
The Effect of Pre-Oxidation on the Reducibility of Chromite Using Hydrogen: A Preliminary Study

 , ,
, ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Sizing of Chromite
2.3. Reduction Procedure
2.4. Pre-Oxidation Procedure
2.5. Analytical Techniques
2.6. Expressing the Extent of Metallization
3. Results and Discussion
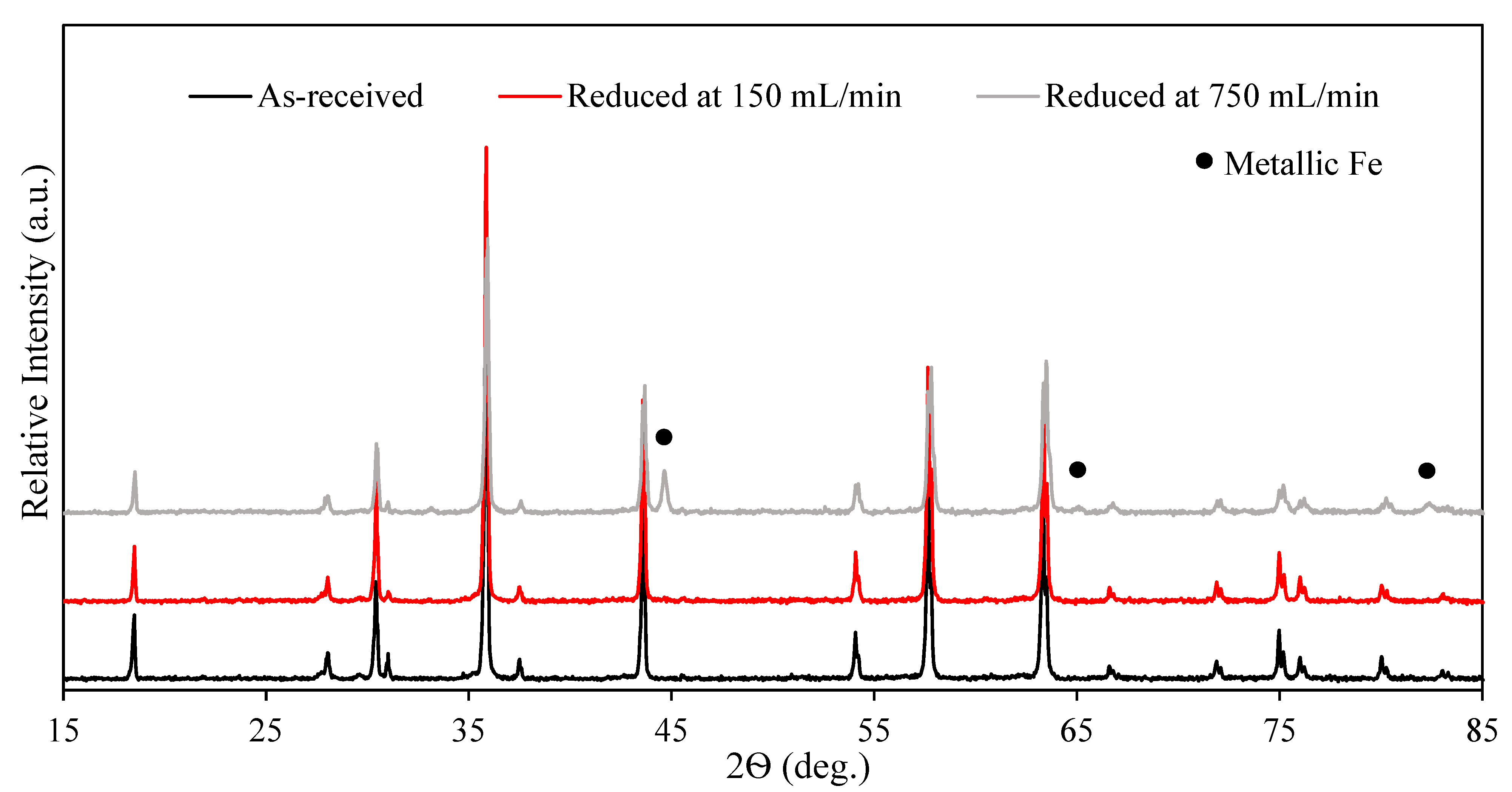
3.1. The Effect of Hydrogen Availability
3.2. Effect of Reduction Time
3.3. Effect of Pre-Oxidation
3.4. Effect of Pre-Oxidation Time
3.5. Effect of Pre-Oxidation and Leaching on Particle Integrity
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Feary, J.; Cullinan, P. Heavy Metals. In Reference Module in Biomedical Sciences [Internet]; Elsevier: Amsterdam, The Netherlands, 2019. [Google Scholar]
- Beukes, J.; Dawson, N.; Van Zyl, P. Theoretical and practical aspects of Cr (VI) in the South African ferrochrome industry. J. S. Afr. Inst. Min. Metall. 2010, 110, 743–750. [Google Scholar]
- du Preez, S.P.; Beukes, J.P.; Van Dalen, W.; van Zyl, P.; Paktunc, D.; Loock-Hattingh, M. Aqueous solubility of Cr(VI) compounds in ferrochrome bag filter dust and the implications thereof. Water SA 2017, 43, 298. [Google Scholar] [CrossRef] [Green Version]
- Riekkola-Vanhanen, M. Finnish Expert Report on Best Available Techniques in Ferrochromium Production. 1999. Available online: https://helda.helsinki.fi/bitstream/handle/10138/40531/FE_314.pdf?sequence=1 (accessed on 2 November 2021).
