
TAL Effector Repertoires of Strains of Xanthomonas phaseoli pv. manihotis in Commercial Cassava Crops Reveal High Diversity at the Country Scale

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains, Plant Material, Culture Conditions and Media
2.2. Aggressiveness Assays and Bacterial Growth Curves
2.3. Genomic DNA Extraction, Restriction Fragment Length Polymorphism (RFLP), and Southern Blot Analysis
2.4. Isolation and Sequencing of TALEs
2.5. TALE Characterization and Clustering
2.6. In-Vitro Plant Inoculation and Cassava RNA Extraction
2.7. RNA Sequencing and Transcriptome Analysis
2.8. TALE Target Prediction and Candidate Analysis
2.9. Semi-Quantitative and Quantitative RT-PCR
2.10. Statistical Analysis and Packages
3. Results
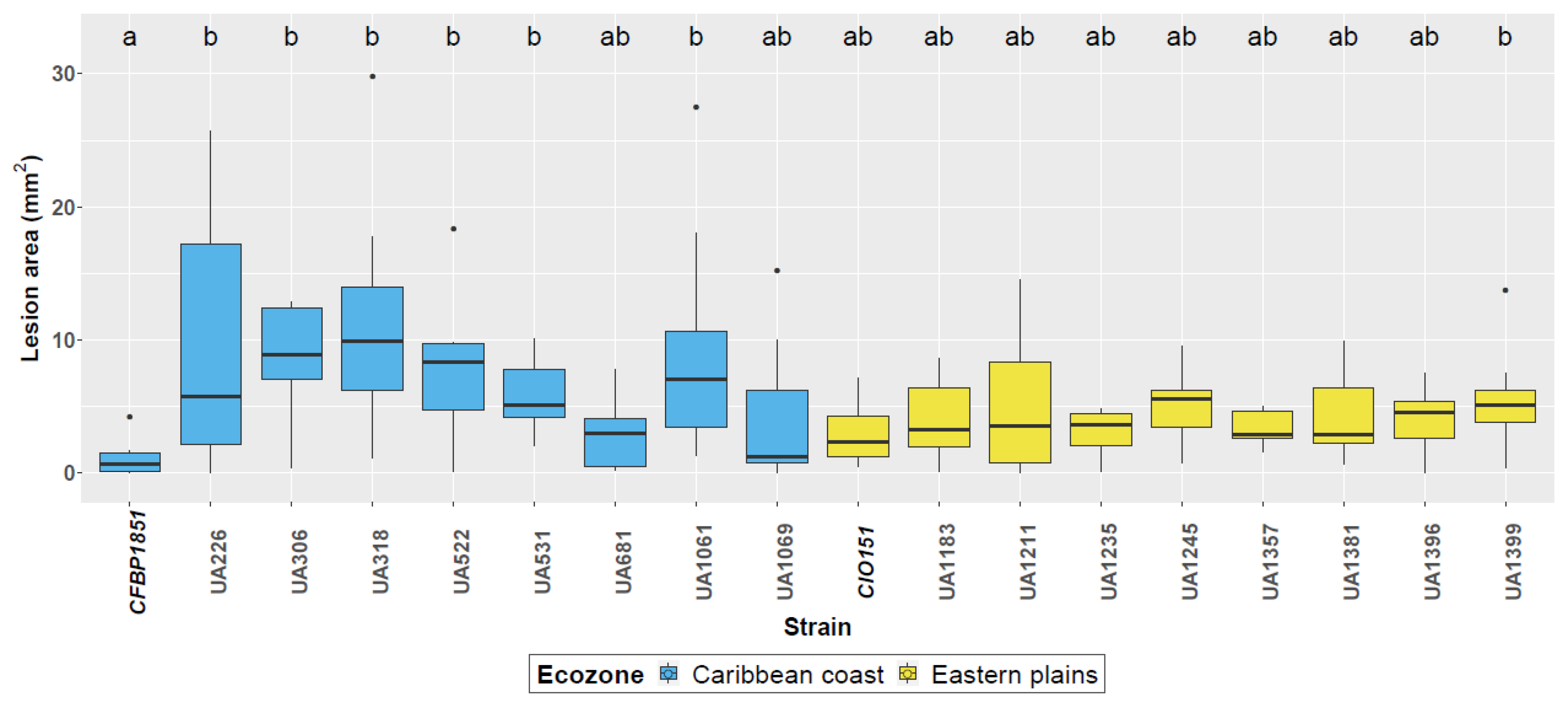
3.1. Aggressiveness of Selected Xpm Strains Is Homogeneous on Cassava Cultivar 60444
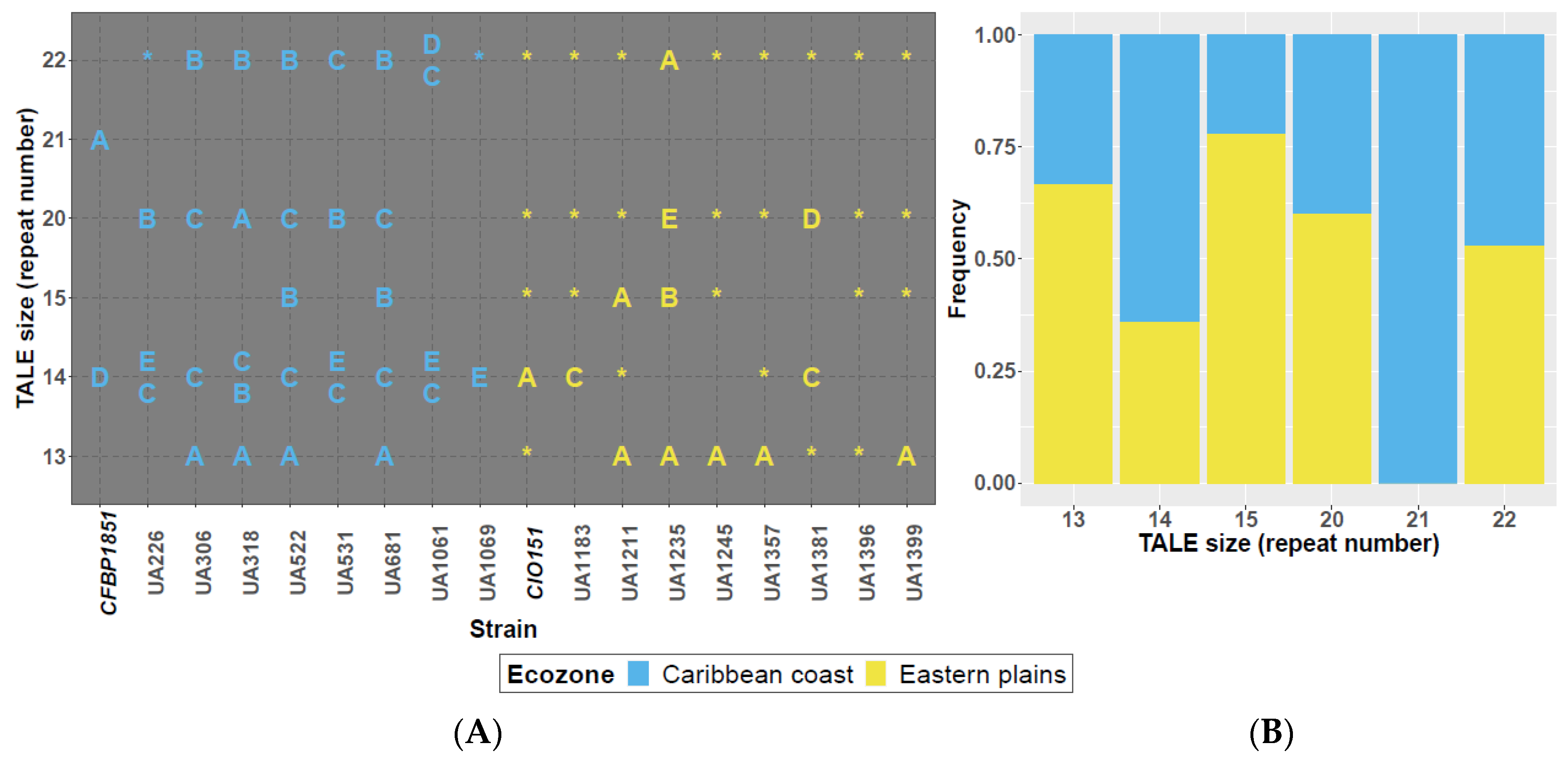
3.2. RFLP Analysis Highlights TALome Patterns with Two to Six TALEs Per Strain and Restricted Size Ranges
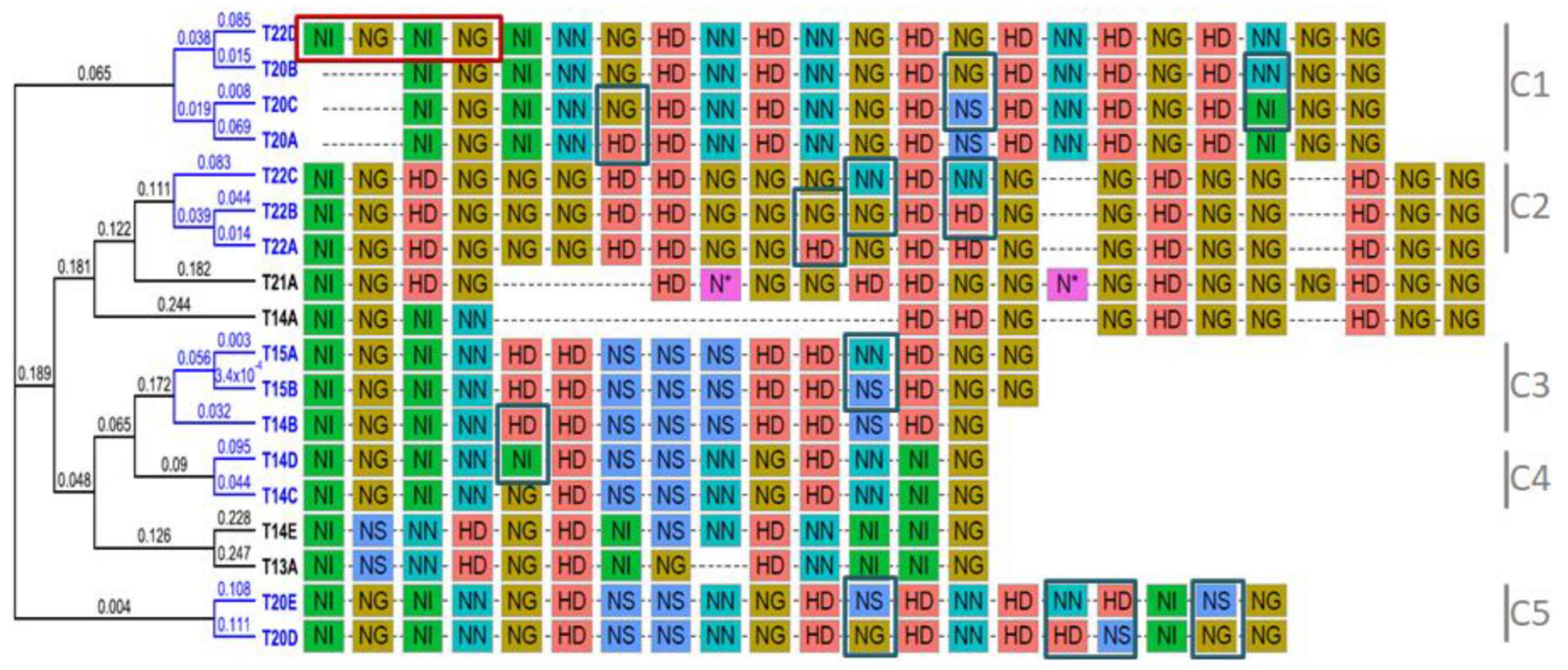
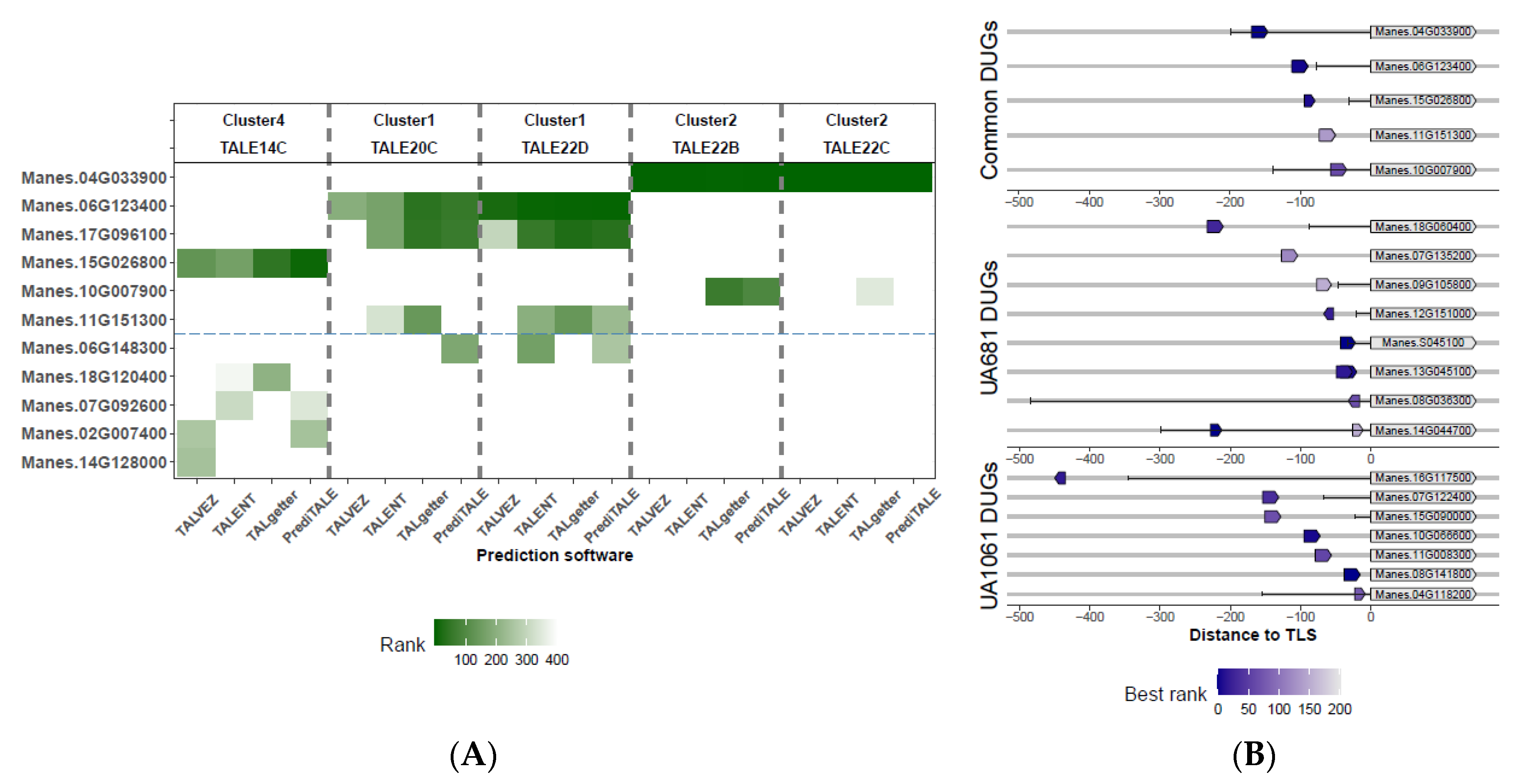
3.3. Five TALE Clusters with Similar Predicted DNA-Binding Affinities
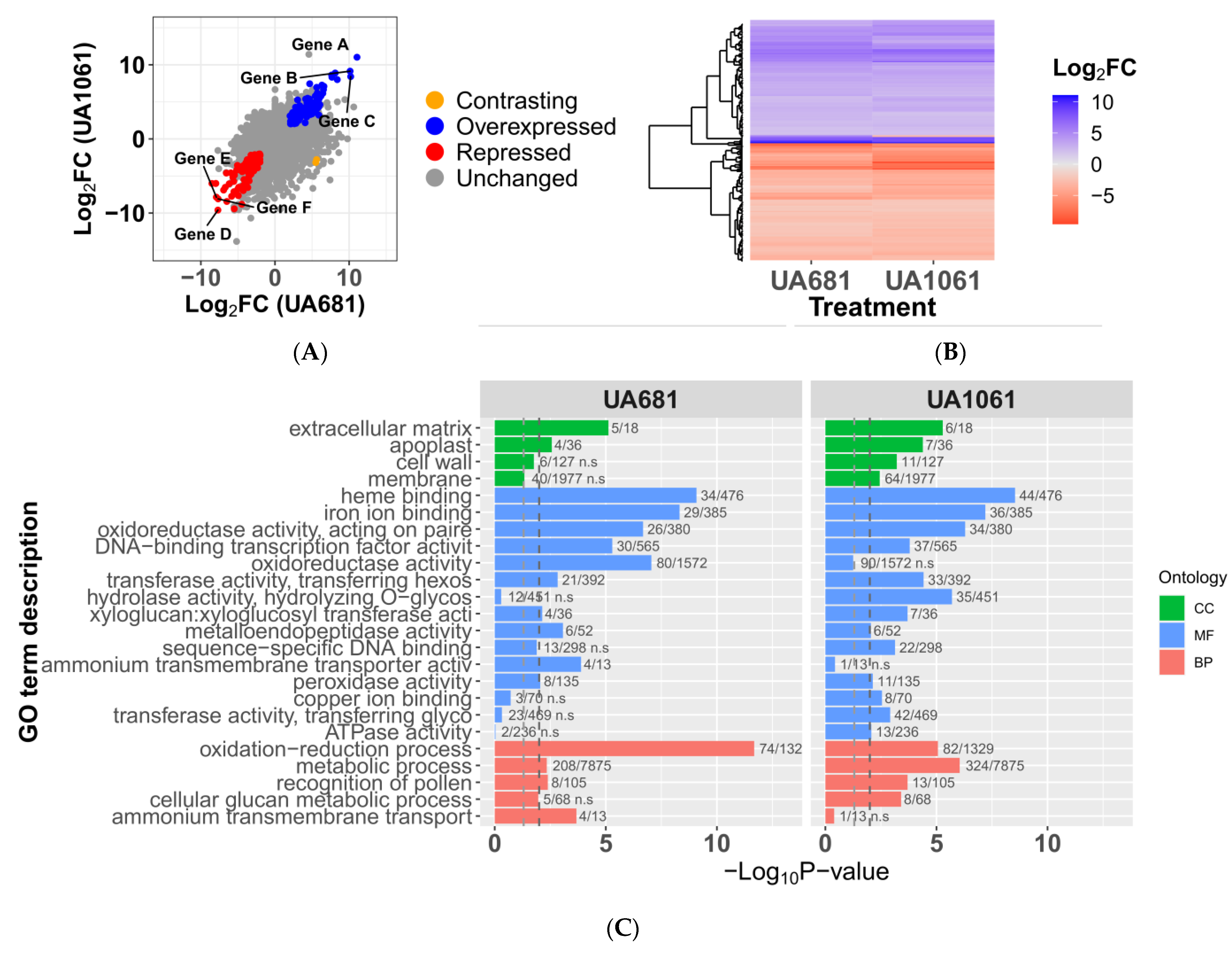
3.4. Expression Profiles of Plants Inoculated with UA681 and UA1061 Are Highly Similar
3.5. Potential TALE Targets Are Involved in the Manipulation of Host Cell Redox Status and Nutrient Transport
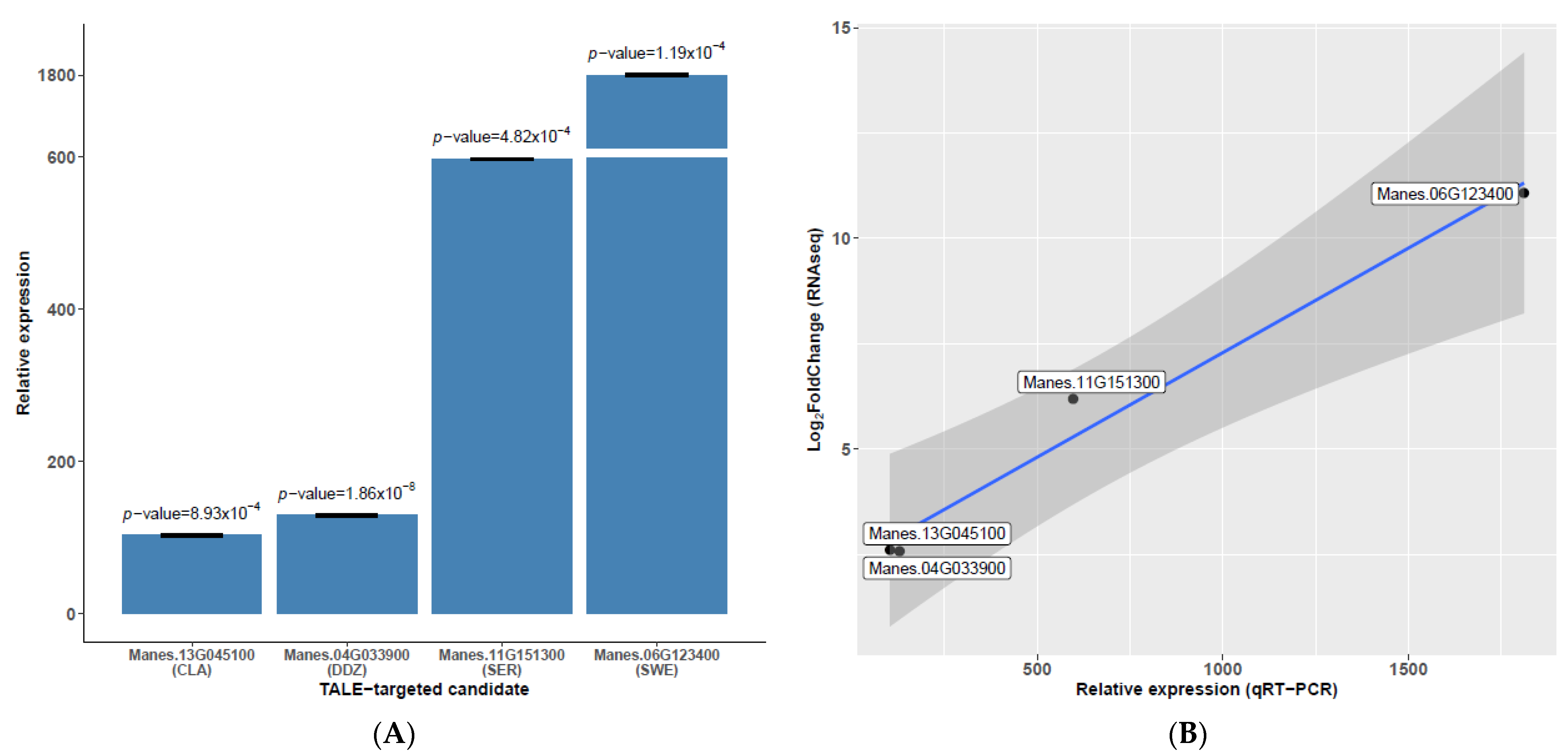
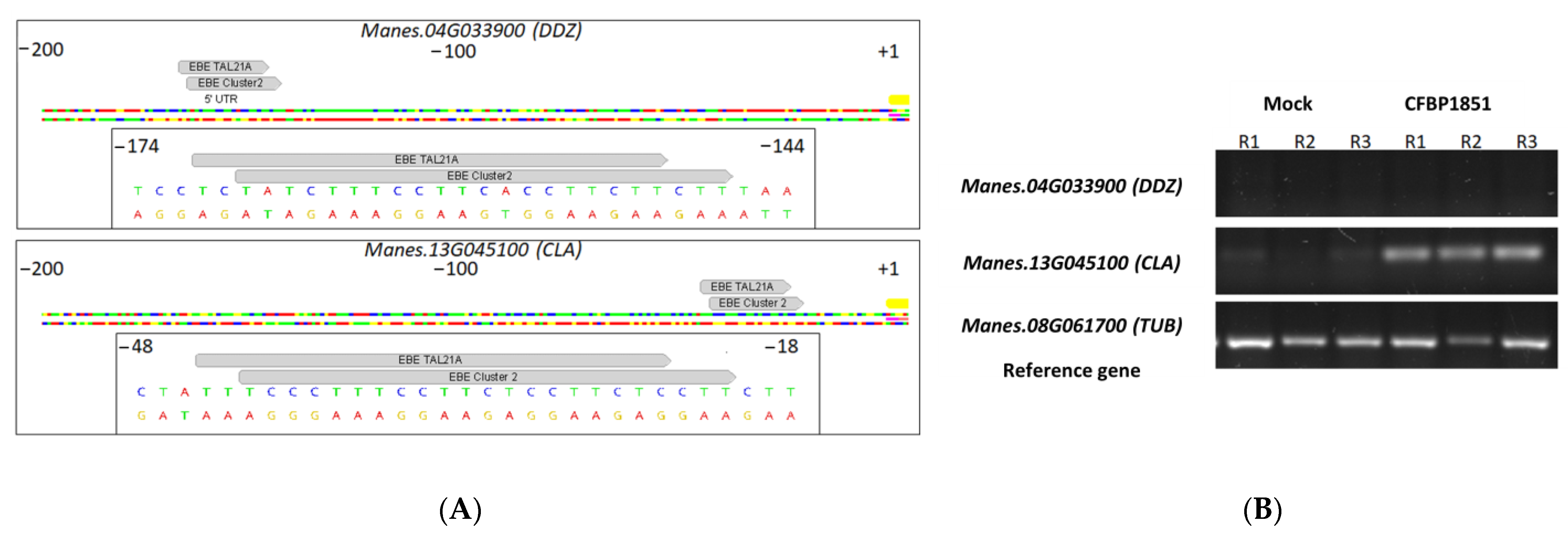
3.6. Candidate Gene Expression Profiles Are in Agreement with Trends Observed in RNAseq and Previous Reports
3.7. Potential Functional Convergence within TALome Members and Among Expanded TALome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Antony, G.; Zhou, J.; Huang, S.; Li, T.; Liu, B.; White, F.; Yang, B. Rice xa13 Recessive Resistance to Bacterial Blight Is Defeated by Induction of the Disease Susceptibility Gene Os-11N3. Plant Cell 2010, 22, 3864–3876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Zhang, J.; Jia, H.; Sosso, D.; Li, T.; Frommer, W.B.; Yang, B.; White, F.F.; Wang, N.; Jones, J.B. Lateral organ boundaries 1 is a disease susceptibility gene for citrus bacterial canker disease. Proc. Natl. Acad. Sci. USA 2014, 111, E521–E529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cernadas, R.A.; Doyle, E.L.; Niño-Liu, D.O.; Wilkins, K.E.; Bancroft, T.; Wang, L.; Schmidt, C.L.; Caldo, R.; Yang, B.; White, F.F.; et al. Code-Assisted Discovery of TAL Effector Targets in Bacterial Leaf Streak of Rice Reveals Contrast with Bacterial Blight and a Novel Susceptibility Gene. PLoS Pathog. 2014, 10, e1003972. [Google Scholar] [CrossRef] [PubMed]
