
Efficient Esterification of Oxidized l-Glutathione and Other Small Peptides

Abstract
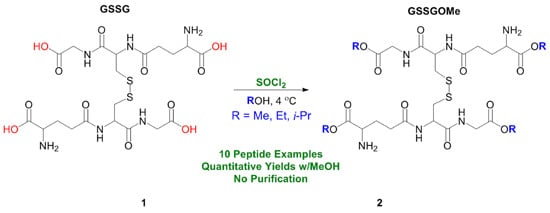
:
1. Introduction

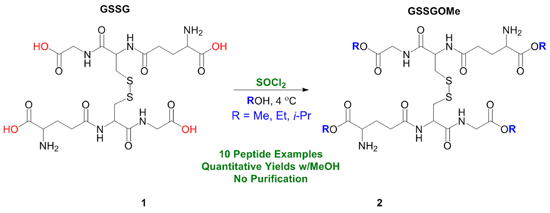
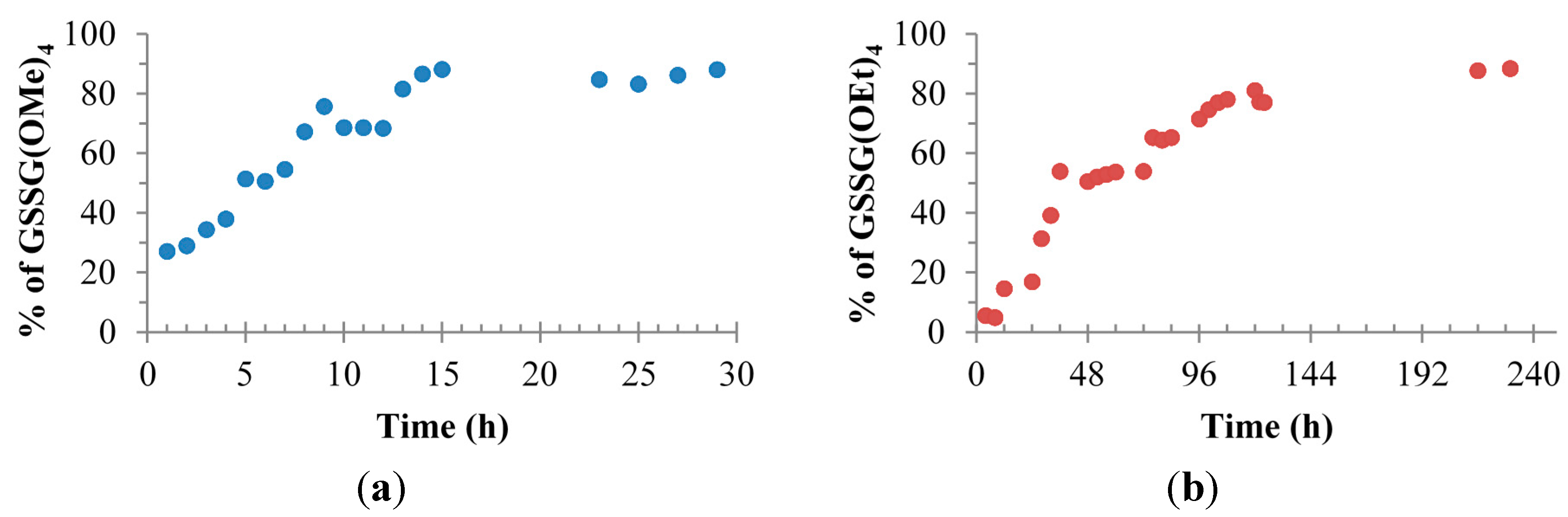
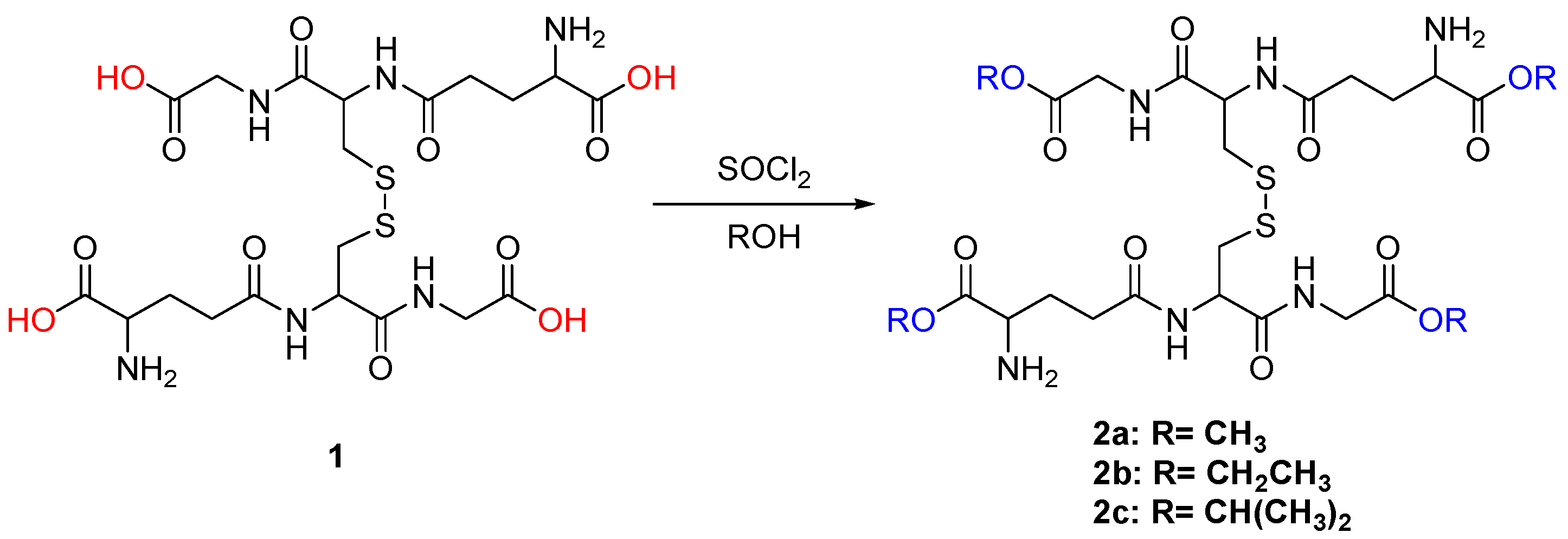
2. Results and Discussion


{kind=link}
{kind=link}
{kind=link}
| Peptide a | Peptide Sequence | Product Entry | Complete Conversion | Estimated % c Conversion |
|---|---|---|---|---|
| l-glutathione oxidized | 2QCG b | 2 | Yes | 100% d |
| l-glutathione reduced | QCG | 3 | No | 26% |
| Fibronectin Analog | GRADSPK | 4 | No | 23% |
| Bradykinin (1–7) | RPPGFSP | 5 | No | 63% |
| Necrofibrin, rat | WTVPTA | 6 | No | 64% |
| [d-Ala2,d-Met5]-Enkephalin | YAGFM | 7 | No | 90% |
| Angiotensin II, human | DRVYIHPF | 8 | Yes | 100% |
| Thymopentin (TP-5) | RKDVY | 9 | Yes | 100% |
| Neurotensin (9–13) | RPYIL | 10 | Yes | 100% |
| [Ile3]-Pressinoic acid | CYIQNC b | 11 | No | 94% |
3. Experimental Section
3.1. General Experimental
3.2. Synthesis of GSSG and GSH Peptides in Alcohol Solutions
3.2.1. Synthesis of Compound 2
3.2.2. Synthesis of Compound 2b
3.2.3. Synthesis of Compound 2c
3.2.4. Synthesis of Compound 3
3.3. General Synthesis of Peptides in Methanol Solutions
3.3.1. Synthesis of Compound 4
3.3.2. Synthesis of Compound 5
3.3.3. Synthesis of Compound 6
3.3.4. Synthesis of Compound 7
3.3.5. Synthesis of Compound 8
3.3.6. Synthesis of Compound 9
3.3.7. Synthesis of Compound 10
3.3.8. Synthesis of Compound 11