- du Preez, S.P.; Beukes, J.P.; van Zyl, P.G.; Tangstad, M.; Tiedt, L.R. Silicon Carbide Formation Enhanced by In-Situ-Formed Silicon Nitride: An Approach to Capture Thermal Energy of CO-Rich Off-Gas Combustion. Met. Mater. Trans. A 2018, 49, 3151–3163. [Google Scholar] [CrossRef] [Green Version]
- Beukes, J.P.; du Preez, S.; van Zyl, P.; Paktunc, D.; Fabritius, T.; Päätalo, M.; Cramer, M. Review of Cr(VI) environmental practices in the chromite mining and smelting industry—Relevance to development of the Ring of Fire, Canada. J. Clean. Prod. 2017, 165, 874–889. [Google Scholar] [CrossRef]
- Basson, J.; Daavittila, J. High carbon ferrochrome technology. In Handbook of Ferroalloys; Elsevier: Amsterdam, The Netherlands, 2013; pp. 317–363. [Google Scholar]
- Barnes, A.; Finn, C.; Algie, S. The prereduction and smelting of chromite concentrate of low chromium-to-iron ratio. J. S. Afr. Inst. Min. Metall. 1983, 83, 49–54. [Google Scholar]
- Erwee, M.; Swanepoel, S.; Reynolds, Q. The importance of controlling the chemistry of pre-oxidized chromite pellets for Submerged Arc Furnace FeCr smelting: A study on furnace Si control. In Proceedings of the 16th International Ferro-Alloys Congress (INFACON XVI), Virtual, 27–29 September 2021. [Google Scholar]
- Alfredsen, K.; Amundsen, P.-A.; Hahn, L.; Harrison, P.M.; Helland, I.P.; Martins, E.G.; Twardek, W.M.; Power, M. A synoptic history of the development, production and environmental oversight of hydropower in Brazil, Canada, and Norway. Hydrobiologia 2022, 849, 269–280. [Google Scholar] [CrossRef]
- du Preez, S.P.; Beukes, J.P.; van Zyl, P. Cr(VI) Generation During Flaring of CO-Rich Off-Gas from Closed Ferrochromium Submerged Arc Furnaces. Met. Mater. Trans. A 2015, 46, 1002–1010. [Google Scholar] [CrossRef] [Green Version]
- Du Preez, S. Cr (VI) Generation during Flaring of Off-Gas from Closed Ferrochromium Submerged Arc Furnaces. Ph.D. Thesis, North-West University, Potchefstroom, South Africa, 2014. [Google Scholar]
- Niemelä, P.; Krogerus, H.; Oikarinen, P. Formation, characteristics and utilisation of CO-gas formed in ferrochromium smelting. In Proceedings of the Tenth International Ferro Alloys Congress, Cape Town, South Africa, 1–4 February 2004; pp. 68–77. [Google Scholar]
- Winter, F. Production of Chromium Iron Alloys Directly from Chromite Ore. U.S. Patent 15,027,479, 25 August 2016. [Google Scholar]
- Canaguier, V. Synthesis and Reduction-Carburization of (Fe, Mg)(Cr, Al)2O4 Composite Spinel Solid Solution with CH4. Ph.D. Thesis, Norwegian University of Science and Technology, Trondheim, Norway, 2018. [Google Scholar]
- Yu, D.; Paktunc, D. Calcium Chloride-Assisted Segregation Reduction of Chromite: Influence of Reductant Type and the Mechanism. Minerals 2018, 8, 45. [Google Scholar] [CrossRef] [Green Version]
- Sokhanvaran, S.; Paktunc, D.; Barnes, A. NaOH-assisted direct reduction of Ring of Fire chromite ores, and the associated implications for processing. J. S. Afr. Inst. Min. Metall. 2018, 118, 581–588. [Google Scholar] [CrossRef]
- Anacleto, N.; Ostrovski, O. Solid-state reduction of chromium oxide by methane-containing gas. Met. Mater. Trans. A 2004, 35, 609–615. [Google Scholar] [CrossRef]
