1H, biotin-NHa), 6.42 (s, 1H, biotin-NHb), 6.53 (s, 2H, NH2), 7.10 (s, 1H, H6), 7.79 (t, 3J = 5.69 Hz, 1H, 3″-NH), 8.03 (s, 1H, H2); ESI-MS: m/z (relative intensity): 575.25 (100) [M + H]+, 418.18 (7) [4-amino-5-[1′-(N-biotinyl-3′-aminopropyl)]-7H-pyrrolo[2,3-d]pyrimidine + H]+.
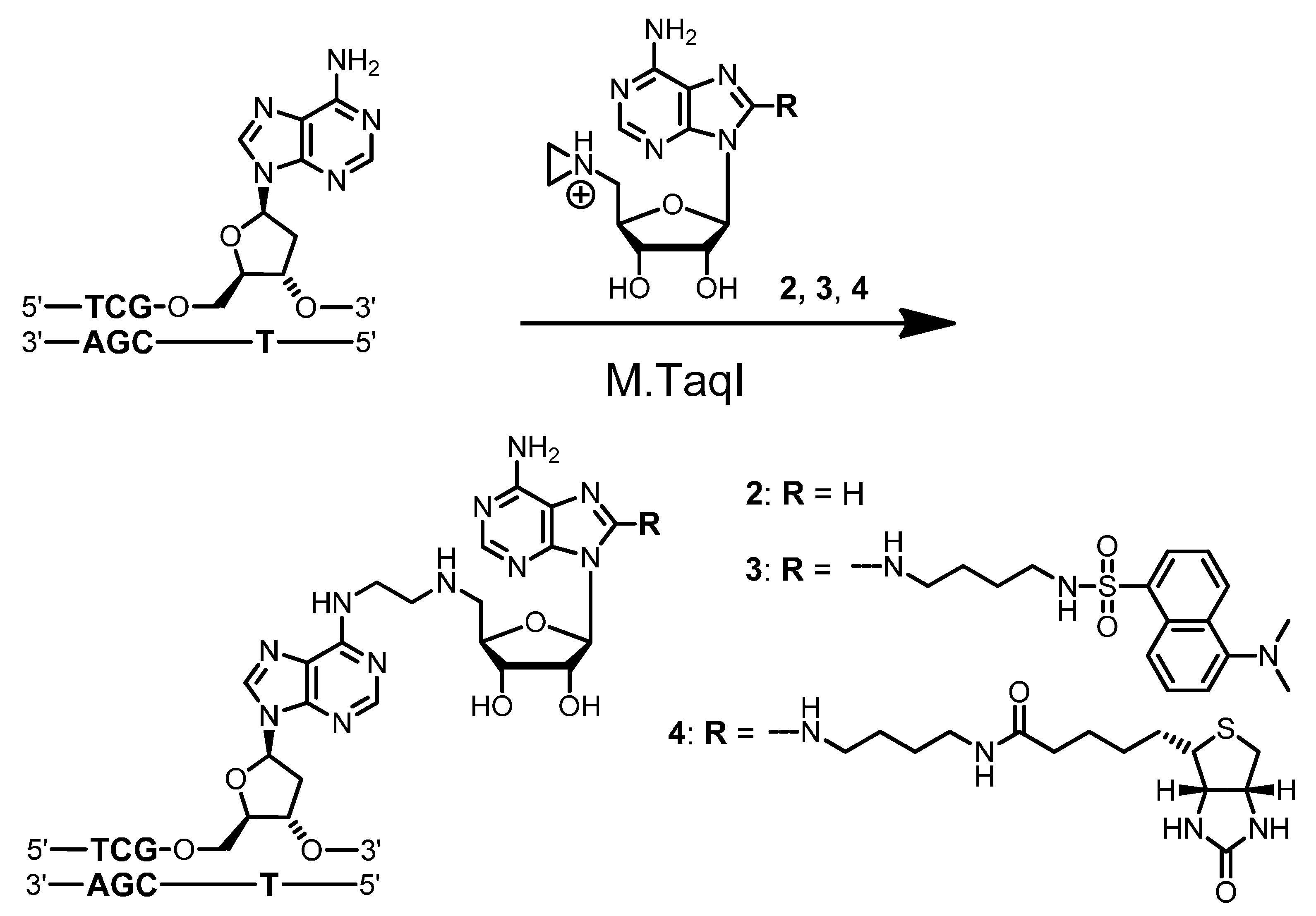
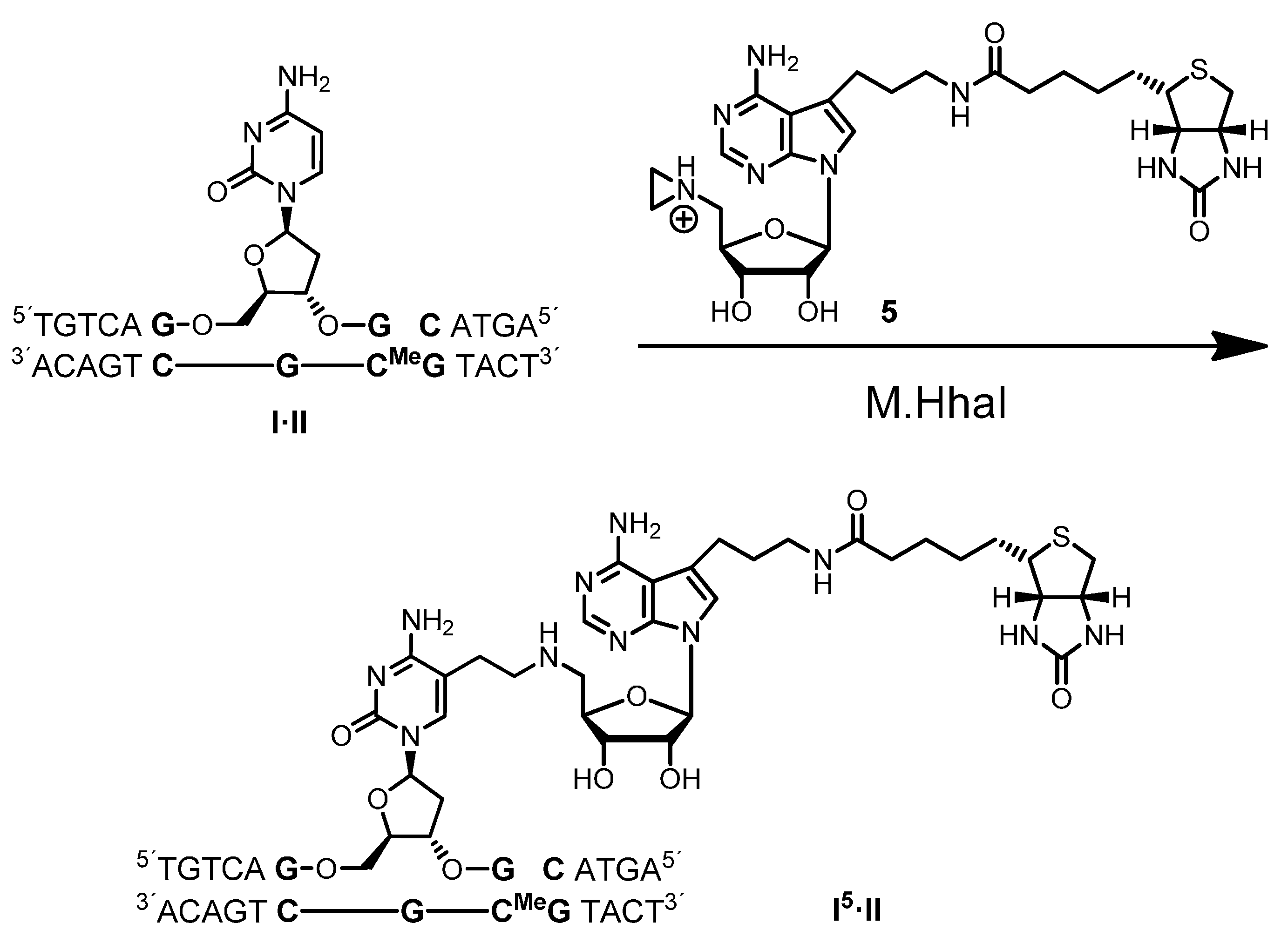
Biotin labeling of duplex ODN
I·II with M.HhaI: Molecular extinction coefficients at 260 nm of ODN
I and
II were calculated according to the nearest neighbor method [
25], and concentrations were determined by UV spectroscopy. Annealing of the complementary strands
I and
II was performed by heating an equimolar solution to 95 °C for 2 min followed by slow cooling to room temperature.