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Ehrlich, K.; Viirlaid, S.; Mahlapuu, R.; Saar, K.; Kullisaar, T.; Zilmer, M.; Langel, Ü.; Soomets, U. Design, synthesis and properties of novel powerful antioxidants glutathione analouges. Free Radic. Res. 2007, 41, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Gatterdam, V.; Stoess, T.; Menge, C.; Heckel, A.; Tampé, R. Caged Glutathione–Triggering Protein Interaction by Light. Angew. Chem. Int. Ed. 2012, 51, 3960–3963. [Google Scholar] [CrossRef] [PubMed]
- Lecchi, P.; Olson, M.; Brancia, F.L. The Role of Esterification on Detection of Protonated and Deprotonated Peptide Ions in Matrix Assisted Laser Desorption/Ionization (MALDI) Mass Spectrometry (MS). J. Am. Soc. Mass Spectrom. 2005, 16, 1269–1274. [Google Scholar] [CrossRef] [PubMed]
- Falck, J.R.; Sangras, B.; Capdevila, J.H. Preparation of N-tBoc l-glutathione dimethyl and di-tert-butyl esters: Versatile synthetic building blocks. Bioorg. Med. Chem. 2007, 15, 1062–1066. [Google Scholar] [CrossRef] [PubMed]
- Stepanov, V.M.; Muratova, G.L. Partial esterification of some amino acids and glutathione. Izv. Akad. Nauk SSSR Ser. Khim. 1961, 10, 1677–1680. [Google Scholar] [CrossRef]
- Thornalley, P.K. Esterification of reduced glutathione. Biochem. J. 1991, 275, 535–539. [Google Scholar] [PubMed]
- Aurora Fine Chemicals Catalog. Available online: http://online.aurorafinechemicals.com (accessed on 27 April 2015). Product Number K11.805.561.