- Suzuki, A.M.; Yasuda, A.; Ozawa, K. Cr and Al diffusion in chromite spinel: Experimental determination and its implication for diffusion creep. Phys. Chem. Miner. 2008, 35, 433–445. [Google Scholar] [CrossRef]
- Davies, J.; Paktunc, D.; Ramos-Hernandez, J.J.; Tangstad, M.; Ringdalen, E.; Beukes, J.P.; Bessarabov, D.G.; Du Preez, S.P. The Use of Hydrogen as a Potential Reductant in the Chromite Smelting Industry. Minerals 2022, 12, 534. [Google Scholar] [CrossRef]
- Tathavakar, V.D.; Antony, M.P.; Jha, A. The physical chemistry of thermal decomposition of South African chromite minerals. Met. Mater. Trans. A 2005, 36, 75–84. [Google Scholar] [CrossRef]
- Kapure, G.; Tathavadkar, V.; Rao, C.; Rao, S.; Raju, K. Coal based direct reduction of preoxidized chromite ore at high temperature. In Proceedings of the 12th International Ferro-Alloys Congress (INFACON XII), Helsinki, Finland, 6–9 June 2010; pp. 293–301. [Google Scholar]
- Zhao, B.; Hayes, P. Effects of oxidation on the microstructure and reduction of chromite pellets. In Proceedings of the 12th International Ferro-Alloys Congress (INFACON XII), Helsinki, Finland, 6–9 June 2010; pp. 263–273. [Google Scholar]
- Borra, C.R.; Kapure, G.; Tathavadkar, V. Investigation of pre-oxidation of Sukinda Chromite Ore. Tata Search 2010, 1, 145–148. [Google Scholar]
- Kleynhans, E.; Neizel, B.; Beukes, J.; van Zyl, P. Utilisation of pre-oxidised ore in the pelletised chromite pre-reduction process. Miner. Eng. 2016, 92, 114–124. [Google Scholar] [CrossRef]
- Biswas, A.; Konar, B.; Kapure, G.U.; Sahu, N.; Paliwal, M. Pre-oxidation treatment of Indian chromite ores: Kinetics and phase transformation behavior relevant to ferrochrome manufacturing and pelletization. Miner. Process. Extr. Met. 2018, 130, 31–41. [Google Scholar] [CrossRef]
- du Preez, S.P.; Beukes, J.P.; Paktunc, D.; van Zyl, P.; Jordaan, A. Recycling pre-oxidized chromite fines in the oxidative sintered pellet production process. J. S. Afr. Inst. Min. Metall. 2019, 119, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Pariser, H.; Backeberg, N.; Masson, O.; Bedder, J. Changing nickel and chromium stainless steel markets—A review. J. S. Afr. Inst. Min. Metall. 2018, 118, 563–568. [Google Scholar] [CrossRef] [Green Version]
- Glastonbury, R.; Beukes, J.P.; van Zyl, P.; Sadikit, L.; Jordaan, A.; Campbell, Q.; Stewart, H.; Dawson, N. Comparison of physical properties of oxidative sintered pellets produced with UG2 or metallurgical-grade South African chromite: A case study. J. S. Afr. Inst. Min. Metall. 2015, 115, 699–706. [Google Scholar] [CrossRef]
- Ringdalen, E.; Olsen, S.E. The effect of chromite ore mineralogy on reduction mechanisms and reducibility. Infacon 1998, 8, 147–152. [Google Scholar]
- Balat, M. Potential importance of hydrogen as a future solution to environmental and transportation problems. Int. J. Hydrogen Energy 2008, 33, 4013–4029. [Google Scholar] [CrossRef]
- Pandey, B.; Prajapati, Y.K.; Sheth, P.N. Recent progress in thermochemical techniques to produce hydrogen gas from biomass: A state of the art review. Int. J. Hydrogen Energy 2019, 44, 25384–25415. [Google Scholar] [CrossRef]