- Perez-Quintero, A.L.; Szurek, B. A Decade Decoded: Spies and Hackers in the History of TAL Effectors Research. Annu. Rev. Phytopathol. 2019, 57, 459–481. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Sugio, A.; White, F.F. Os8N3 is a host disease-susceptibility gene for bacterial blight of rice. Proc. Natl. Acad. Sci. USA 2006, 103, 10503–10508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, T.T.; Pérez-Quintero, A.L.; Wonni, I.; Carpenter, S.C.D.; Yu, Y.; Wang, L.; Leach, J.E.; Verdier, V.; Cunnac, S.; Bogdanove, A.J.; et al. Functional analysis of African Xanthomonas oryzae pv. oryzae TALomes reveals a new susceptibility gene in bacterial leaf blight of rice. PLoS Pathog. 2018, 14, e1007092. [Google Scholar] [CrossRef]
- Yu, Y.; Streubel, J.; Balzergue, S.; Champion, A.; Boch, J.; Koebnik, R.; Feng, J.; Verdier, V.; Szurek, B. Colonization of Rice Leaf Blades by an African Strain of Xanthomonas oryzae pv. oryzae Depends on a New TAL Effector That Induces the Rice Nodulin-3 Os11N3 Gene. Mol. Plant-Microbe Interact. 2011, 24, 1102–1113. [Google Scholar] [CrossRef] [Green Version]
- Cohn, M.; Bart, R.S.; Shybut, M.; Dahlbeck, D.; Gomez, M.; Morbitzer, R.; Hou, B.-H.; Frommer, W.B.; Lahaye, T.; Staskawicz, B.J. Xanthomonas axonopodis Virulence Is Promoted by a Transcription Activator-Like Effector–Mediated Induction of a SWEET Sugar Transporter in Cassava. Mol. Plant-Microbe Interact. 2014, 27, 1186–1198. [Google Scholar] [CrossRef] [Green Version]
- Sugio, A.; Yang, B.; Zhu, T.; White, F.F. Two type III effector genes of Xanthomonas oryzae pv. oryzae control the induction of the host genes OsTFIIAγ1 and OsTFX1 during bacterial blight of rice. Proc. Natl. Acad. Sci. USA 2007, 104, 10720–10725. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, A.R.; Morbitzer, R.; Lahaye, T.; Staskawicz, B.J. TALE-induced bHLH transcription factors that activate a pectate lyase contribute to water soaking in bacterial spot of tomato. Proc. Natl. Acad. Sci. USA 2017, 114, E897–E903. [Google Scholar] [CrossRef] [Green Version]
- Cox, K.L.; Meng, F.; Wilkins, K.E.; Li, F.; Wang, P.; Booher, N.J.; Carpenter, S.C.D.; Chen, L.-Q.; Zheng, H.; Gao, X.; et al. TAL effector driven induction of a SWEET gene confers susceptibility to bacterial blight of cotton. Nat. Commun. 2017, 8, 15588. [Google Scholar] [CrossRef] [PubMed]
- Kay, S.; Hahn, S.; Marois, E.; Hause, G.; Bonas, U. A Bacterial Effector Acts as a Plant Transcription Factor and Induces a Cell Size Regulator. Science 2007, 318, 648–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boch, J.; Bonas, U. Xanthomonas AvrBs3 Family-Type III Effectors: Discovery and Function. Annu. Rev. Phytopathol. 2010, 48, 419–436. [Google Scholar] [CrossRef] [PubMed]
- Bogdanove, A.J.; Schornack, S.; Lahaye, T. TAL effectors: Finding plant genes for disease and defense. Curr. Opin. Plant Biol. 2010, 13, 394–401. [Google Scholar] [CrossRef]