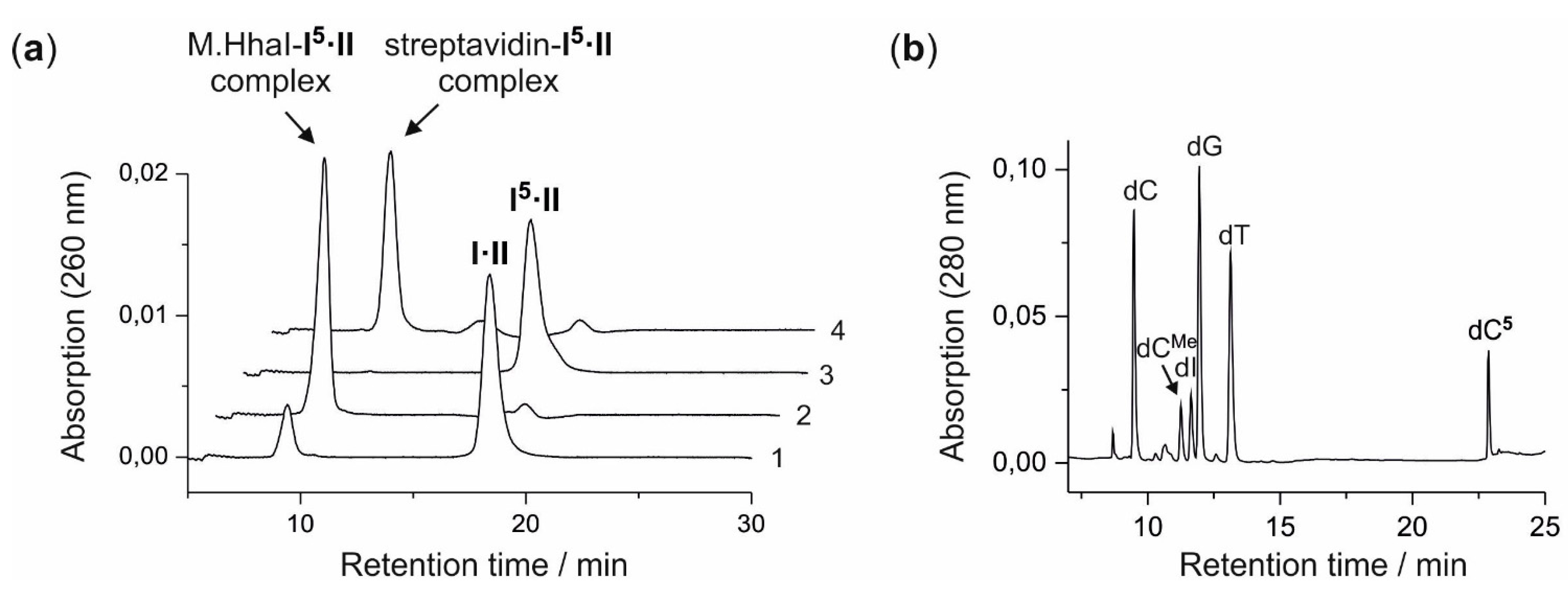
A solution (400 µL) of aziridine cofactor 5 (32 nmol, 80 µM), duplex ODN I·II (4 nmol, 10 µM) and M.HhaI (4.4 nmol, 11 µM) in buffer (10 mM Tris hydrochloride, pH 7.4, 50 mM sodium chloride, 0.05 mM ethylenediaminetetraacetic acid and 2 mM β-mercaptoethanol) were incubated at 37 °C. The progress of the reaction was monitored by anion exchange HPLC (Poros 10 HQ, 10 µm, 4.6 × 10 mm, Applied Biosystems). Compounds were eluted with aqueous potassium chloride (0.2 M for 5 min, followed by linear gradients to 0.4 M in 5 min, to 0.6 M in 20 min and to 1 M in 5 min) at a flow rate of 4 mL/min. The labeled duplex ODN I5·II was released from the protein-DNA complex by incubation at 65 °C for 30 min, and complete release was verified by anion exchange chromatography (as described above). Residual aziridine cofactor 5 was removed by gel filtration using a NAP-5 column (Amersham, Biosciences, Freiburg, Germany) following the instructions of the supplier. The presence and functionality of biotin in the duplex ODN I5·II was verified by the addition of streptavidin (25 µg) to the I5·II solution (25 µL, 0.25 nmol) and incubation at 37 °C for 30 min. Binding of streptavidin to I5·II was confirmed by anion exchange HPLC (as described above).