- Canavesio, C.; Nassini, D.; Nassini, H.E.; Bohé, A.E. Study on an original cobalt-chlorine thermochemical cycle for nuclear hydrogen production. Int. J. Hydrogen Energy 2019, 45, 26090–26103. [Google Scholar] [CrossRef]
- Falch, A.; Kriek, R.J. Laser induced H2 production employing Pt-TiO2 photocatalysts. J. Photochem. Photobiol. A Chem. 2013, 271, 117–123. [Google Scholar] [CrossRef]
- Zhang, J.; Xing, C.; Shi, F. MoS2/Ti3C2 heterostructure for efficient visible-light photocatalytic hydrogen generation. Int. J. Hydrogen Energy 2020, 45, 6291–6301. [Google Scholar] [CrossRef]
- Ziming, C.; Fuqiang, W.; Huaxu, L.; Shengpeng, H.; Bo, L.; Jianyu, T.; Hongyang, L. Photon-absorption-based explanation of ultrasonic-assisted solar photochemical splitting of water to improve hydrogen production. Int. J. Hydrogen Energy 2018, 43, 14439–14450. [Google Scholar] [CrossRef]
- Liu, W.-S.; Perng, T.-P. Ta2O5 hollow fiber composed of internal interconnected mesoporous nanotubes and its enhanced photochemical H2 evolution. Int. J. Hydrogen Energy 2019, 44, 17688–17696. [Google Scholar] [CrossRef]
- Patel, P.P.; Ghadge, S.D.; Hanumantha, P.J.; Datta, M.K.; Gattu, B.; Shanthi, P.M.; Kumta, P.N. Active and robust novel bilayer photoanode architectures for hydrogen generation via direct non-electric bias induced photo-electrochemical water splitting. Int. J. Hydrogen Energy 2018, 43, 13158–13176. [Google Scholar] [CrossRef]
- Manwar, N.R.; Borkar, R.G.; Khobragade, R.; Rayalu, S.S.; Jain, S.L.; Bansiwal, A.K.; Labhsetwar, N.K. Efficient solar photo-electrochemical hydrogen generation using nanocrystalline CeFeO3 synthesized by a modified microwave assisted method. Int. J. Hydrogen Energy 2017, 42, 10931–10942. [Google Scholar] [CrossRef]
- Du Preez, S.P. Hydrogen Generation by Means of Hydrolysis Using Activated Al-In-Bi-Sn Composites for Electrochemical Energy Applications. Int. J. Electrochem. Sci. 2017, 12, 8663–8682. [Google Scholar] [CrossRef]
- du Preez, S.P.; Bessarabov, D. Hydrogen generation of mechanochemically activated Al Bi In composites. Int. J. Hydrogen Energy 2017, 42, 16589–16602. [Google Scholar] [CrossRef]
- du Preez, S.P.; Bessarabov, D.G. Hydrogen generation by the hydrolysis of mechanochemically activated aluminum-tin-indium composites in pure water. Int. J. Hydrogen Energy 2018, 43, 21398–21413. [Google Scholar] [CrossRef]
- du Preez, S.; Bessarabov, D.G. The effects of bismuth and tin on the mechanochemical processing of aluminum-based composites for hydrogen generation purposes. Int. J. Hydrogen Energy 2019, 44, 21896–21912. [Google Scholar] [CrossRef]
- Du Preez, S. Material Aspects Pertaining to Hydrogen Production from Aluminum: Opinion. Res. Dev. Mater. Sci. 2019, 44, 21896–21912. [Google Scholar] [CrossRef]
- Du Preez, S.P. Hydrogen Generation by the Reaction of Mechanochemically Activated Aluminium and Water. Ph.D. Thesis, North-West University, Potchefstroom, South Africa, 2019. [Google Scholar]
- du Preez, S.; Bessarabov, D. On-demand hydrogen generation by the hydrolysis of ball-milled aluminum composites: A process overview. Int. J. Hydrogen Energy 2021, 46, 35790–35813. [Google Scholar] [CrossRef]