- Szurek, B.; Rossier, O.; Hause, G.; Bonas, U. Type III-dependent translocation of the Xanthomonas AvrBs3 protein into the plant cell. Mol. Microbiol. 2002, 46, 13–23. [Google Scholar] [CrossRef]
- Yang, Y.; Gabriel, D.W. Xanthomonas avirulence/pathogenicity gene family encodes functional plant nuclear targeting signals. Mol. Plant. Microbe Interact. 1995, 8, 627–631. [Google Scholar] [CrossRef]
- Zhu, W.; Yang, B.; Chittoor, J.M.; Johnson, L.B.; White, F.F. AvrXa10 Contains an Acidic Transcriptional Activation Domain in the Functionally Conserved C Terminus. Mol. Plant-Microbe Interact. 1998, 11, 824–832. [Google Scholar] [CrossRef] [Green Version]
- Deng, D.; Yan, C.; Wu, J.; Pan, X.; Yan, N. Revisiting the TALE repeat. Protein Cell 2014, 5, 297–306. [Google Scholar] [CrossRef] [Green Version]
- Schandry, N.; Jacobs, J.M.; Szurek, B.; Perez-Quintero, A.L. A cautionary TALE: How plant breeding may have favoured expanded TALE repertoires in Xanthomonas. Mol. Plant Pathol. 2018, 19, 1297–1301. [Google Scholar] [CrossRef] [Green Version]
- Mak, A.N.-S.; Bradley, P.; Bogdanove, A.J.; Stoddard, B.L. TAL effectors: Function, structure, engineering and applications. Curr. Opin. Struct. Biol. 2013, 23, 93–99. [Google Scholar] [CrossRef] [Green Version]
- Mak, A.N.-S.; Bradley, P.; Cernadas, R.A.; Bogdanove, A.J.; Stoddard, B.L. The Crystal Structure of TAL Effector PthXo1 Bound to Its DNA Target. Science 2012, 335, 716–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, D.; Yan, C.; Pan, X.; Mahfouz, M.; Wang, J.; Zhu, J.-K.; Shi, Y.; Yan, N. Structural basis for sequence-specific recognition of DNA by TAL effectors. Science 2012, 335, 720–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moscou, M.J.; Bogdanove, A.J. A Simple Cipher Governs DNA Recognition by TAL Effectors. Science 2009, 326, 1501. [Google Scholar] [CrossRef] [PubMed]
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 2009, 326, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Ke, Y.; Huang, R.; Ma, L.; Yang, Z.; Chu, Z.; Xiao, J.; Li, X.; Wang, S. A host basal transcription factor is a key component for infection of rice by TALE-carrying bacteria. eLife 2016, 5, e19605. [Google Scholar] [CrossRef]
- Peng, Z.; Hu, Y.; Zhang, J.; Huguet-Tapia, J.C.; Block, A.K.; Park, S.; Sapkota, S.; Liu, Z.; Liu, S.; White, F.F. Xanthomonas translucens commandeers the host rate-limiting step in ABA biosynthesis for disease susceptibility. Proc. Natl. Acad. Sci. USA 2019, 116, 20938–20946. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.-Q.; Hou, B.-H.; Lalonde, S.; Takanaga, H.; Hartung, M.L.; Qu, X.-Q.; Guo, W.-J.; Kim, J.-G.; Underwood, W.; Chaudhuri, B.; et al. Sugar transporters for intercellular exchange and nutrition of pathogens. Nature 2010, 468, 527–532. [Google Scholar] [CrossRef] [Green Version]
- El Kasmi, F.; Horvath, D.; Lahaye, T. Microbial effectors and the role of water and sugar in the infection battle ground. Curr. Opin. Plant Biol. 2018, 44, 98–107. [Google Scholar] [CrossRef]