For enzymatic fragmentation, desalted duplex ODN I5·II (10 nmol) was dried by lyophilization and the residue dissolved in buffer (200 µL, 10 mM potassium phosphate, pH 7.0, 10 mM magnesium chloride). DNase I (7.7 U), phosphodiesterase from Crotalus adamanteus (576 mU), phosphodiesterase from calf spleen (768 mU) and alkaline phosphatase (4.8 U) were added and the solution incubated at 37 °C for 24 h. The nucleoside composition of the fragmentation reaction was analyzed by reverse-phase HPLC (Symmetry C-18, 5 µm, 100 Å, 100 × 4.6 mm, Waters, Eschborn, Germany). Compounds were eluted with acetonitrile (linear gradient from 0%–7% in 7 min, followed by linear gradients to 8% in 10 min and to 100% in 13 min) at a flow rate of 1 mL/min. The modified nucleoside dC5 (retention time 22.9 min, detection at 254 nm and 280 nm) was collected, dried by lyophilization and analyzed by mass spectrometry. ESI-MS m/z (relative intensity): 824.4 (100) [M + Na]+.
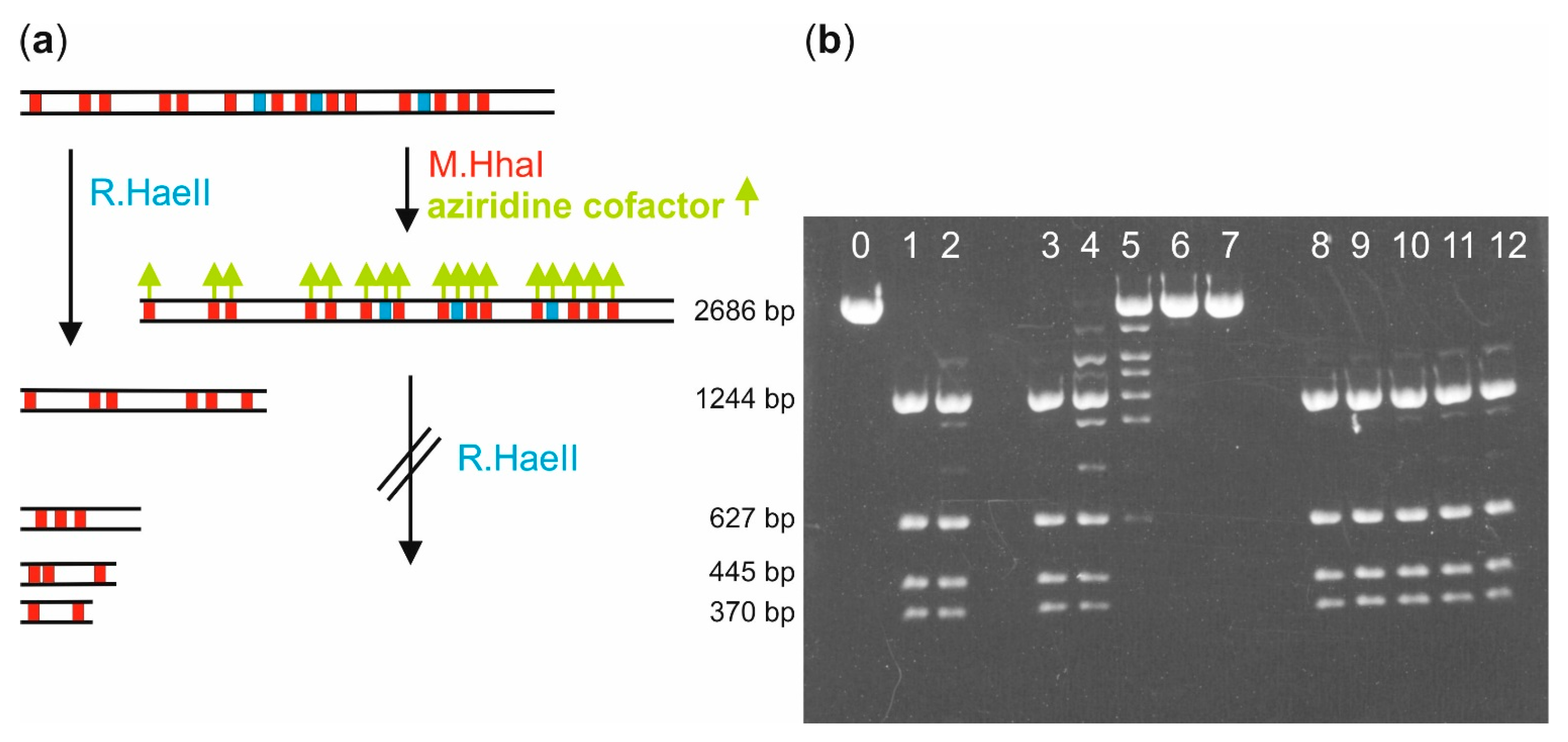