- Davies, J.; Du Preez, S.P.; Bessarabov, D.G. The Hydrolysis of Ball-Milled Aluminum–Bismuth–Nickel Composites for On-Demand Hydrogen Generation. Energies 2022, 15, 2356. [Google Scholar] [CrossRef]
- Davies, J.; du Preez, S.P.; Bessarabov, D.G. On-Demand Hydrogen Generation by the Hydrolysis of Ball-Milled Aluminum–Bismuth–Zinc Composites. Materials 2022, 15, 1197. [Google Scholar] [CrossRef]
- Bessarabov, D.; Human, G.; Kruger, A.J.; Chiuta, S.; Modisha, P.M.; du Preez, S.P.; Oelofse, S.P.; Vincent, I.; Van Der Merwe, J.; Langmi, H.W.; et al. South African hydrogen infrastructure (HySA infrastructure) for fuel cells and energy storage: Overview of a projects portfolio. Int. J. Hydrogen Energy 2017, 42, 13568–13588. [Google Scholar] [CrossRef]
- Phillips, R.; Dunnill, C.W. Zero gap alkaline electrolysis cell design for renewable energy storage as hydrogen gas. RSC Adv. 2016, 6, 100643–100651. [Google Scholar] [CrossRef] [Green Version]
- Falch, A.; Badets, V.A.; Labrugère, C.; Kriek, R.J. Co-sputtered PtxPdyAlz thin film electrocatalysts for the production of hydrogen via SO2(aq) electro-oxidation. Electrocatalysis 2016, 7, 376–390. [Google Scholar] [CrossRef]
- Vincent, I.; Bessarabov, D. Low cost hydrogen production by anion exchange membrane electrolysis: A review. Renew. Sustain. Energy Rev. 2018, 81, 1690–1704. [Google Scholar] [CrossRef]
- Vincent, I.; Kruger, A.; Bessarabov, D. Development of efficient membrane electrode assembly for low cost hydrogen production by anion exchange membrane electrolysis. Int. J. Hydrogen Energy 2017, 42, 10752–10761. [Google Scholar] [CrossRef]
- Merwe, J.v.d.; Uren, K.; Schoor, G.v.; Bessarabov, D. A study of the loss characteristics of a single cell PEM electrolyser for pure hydrogen production. In Proceedings of the 2013 IEEE International Conference on Industrial Technology (ICIT), Cape Town, South Africa, 25–28 February 2013; pp. 668–672. [Google Scholar]
- Chiuta, S.; Everson, R.C.; Neomagus, H.; Bessarabov, D.G. Hydrogen production from ammonia decomposition over a commercial Ru/Al2O3 catalyst in a microchannel reactor: Experimental validation and CFD simulation. Int. J. Hydrogen Energy 2016, 41, 3774–3785. [Google Scholar] [CrossRef]
- Chiuta, S.; Everson, R.C.; Neomagus, H.W.; Bessarabov, D.G. Performance evaluation of a high-throughput microchannel reactor for ammonia decomposition over a commercial Ru-based catalyst. Int. J. Hydrogen Energy 2015, 40, 2921–2926. [Google Scholar] [CrossRef]
- Chiuta, S.; Everson, R.C.; Neomagus, H.W.; van der Gryp, P.; Bessarabov, D.G. Reactor technology options for distributed hydrogen generation via ammonia decomposition: A review. Int. J. Hydrogen Energy 2013, 38, 14968–14991. [Google Scholar] [CrossRef]
- Sekoai, P.T.; Ouma, C.N.M.; du Preez, S.P.; Modisha, P.; Engelbrecht, N.; Bessarabov, D.G.; Ghimire, A. Application of nanoparticles in biofuels: An overview. Fuel 2019, 237, 380–397. [Google Scholar] [CrossRef]
- Sekoai, P.T.; Engelbrecht, N.; du Preez, S.P.; Bessarabov, D. Thermophilic Biogas Upgrading via ex Situ Addition of H2 and CO2 Using Codigested Feedstocks of Cow Manure and the Organic Fraction of Solid Municipal Waste. ACS Omega 2020, 5, 17367–17376. [Google Scholar] [CrossRef]