- Duan, S.; Jia, H.; Pang, Z.; Teper, D.; White, F.; Jones, J.; Zhou, C.; Wang, N. Functional characterization of the citrus canker susceptibility gene CsLOB1. Mol. Plant Pathol. 2018, 19, 1908–1916. [Google Scholar] [CrossRef] [Green Version]
- Constantin, E.C.; Cleenwerck, I.; Maes, M.; Baeyen, S.; Van Malderghem, C.; De Vos, P.; Cottyn, B. Genetic characterization of strains named as Xanthomonas axonopodis pv. dieffenbachiae leads to a taxonomic revision of the X. axonopodis species complex. Plant Pathol. 2016, 65, 792–806. [Google Scholar] [CrossRef]
- CABI. Xanthomonas axonopodis pv. manihotis (cassava Bacterial Blight). Available online: www.cabi.org/isc (accessed on 1 October 2020).
- Bart, R.; Cohn, M.; Kassen, A.; McCallum, E.J.; Shybut, M.; Petriello, A.; Krasileva, K.; Dahlbeck, D.; Medina, C.; Alicai, T.; et al. High-throughput genomic sequencing of cassava bacterial blight strains identifies conserved effectors to target for durable resistance. Proc. Natl. Acad. Sci. USA 2012, 109, E1972–E1979. [Google Scholar] [CrossRef] [Green Version]
- Restrepo, S.; Valle, T.L.; Duque, M.C.; Verdier, V. Assessing genetic variability among Brazilian strains of Xanthomonas axonopodis pv. manihotis through restriction fragment length polymorphism and amplified fragment length polymorphism analyses. Can. J. Microbiol. 1999, 45, 754–763. [Google Scholar] [CrossRef]
- Restrepo, S.; Vélez, C.M.; Verdier, V. Measuring the Genetic Diversity of Xanthomonas axonopodis pv. manihotis Within Different Fields in Colombia. Phytopathology 2000, 90, 683–690. [Google Scholar] [CrossRef] [Green Version]
- Restrepo, S.; Verdier, V. Geographical Differentiation of the Population of Xanthomonas axonopodis pv. manihotis in Colombia. Appl. Environ. Microbiol. 1997, 63, 4427–4434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdier, V.; Restrepo, S.; Mosquera, G.; Duque, M.C.; Gerstl, A.; Laberry, R. Genetic and pathogenic variation of Xanthomonas axonopodis pv. manihotis in Venezuela. Plant Pathol. 2001, 47, 601–608. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, C.; Restrepo, S.; Tohme, J.; Verdier, V. Characterization of pathogenic and nonpathogenic strains of Xanthomonas axonopodis pv. manihotis by PCR-based DNA fingerprinting techniques. Fems Microbiol. Lett. 2002, 215, 23–31. [Google Scholar] [CrossRef]
- Arrieta-Ortiz, M.L.; Rodríguez-R, L.M.; Pérez-Quintero, Á.L.; Poulin, L.; Díaz, A.C.; Arias Rojas, N.; Trujillo, C.; Restrepo Benavides, M.; Bart, R.; Boch, J.; et al. Genomic survey of pathogenicity determinants and VNTR markers in the cassava bacterial pathogen Xanthomonas axonopodis pv. manihotis strain CIO151. PLoS ONE 2013, 8, e79704. [Google Scholar] [CrossRef]