Biotin labeling of pUC19 plasmid DNA with M.HhaI: Linearization of pUC19 plasmid DNA (10 µg) was carried out by incubation with the restriction endonuclease R.XmnI (100 U) in buffer (40 µL, 10 mM Tris hydrochloride, pH 7.9, 10 mM magnesium chloride, 50 mM sodium chloride, 1 mM 1,4-dithiothreitol and 1 mg/mL bovine serum albumin) at 37 °C for 1 h. The linearized pUC19 (LpUC19) was either directly used for following experiments or stored at 4 °C.
Solutions of LpUC19 (1 µg, 0.56 pmol, 9.6 pmol M.HhaI recognition sites), aziridine cofactor 5 or 4 (80 µM) and M.HhaI (18.5 pmol) in buffer (40 µL, 10 mM Tris hydrochloride, pH 7.4, 50 mM sodium chloride, 0.05 mM ethylenediaminetetraacetic acid and 2 mM β-mercaptoethanol) were incubated at 37 °C. Aliquots (8 µL) were taken after different incubation times and the labeling reaction quenched by heating to 65 °C for 20 min. Each aliquot was supplemented with 10 × NEB2 buffer (1 µL, 100 mM Tris hydrochloride, pH 7.9, 100 mM magnesium chloride, 500 mM sodium chloride, 10 mM 1,4-dithiothreitol, New England Biolabs) and R.HaeII (1 µL, 4 U) and incubated at 37 °C for 1 h. Fragmentation was analyzed by agarose gel (1%) electrophoresis.