- Sekoai, P.T.; Daramola, M.O.; Mogwase, B.; Engelbrecht, N.; Yoro, K.O.; du Preez, S.P.; Mhlongo, S.; Ezeokoli, O.T.; Ghimire, A.; Ayeni, A.O.; et al. Revising the dark fermentative H2 research and development scenario—An overview of the recent advances and emerging technological approaches. Biomass Bioenergy 2020, 140, 105673. [Google Scholar] [CrossRef]
- du Toit, M.H.; Avdeenkov, A.V.; Bessarabov, D. Reviewing H2 Combustion: A Case Study for Non-Fuel-Cell Power Systems and Safety in Passive Autocatalytic Recombiners. Energy Fuels 2018, 32, 6401–6422. [Google Scholar] [CrossRef]
- du Preez, S.; Jones, D.; Warwick, M.; Falch, A.; Sekoai, P.; Quaresma, C.M.D.N.; Bessarabov, D.; Dunnill, C. Thermally stable Pt/Ti mesh catalyst for catalytic hydrogen combustion. Int. J. Hydrogen Energy 2020, 45, 16851–16864. [Google Scholar] [CrossRef]
- du Preez, S.; Jones, D.; Bessarabov, D.; Falch, A.; Quaresma, C.M.D.N.; Dunnill, C. Development of a Pt/stainless steel mesh catalyst and its application in catalytic hydrogen combustion. Int. J. Hydrogen Energy 2019, 44, 27094–27106. [Google Scholar] [CrossRef]
- Lughi, V.; Lenaz, D.; Bonifacio, A.; Princivalle, F.; Sergo, V.; Parisi, F. A Raman spectroscopy study of the oxidation processes in synthetic chromite FeCr2O4. Cer. Int. 2020, 46, 29382–29387. [Google Scholar] [CrossRef]
- Algie, S.H.; Finn, C.W.P. Reaction Mechanisms in the Reduction of Winterveld Chrome Spinel with Graphite and Carbon; Council for Mineral Technology: Randburg, South Africa, 1984. [Google Scholar]
- Hazar-Yoruç, A.B. Reduction mechanism of chromite spinel with carbon. Min. Metall. Explor. 2007, 24, 115–120. [Google Scholar] [CrossRef]
- Shepherd, P.H. Chromite Crystal Structure and Chemistry Applied as an Exploration Tool. Master’s Thesis, The University of Western Ontario, London, ON, Canada, 2015. Available online: https://ir.lib.uwo.ca/etd/2685 (accessed on 27 December 2021).
- Mehio, N.; Dai, S.; Jiang, D.-E. Quantum Mechanical Basis for Kinetic Diameters of Small Gaseous Molecules. J. Phys. Chem. A 2014, 118, 1150–1154. [Google Scholar] [CrossRef]
- Soykan, O.; Eric, R.H.; King, R.P. Kinetics of the reduction of bushveld complex chromite ore at 1416 °C. Metall. Mater. Trans. A 1991, 22, 801–810. [Google Scholar] [CrossRef]
- Boericke, F.; Bangert, W. Equilibrium in the Reduction of Ferrous Chromite by Hydrogen and Energy Requirements in the Selective Reduction of Iron in Chromite; US Department of the Interior, Bureau of Mines: Washington, DC, USA, 1945; Volume 3813. [Google Scholar]
- Chakraborty, D.; Ranganathan, S.; Sinha, S. Carbothermic Reduction of Chromite Ore Under Different Flow Rates of Inert Gas. Metall. Mater. Trans. A 2009, 41, 10–18. [Google Scholar] [CrossRef]
- Chu, W.F.; Rahmel, A. The kinetics of the reduction of chromium oxide by hydrogen. Metall. Mater. Trans. A 1979, 10, 401–407. [Google Scholar] [CrossRef]
- Park, E.; Ostrovski, O.; Zhang, J.; Thomson, S.; Howe, R. Characterization of phases formed in the iron carbide process by X-ray diffraction, Mössbauer, X-ray photoelectron spectroscopy, and Raman spectroscopy analyses. Metall. Mater. Trans. A 2001, 32, 839–845. [Google Scholar] [CrossRef]