- Cohn, M.; Morbitzer, R.; Lahaye, T.; Staskawicz, B.J. Comparison of gene activation by two TAL effectors from Xanthomonas axonopodis pv. manihotis reveals candidate host susceptibility genes in cassava. Mol. Plant Pathol. 2016, 875–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castiblanco, L.F.; Gil, J.; Rojas, A.; Osorio, D.; Gutiérrez, S.; Muñoz-Bodnar, A.; Perez-Quintero, A.L.; Koebnik, R.; Szurek, B.; López, C.; et al. TALE1 from Xanthomonas axonopodis pv. manihotis acts as a transcriptional activator in plant cells and is important for pathogenicity in cassava plants. Mol. Plant Pathol. 2012, 14, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Bodnar, A.; Pérez-Quintero, A.; Gomez-Cano, F.; Gil, J.; Michelmore, R.; Bernal, A.; Szurek, B.; Lopez, C. RNAseq analysis of cassava reveals similar plant responses upon infection with pathogenic and non-pathogenic strains of Xanthomonas axonopodis pv. manihotis. Plant Cell Rep. 2014, 33, 1901–1912. [Google Scholar] [CrossRef]
- Aristizábal, J.; Sánchez, T. Guía Técnica para Producción y Análisis de Almidón de Yuca; FAO: Roma, Italy, 2007. [Google Scholar]
- FAO. Save and Grow: Cassava, a Guide to Sustainable Production Intensification; FAO: Roma, Italy, 2013; ISBN 978-92-5-107641-5. [Google Scholar]
- Parmar, A.; Sturm, B.; Hensel, O. Crops that feed the world: Production and improvement of cassava for food, feed, and industrial uses. Food Secur. 2017, 9, 907–927. [Google Scholar] [CrossRef]
- Lozano, J.C. Cassava Bacterial Blight: A Manageable Disease. Plant Dis. 1986, 70, 1089. [Google Scholar] [CrossRef]
- Wydra, K.; Verdier, V. Occurrence of cassava diseases in relation to environmental, agronomic and plant characteristics. Agric. Ecosyst. Environ. 2002, 93, 211–226. [Google Scholar] [CrossRef]
- Abaca, A.; Kiryowa, M.; Awori, E.; Andema, A.; Dradiku, F.; Moja, A.; Mukalazi, J. Cassava Pests and Diseases’ Prevalence and Performance as Revealed by Adaptive Trial Sites in North Western Agro-Ecological Zone of Uganda. J. Agric. Sci. 2013, 6. [Google Scholar] [CrossRef]
- López, C.E.; Bernal, A.J. Cassava Bacterial Blight: Using Genomics for the Elucidation and Management of an Old Problem. Trop. Plant Biol. 2012, 5, 117–126. [Google Scholar] [CrossRef]
- Trujillo, C.; Arias-Rojas, N.; Poulin, L.; Medina, C.; Tapiero, A.; Restrepo, S.; Koebnik, R.; Bernal, A. Population typing of the causal agent of cassava bacterial blight in the Eastern Plains of Colombia using two types of molecular markers. BMC Microbiol. 2014, 14, 161. [Google Scholar] [CrossRef] [Green Version]
- Trujillo, C.; Ochoa, J.C.; Mideros, M.F.; Restrepo, S.; Lopez, C.; Bernal, A. A complex population structure of the cassava pathogen Xanthomonas axonopodis pv. manihotis in recent years in the Caribbean Region of Colombia. Microb. Ecol. 2014, 68, 155–167. [Google Scholar] [CrossRef]