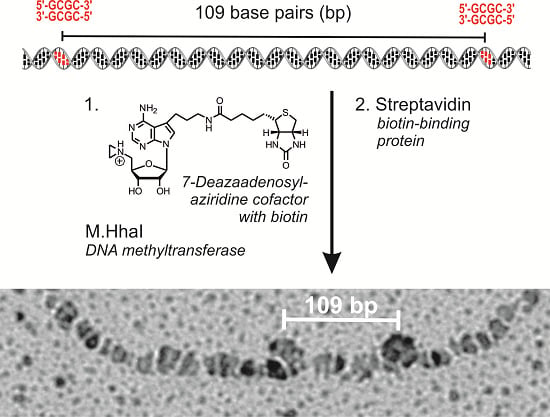
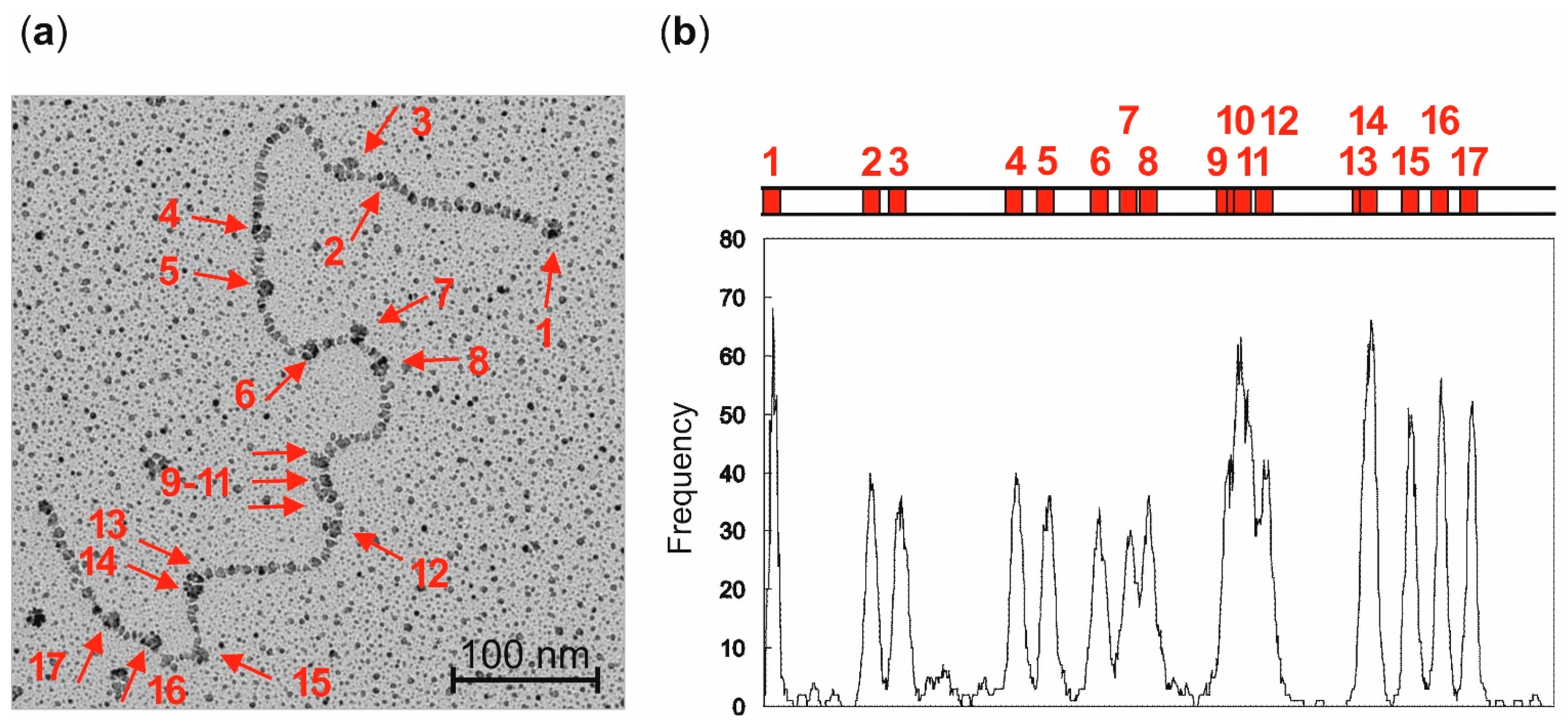
Localization of streptavidin-biotin complexes on plasmid DNA by electron microscopy: LpUC19 plasmid DNA was biotinylated using aziridine cofactor
5 and M.HhaI as described above and heated at 65 °C for 20 min. Labeled LpUC19 was isolated using the QIAquick PCR Purification Kit from QIAGEN (Hilden, Germany) and eluted with buffer (50 µL, 10 mM Tris hydrochloride, pH 8.5). Streptavidin (20 µg protein/1 µg plasmid DNA) was added and the solution incubated at room temperature for 1 h. The excess of streptavidin was removed by gel filtration on sepharose CL-4B (Pharmacia, Uppsala, Sweden). The DNA was diluted in buffer (10 mM triethylammonium hydrochloride, pH 7.9, 10 mM magnesium chloride) to a final concentration of 1 ng/µL and directly absorbed on a glimmer surface [