- Leikola, M.; Taskinen, P.; Eric, R. Reduction of Kemi chromite with methane. J. S. Afr. Inst. Min. Metall. 2018, 118, 575–580. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.; Yang, C.; Zhu, D. Solid State Reduction of Preoxidized Chromite-iron Ore Pellets by Coal. ISIJ Int. 2015, 55, 727–735. [Google Scholar] [CrossRef] [Green Version]
- Bondioli, F.; Ferrari, A.M.; Leonelli, C.; Manfredini, T.; Linati, L.; Mustarelli, P. Reaction Mechanism in Alumina/Chromia (Al2O3-Cr2O3) Solid Solutions Obtained by Coprecipitation. J. Am. Ceram. Soc. 2000, 83, 2036–2040. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Size Equivalent | As-Received | Sieve Sizes (µm) | |||
|---|---|---|---|---|---|
| <106 | 106 to 212 | 212 to 425 | 425 to 850 | ||
| d90 | 455 | 133 | 260 | 467 | 778 |
| d50 | 253 | 84 | 178 | 335 | 569 |
| d10 | 132 | 51 | 122 | 238 | 416 |
| As-Received | Size-Fractioned Chromite Ore | ||||
|---|---|---|---|---|---|
| Size equivalent d90 (µm) | 455 | 778 | 467 | 260 | 133 |
| g Fe/g chromite ore | 0.186 | 0.189 | 0.184 | 0.196 | 0.199 |
| g Cr/g chromite ore | 0.302 | 0.313 | 0.295 | 0.313 | 0.289 |
| Spectrum | Designation | Detected Element (wt%) | Cr/Fe | Hydrogen Flow Rate (mL/min) | ||||
|---|---|---|---|---|---|---|---|---|
| O | Mg | Al | Cr | Fe | ||||
| 1 | n/a | 38.60 | 6.01 | 7.76 | 30.06 | 17.57 | 1.71 | 150 |
| 2 | 38.39 | 6.02 | 7.63 | 30.30 | 17.66 | 1.72 | ||
| 3 | 40.72 | 6.24 | 7.8 | 28.55 | 16.69 | 1.71 | ||
| 4 | Area 1 | 8.81 | 0.39 | 5.99 | 5.10 | 79.71 | 0.06 | 750 |
| 5 | 1.42 | 0.05 | 0.02 | 5.71 | 92.8 | 0.06 | ||
| 6 | 16.78 | 2.94 | 3.4 | 11.06 | 65.83 | 0.17 | ||
| 7 | Area 2 | 41.25 | 7.85 | 7.93 | 34.15 | 8.82 | 3.87 | |
| 8 | 40.59 | 8.05 | 8.32 | 34.4 | 8.65 | 3.98 | ||
| 9 | Area 3 | 40.33 | 5.8 | 7.63 | 29.18 | 17.06 | 1.71 | |
| Spectrum | Description | Detected Element (wt%) | ||||||
|---|---|---|---|---|---|---|---|---|
| O | Mg | Al | Ti | Cr | Fe | Cr/Fe | ||
| 1 | White area | 9.37 | 2.35 | 2.38 | - | 8.4 | 77.5 | 0.12 |
| 2 | 6.2 | 0.89 | 1.63 | - | 5.55 | 85.73 | 0.06 | |
| 3 | 8.15 | 1.21 | 2.61 | - | 7.31 | 80.71 | 0.09 | |
| 4 | Grey area | 42.28 | 4.09 | 12.46 | 1.04 | 34.36 | 5.77 | 5.95 |
| 5 | 42.08 | 10.21 | 9.71 | - | 30.66 | 7.34 | 4.18 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davies, J.; Tangstad, M.; Ringdalen, E.; Beukes, J.P.; Bessarabov, D.; du Preez, S.P. The Effect of Pre-Oxidation on the Reducibility of Chromite Using Hydrogen: A Preliminary Study. Minerals 2022, 12, 911. https://doi.org/10.3390/min12070911
Davies J, Tangstad M, Ringdalen E, Beukes JP, Bessarabov D, du Preez SP. The Effect of Pre-Oxidation on the Reducibility of Chromite Using Hydrogen: A Preliminary Study. Minerals. 2022; 12(7):911. https://doi.org/10.3390/min12070911
Chicago/Turabian StyleDavies, Jamey, Merete Tangstad, Eli Ringdalen, Johan Paul Beukes, Dmitri Bessarabov, and Stephanus Petrus du Preez. 2022. "The Effect of Pre-Oxidation on the Reducibility of Chromite Using Hydrogen: A Preliminary Study" Minerals 12, no. 7: 911. https://doi.org/10.3390/min12070911