- Blanvillain-Baufumé, S.; Reschke, M.; Solé, M.; Auguy, F.; Doucoure, H.; Szurek, B.; Meynard, D.; Portefaix, M.; Cunnac, S.; Guiderdoni, E.; et al. Targeted promoter editing for rice resistance to Xanthomonas oryzae pv. oryzae reveals differential activities for SWEET14-inducing TAL effectors. Plant Biotechnol. J. 2017, 15, 306–317. [Google Scholar] [CrossRef] [Green Version]
- Oliva, R.; Ji, C.; Atienza-Grande, G.; Huguet-Tapia, J.C.; Perez-Quintero, A.; Li, T.; Eom, J.-S.; Li, C.; Nguyen, H.; Liu, B.; et al. Broad-spectrum resistance to bacterial blight in rice using genome editing. Nat. Biotechnol. 2019, 37, 1344–1350. [Google Scholar] [CrossRef] [Green Version]
- Hutin, M.; Pérez-Quintero, A.L.; Lopez, C.; Szurek, B. MorTAL Kombat: The story of defense against TAL effectors through loss-of-susceptibility. Front. Plant Sci. 2015, 6, 535. [Google Scholar] [CrossRef] [Green Version]
- Shantharaj, D.; Römer, P.; Figueiredo, J.F.L.; Minsavage, G.V.; Krönauer, C.; Stall, R.E.; Moore, G.A.; Fisher, L.C.; Hu, Y.; Horvath, D.M.; et al. An engineered promoter driving expression of a microbial avirulence gene confers recognition of TAL effectors and reduces growth of diverse Xanthomonas strains in citrus. Mol. Plant Pathol. 2017, 18, 976–989. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yin, Z.; White, F. TAL effectors and the executor R genes. Front. Plant Sci. 2015, 6, 641. [Google Scholar] [CrossRef] [PubMed]
- Mora, R.; Rodriguez, M.; Gayosso, L.; López, C. Using in vitro plants to study the cassava response to Xanthomonas phaseoli pv. manihotis infection. Trop. Plant Pathol. 2019. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Medina, C.A.; Reyes, P.A.; Trujillo, C.A.; Gonzalez, J.L.; Bejarano, D.A.; Montenegro, N.A.; Jacobs, J.M.; Joe, A.; Restrepo, S.; Alfano, J.R.; et al. The role of type III effectors from Xanthomonas axonopodis pv. manihotis in virulence and suppression of plant immunity. Mol. Plant Pathol. 2018, 19, 593–606. [Google Scholar] [CrossRef] [Green Version]
- Sambrook, J.R.D.W. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2001. [Google Scholar]
- Eisel, D.; Seth, O.; Grünewald-Janho, S.; Kruchen, B.; Rüger, B. DIG Application Manual for Filter Hybridization; Roche Applied Science: Mannheim, Germany, 2008. [Google Scholar]
- Pérez-Quintero, A.L.; Lamy, L.; Gordon, J.L.; Escalon, A.; Cunnac, S.; Szurek, B.; Gagnevin, L. QueTAL: A suite of tools to classify and compare TAL effectors functionally and phylogenetically. Front. Plant Sci. 2015, 6, 545. [Google Scholar] [CrossRef] [Green Version]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Rahnenfuhrer, J.; Alexa, A. TopGO: Enrichment Analysis for Gene Ontology; R Package Version 2.38.1. 2019. Available online: https://bioconductor.org/packages/release/bioc/manuals/topGO/man/topGO.pdf (accessed on 1 October 2020).