- Li, J.; Sha, Y. A Convenient Synthesis of Amino Acid Methyl Esters. Molecules 2008, 13, 1111–1119. [Google Scholar] [CrossRef] [PubMed]
- Pozgan, F.; Lukman, K.; Kocevar, M. An efficient synthesis of methyl esters of heterocyclic α,β-didehydro-α-amino acid derivatives. Heterocycles 2010, 82, 543–554. [Google Scholar] [CrossRef]
- Di Gioia, M.L.; Leggio, A.; le Pera, A.; Liguori, A.; Napoli, A.; Siciliano, C.; Sindona, G. “One-Pot” Methylation of N-Nosyl-α-amino Acid Methyl Esters with Diazomethane and Their Coupling To Prepare N-Methyl Dipeptides. J. Org. Chem. 2003, 68, 7416–7421. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Kutz-Naber, K.K.; Li, L. Methyl Esterification Assisted MALDI FTMS Characterization of the Orcokinin Neuropeptide Family. Anal. Chem. 2007, 79, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.Y.; Brun, Y.V.; Reilly, J.P. Effects of Tryptic Peptide Esterification in MALDI Mass Spectrometry. Anal. Chem. 2005, 77, 4185–4193. [Google Scholar] [CrossRef] [PubMed]
- Simon, E.S.; Young, M.; Chan, A.; Bao, Z.Q.; Andrews, P.C. Improved enrichment strategies for phosphorylated peptides on titanium dioxide using methyl esterification and pH gradient elution. Anal. Biochem. 2008, 377, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Su, D.; Ren, X.; You, D.; Li, D.; Mu, Y.; Yan, G.; Zhang, Y.; Luo, Y.; Xue, Y.; Shen, J.; et al. Generation of Three Selenium-Containing Catalytic Antibodies with High Catalytic Efficiency Using a Novel Hapten Design Method. Arch. Biochem. Biophys. 2001, 395, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Cerny, C.; Guntz-Dubini, R. Formation of cysteine-S-conjugates in the Maillard reaction of cysteine and xylose. Food Chem. 2013, 141, 1078–1086. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, K.; Inoue, K.; Kinoshita, K.; Ueda, Y.; Murao, H. Processes for Producing β-Halogeno-α-amino-carboxylic Acids and S-Phenylcysteine Derivatives and Intermediates Thereof. WO9933785A1, 8 July 1999. [Google Scholar]
- Shen, L.; Guan, L.; Liu, F.; Cao, Y.; Zhou, J. Method for Preparing Levetiracetam. CN101550100A, 7 October 2009. [Google Scholar]
- Gatterdam, V.; Ramadass, R.; Stoess, T.; Fichte, M.A.H.; Wachtveitl, J.; Heckel, A.; Tampé, R. Three-Dimensional Protein Networks Assembled by Two-Photon Activation. Angew. Chem. Int. Ed. 2014, 53, 5680–5684. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, M.W.B.; Coombs, G.S.; Banerjee, N.; Bugni, T.S.; Cannon, K.M.; Harper, M.K.; Veltri, C.A.; Virshup, D.M.; Ireland, C.M. Psammaplin A as a general activator of cell-based signaling assays via HDAC inhibition and studies on some bromotyrosine derivatives. Bioorg. Med. Chem. 2009, 17, 2189–2198. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vogel, E.R.; Jackson, W.; Masterson, D.S. Efficient Esterification of Oxidized l-Glutathione and Other Small Peptides. Molecules 2015, 20, 10487-10495. https://doi.org/10.3390/molecules200610487
Vogel ER, Jackson W, Masterson DS. Efficient Esterification of Oxidized l-Glutathione and Other Small Peptides. Molecules. 2015; 20(6):10487-10495. https://doi.org/10.3390/molecules200610487
Chicago/Turabian StyleVogel, Emily R., William Jackson, and Douglas S. Masterson. 2015. "Efficient Esterification of Oxidized l-Glutathione and Other Small Peptides" Molecules 20, no. 6: 10487-10495. https://doi.org/10.3390/molecules200610487