26]. Positions of the streptavidin-biotin complexes of 84 plasmids were determined from electron micrographs taken statistically with an EM400 (Philips, Eindhoven, The Netherlands). Contour lengths were measured with a LM4 (Brühl, Nürnberg, Germany) and the data analyzed with a computer program [
27].
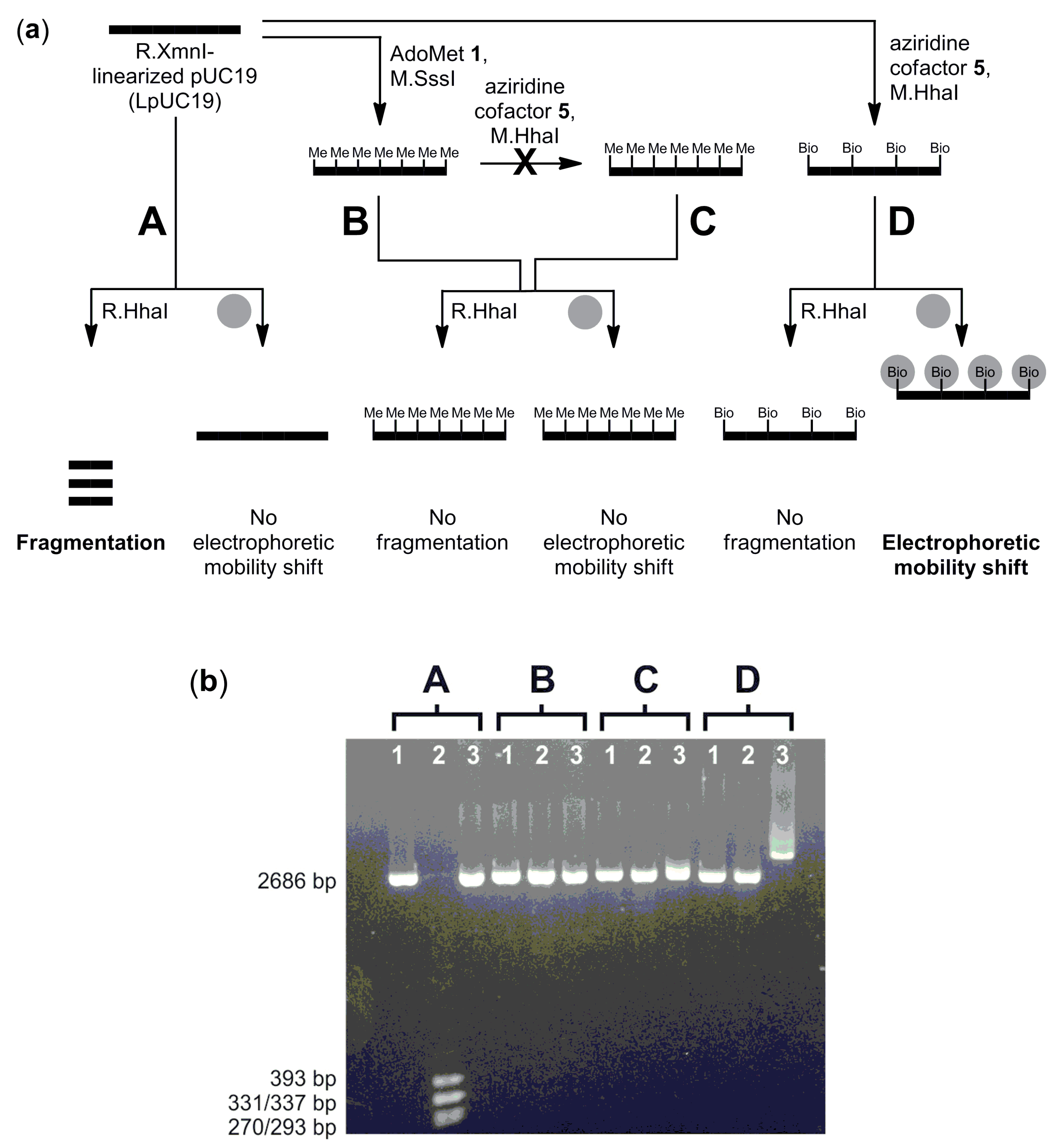
CpG-methylation detection: CpG-methylation of LpUC19 (6 µg) was carried out with M.SssI (54 pmol) and AdoMet 1 (160 µM) in buffer (40 µL, 10 mM Tris hydrochloride, pH 7.9, 6 mM magnesium chloride, 50 mM sodium chloride, 1 mM 1,4-dithiothreitol and 0.6 mg/mL bovine serum albumin) at 37 °C for 1 h. The reaction was stopped by heating to 65 °C for 20 min. Methylated LpUC19 was purified using the QIAquick PCR Purification Kit (QIAGEN GmbH) according to the instructions of the supplier and eluted with buffer (10 mM Tris hydrochloride, pH 8.5).
Unmodified or CpG-methylated LpUC19 (2 µg, 1.12 pmol, 19.2 pmol M.HhaI recognition sites), aziridine cofactor 5 (80 µM) and M.HhaI (38 pmol) in buffer (100 µL, 10 mM Tris hydrochloride, pH 7.4, 50 mM sodium chloride, 0.05 mM ethylenediaminetetraacetic acid and 2 mM β-mercaptoethanol) were incubated at 37 °C for 2 h and then at 65 °C for 20 min. Plasmid DNA was purified using the QIAquick PCR Purification Kit (QIAGEN GmbH) and eluted with buffer (10 mM Tris hydrochloride, pH 8.5).
Unmodified, CpG-methylated or biotinylated LpUC19 (200 ng) in buffer (10 µL, 10 mM Tris hydrochloride, pH 7.9, 10 mM magnesium chloride, 50 mM sodium chloride, 1 mM 1,4-dithiothreitol and 1 mg/mL bovine serum albumin) were incubated with R.HhaI (2 U) at 37 °C for 1 h. DNA fragmentation was analyzed by agarose gel (1%) electrophoresis.
Unmodified, CpG-methylated or biotinylated LpUC19 (200 ng) in buffer (10 µL, 10 mM Tris hydrochloride, pH 8.5) were incubated with streptavidin (5 µg) at 37 °C for 1 h, and binding of streptavidin was analyzed by the electromobility shift assay (EMSA) using agarose gel (1%) electrophoresis.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