- Grau, J.; Wolf, A.; Reschke, M.; Bonas, U.; Posch, S.; Boch, J. Computational Predictions Provide Insights into the Biology of TAL Effector Target Sites. PLoS Comput. Biol. 2013, 9, e1002962. [Google Scholar] [CrossRef] [Green Version]
- Erkes, A.; Mücke, S.; Reschke, M.; Boch, J.; Grau, J. PrediTALE: A novel model learned from quantitative data allows for new perspectives on TALE targeting. PLoS Comput. Biol. 2019, 15, e1007206. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Rinaldi, F.C.; Singh, P.; Doyle, E.L.; Dubrow, Z.E.; Tran, T.T.; Pérez-Quintero, A.L.; Szurek, B.; Bogdanove, A.J. TAL Effectors Drive Transcription Bidirectionally in Plants. Mol. Plant 2017, 10, 285–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuon, J.-E.; Qi, W.; Schläpfer, P.; Hirsch-Hoffmann, M.; von Bieberstein, P.R.; Patrignani, A.; Poveda, L.; Grob, S.; Keller, M.; Shimizu-Inatsugi, R.; et al. Haplotype-resolved genomes of geminivirus-resistant and geminivirus-susceptible African cassava cultivars. BMC Biol. 2019, 17, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.-C.; Chang, Y.-A.; Chang, H. A pthA homolog from a variant of Xanthomonas axonopodis pv. citri enhances virulence without inducing canker symptom. Eur. J. Plant Pathol. 2013, 137, 677–688. [Google Scholar] [CrossRef]
- Kassambara, A. ggpubr: “ggplot2” Based Publication Ready Plots; R Package Version 0.2.4. 2019. Available online: https://cran.r-project.org/web/packages/ggpubr/index.html (accessed on 1 October 2020).
- Ahmed, M.; Kim, D.R. pcr: An R package for quality assessment, analysis and testing of qPCR data. PeerJ 2018, 6, e4473. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Warnes, G.R.; Bolker, B.; Bonebakker, L.; Gentleman, R.; Huber, W.; Liaw, A.; Lumley, T.; Maechler, M.; Magnusson, A.; Moeller, S.; et al. gplots: Various R Programming Tools for Plotting Data; R Package Version 3.0.1.2. 2020. Available online: https://cran.r-project.org/web/packages/gplots/index.html (accessed on 1 October 2020).
- Wilkins, D. gggenes: Draw Gene Arrow Maps in “ggplot2”; R Package Version 0.4.0. 2019. Available online: http://cran.wustl.edu/web/packages/gggenes/gggenes.pdf (accessed on 1 October 2020).
- Soto-Sedano, J.C.; Mora-Moreno, R.E.; Mathew, B.; Léon, J.; Gómez-Cano, F.A.; Ballvora, A.; López-Carrascal, C.E. Major Novel QTL for Resistance to Cassava Bacterial Blight Identified through a Multi-Environmental Analysis. Front. Plant Sci. 2017, 8, 1169. [Google Scholar] [CrossRef] [PubMed]
- Bredeson, J.V.; Lyons, J.B.; Prochnik, S.E.; Wu, G.A.; Ha, C.M.; Edsinger-Gonzales, E.; Grimwood, J.; Schmutz, J.; Rabbi, I.Y.; Egesi, C.; et al. Sequencing wild and cultivated cassava and related species reveals extensive interspecific hybridization and genetic diversity. Nat. Biotechnol. 2016, 34, 562–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Quintero, A.L.; Rodriguez-R, L.M.; Dereeper, A.; López, C.; Koebnik, R.; Szurek, B.; Cunnac, S. An Improved Method for TAL Effectors DNA-Binding Sites Prediction Reveals Functional Convergence in TAL Repertoires of Xanthomonas oryzae Strains. PLoS ONE 2013, 8, e68464. [Google Scholar] [CrossRef]
- Doyle, E.L.; Booher, N.J.; Standage, D.S.; Voytas, D.F.; Brendel, V.P.; VanDyk, J.K.; Bogdanove, A.J. TAL Effector-Nucleotide Targeter (TALE-NT) 2.0: Tools for TAL effector design and target prediction. Nucleic Acids Res. 2012, 40, W117–W122. [Google Scholar] [CrossRef] [Green Version]
- Ye, G.; Hong, N.; Zou, L.-F.; Zou, H.-S.; Zakria, M.; Wang, G.-P.; Chen, G.-Y. tale-Based Genetic Diversity of Chinese Isolates of the Citrus Canker Pathogen Xanthomonas citri subsp. citri. Plant Dis. 2013, 97, 1187–1194. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, R.M.; de Oliveira, A.C.P.; Moreira, L.M.; Belasque, J.; Gourbeyre, E.; Siguier, P.; Ferro, M.I.T.; Ferro, J.A.; Chandler, M.; Varani, A.M. A TALE of Transposition: Tn3-Like Transposons Play a Major Role in the Spread of Pathogenicity Determinants of Xanthomonas citri and Other Xanthomonads. mBio 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Ruh, M.; Briand, M.; Bonneau, S.; Jacques, M.-A.; Chen, N.W.G. Xanthomonas adaptation to common bean is associated with horizontal transfers of genes encoding TAL effectors. BMC Genom. 2017, 18, 670. [Google Scholar] [CrossRef] [PubMed]
- Denancé, N.; Szurek, B.; Doyle, E.L.; Lauber, E.; Fontaine-Bodin, L.; Carrère, S.; Guy, E.; Hajri, A.; Cerutti, A.; Boureau, T.; et al. Two ancestral genes shaped the Xanthomonas campestris TAL effector gene repertoire. New Phytol. 2018, 219, 391–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erkes, A.; Reschke, M.; Boch, J.; Grau, J. Evolution of Transcription Activator-Like Effectors in Xanthomonas oryzae. Genome Biol. Evol. 2017, 9, 1599–1615. [Google Scholar] [CrossRef] [PubMed]
- Schandry, N.; de Lange, O.; Prior, P.; Lahaye, T. TALE-Like Effectors Are an Ancestral Feature of the Ralstonia solanacearum Species Complex and Converge in DNA Targeting Specificity. Front. Plant Sci. 2016, 7, 1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meckler, J.F.; Bhakta, M.S.; Kim, M.-S.; Ovadia, R.; Habrian, C.H.; Zykovich, A.; Yu, A.; Lockwood, S.H.; Morbitzer, R.; Elsäesser, J.; et al. Quantitative analysis of TALE–DNA interactions suggests polarity effects. Nucleic Acids Res. 2013, 41, 4118–4128. [Google Scholar] [CrossRef] [Green Version]
- Wan, H.; Hu, J.; Li, K.; Tian, X.; Chang, S. Molecular Dynamics Simulations of DNA-Free and DNA-Bound TAL Effectors. PLoS ONE 2013, 8, e76045. [Google Scholar] [CrossRef] [Green Version]
- Guilinger, J.P.; Pattanayak, V.; Reyon, D.; Tsai, S.Q.; Sander, J.D.; Joung, J.K.; Liu, D.R. Broad specificity profiling of TALENs results in engineered nucleases with improved DNA-cleavage specificity. Nat. Methods 2014, 11, 429–435. [Google Scholar] [CrossRef] [Green Version]
- Grewal, R.K.; Gupta, S.; Das, S. Xanthomonas oryzae pv. oryzae triggers immediate transcriptomic modulations in rice. BMC Genom. 2012, 13, 49. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; White, F.F. Diverse Members of the AvrBs3/PthA Family of Type III Effectors Are Major Virulence Determinants in Bacterial Blight Disease of Rice. Mol. Plant-Microbe Interact. 2004, 17, 1192–1200. [Google Scholar] [CrossRef] [Green Version]
- Streubel, J.; Pesce, C.; Hutin, M.; Koebnik, R.; Boch, J.; Szurek, B. Five phylogenetically close rice SWEET genes confer TAL effector-mediated susceptibility to Xanthomonas oryzae pv. oryzae. New Phytol. 2013, 200, 808–819. [Google Scholar] [CrossRef]
- Zhou, J.; Peng, Z.; Long, J.; Sosso, D.; Liu, B.; Eom, J.-S.; Huang, S.; Liu, S.; Vera Cruz, C.; Frommer, W.B.; et al. Gene targeting by the TAL effector PthXo2 reveals cryptic resistance gene for bacterial blight of rice. Plant J. 2015, 82, 632–643. [Google Scholar] [CrossRef] [PubMed]
- Betsuyaku, S.; Sawa, S.; Yamada, M. The Function of the CLE Peptides in Plant Development and Plant-Microbe Interactions. Arab. Book/Am. Soc. Plant Biol. 2011, 9, e0149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strabala, T.J.; O’donnell, P.J.; Smit, A.-M.; Ampomah-Dwamena, C.; Martin, E.J.; Netzler, N.; Nieuwenhuizen, N.J.; Quinn, B.D.; Foote, H.C.C.; Hudson, K.R. Gain-of-function phenotypes of many CLAVATA3/ESR genes, including four new family members, correlate with tandem variations in the conserved CLAVATA3/ESR domain. Plant Physiol. 2006, 140, 1331–1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, Y.L.; Ishida, T.; Sawa, S. CLE peptides and their signaling pathways in plant development. J. Exp. Bot. 2016, 67, 4813–4826. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Ni, J.; Denver, R.; Wang, X.; Clark, S.E. Mechanisms of molecular mimicry of plant CLE peptide ligands by the parasitic nematode Globodera rostochiensis. Plant Physiol. 2011, 157, 476–484. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Mitchum, M.G.; Gao, B.; Li, C.; Diab, H.; Baum, T.J.; Hussey, R.S.; Davis, E.L. A parasitism gene from a plant-parasitic nematode with function similar to CLAVATA3/ESR (CLE) of Arabidopsis thaliana. Mol. Plant Pathol. 2005, 6, 187–191. [Google Scholar] [CrossRef]
- Marois, E.; Van den Ackerveken, G.; Bonas, U. The Xanthomonas Type III Effector Protein AvrBs3 Modulates Plant Gene Expression and Induces Cell Hypertrophy in the Susceptible Host. Mol. Plant-Microbe Interact. 2002, 15, 637–646. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene a | Type | Sequence (5′→3′) | Reference |
|---|---|---|---|
| Candidate gene: Manes.06G123400 (MeSWEET10a) | Fw | TCCTCACCTTGACTGCGGTG | [8] |
| Rv | AGCACCATCTGGACAATCCCA | ||
| Candidate gene: Manes.04G033900 (Dof domain, zinc finger) | Fw | AAAGTGCCCAAGAGGTGGTG | This study |
| Rv | GCCTTTCACTTGAAGCTGGG | ||
| Candidate gene: Manes.13G045100 (Clavata3/ESR CLE-related protein) | Fw | CCACGACGAACTTTCACCCA | This study |
| Rv | CGCTGGGAACTTCATGAGCT | ||
| Candidate gene: Manes.11G151300 (Serine carboxypeptidase) | Fw | GCCCCAACTGTTAGATTTGTGG | This study |
| Rv | GGTGACCAGCTTCATACACCTT | ||
| Candidate gene: Manes.15G052000 (Beta-glucosidase) | Fw | TTGAAGATATGCTCAACGACACG | This study |
| Rv | CGTCTGCTCCGTTCCTGATA | ||
| Reference gene: Manes.08G061700.1 (Tubulin beta-6 chain) | Fw | GGAAAGATGAGCACCAAGGA | [56] |
| Rv | ACCAGTATACCAGTGCAAGAAG |
| Strain | Region of Origin (ECZ a) | Location of Origin | Year of Isolation | AFLP-Based Haplotype b | Estimated Number of TALEs c |
|---|---|---|---|---|---|
| CFBP1851 | Caribbean coast (ECZ 1) | nd | 1974 | 3 * | 2 |
| CIO151 | Eastern plains (ECZ 2) | Meta | 1995 | 3 * | 5 |
| UA226 | Caribbean coast (ECZ1) | Chinú | 2008 | 5 * | 3 |
| UA306 | Caribbean coast (ECZ 1) | Palmitos | 2008 | 7 * | 4 |
| UA318 | Caribbean coast (ECZ 1) | Ciénaga de oro | 2008 | 3 * | 4 |
| UA522 | Caribbean coast (ECZ 1) | Chinú | 2009 | 4 * | 5 |
| UA531 | Caribbean coast (ECZ 1) | Chinú | 2009 | 5 * | 3 |
| UA681 | Caribbean coast (ECZ 1) | Chinú | 2009 | 8 * | 5 |
| UA1061 | Caribbean coast (ECZ 1) | Chinú | 2009 | 5 * | 2 |
| UA1069 | Caribbean coast (ECZ 1) | Chinú | 2009 | 5 * | 2 |
| UA1183 | Eastern plains (ECZ 2) | Villavicencio | 2011 | 1 ** | 5 |
| UA1211 | Eastern plains (ECZ 2) | Granada | 2011 | 2 ** | 5 |
| UA1235 | Eastern plains (ECZ 2) | Villavicencio | 2011 | 3 ** | 5 |
| UA1245 | Eastern plains (ECZ 2) | Villavicencio | 2011 | 3 ** | 5 |
| UA1357 | Eastern plains (ECZ 2) | Orocué | 2012 | 8 ** | 4 |
| UA1381 | Eastern plains (ECZ 2) | Orocué | 2012 | 6 ** | 4 |
| UA1396 | Eastern plains (ECZ 2) | Orocué | 2012 | 7 ** | 5 |
| UA1399 | Eastern plains (ECZ 2) | Orocué | 2012 | 7 ** | 5 |
| Variant | RVD Sequence | Occurrences | Strains |
|---|---|---|---|
| TALE13A | NI-NS-NN-HD-NG-HD-NI-NG-HD-NN-NI-NI-NG | 9 | UA306, UA318, UA522, UA681, UA1211, UA1235, UA1245, UA1357, UA1399 |
| TALE14A | NI-NG-NI-NN-HD-HD-NG-NG-HD-NG-NG-HD-NG-NG | 1 | CIO151 |
| TALE14B | NI-NG-NI-NN-HD-HD-NS-NS-NS-HD-HD-NS-HD-NG | 1 | UA318 |
| TALE14C | NI-NG-NI-NN-NG-HD-NS-NS-NN-NG-HD-NN-NI-NG | 9 | UA226, UA306, UA318, UA522, UA531, UA681, UA1061, UA1183, UA1381 |
| TALE14D | NI-NG-NI-NN-NI-HD-NS-NS-NN-NG-HD-NN-NI-NG | 1 | CFBP1851 |
| TALE14E | NI-NS-NN-HD-NG-HD-NI-NS-NN-HD-NN-NI-NI-NG | 4 | UA226, UA531, UA1061, UA1069 |
| TALE15A | NI-NG-NI-NN-HD-HD-NS-NS-NS-HD-HD-NN-HD-NG-NG | 1 | UA1211 |
| TALE15B | NI-NG-NI-NN-HD-HD-NS-NS-NS-HD-HD-NS-HD-NG-NG | 3 | UA522, UA681, UA1235 |
| TALE20A | NI-NG-NI-NN-HD-HD-NN-HD-NN-NG-HD-NS-HD-NN-HD-NG-HD-NI-NG-NG | 1 | UA318 |
| TALE20B | NI-NG-NI-NN-NG-HD-NN-HD-NN-NG-HD-NG-HD-NN-HD-NG-HD-NN-NG-NG | 2 | UA226, UA531 |
| TALE20C | NI-NG-NI-NN-NG-HD-NN-HD-NN-NG-HD-NS-HD-NN-HD-NG-HD-NI-NG-NG | 3 | UA306, UA522, UA681 |
| TALE20D | NI-NG-NI-NN-NG-HD-NS-NS-NN-NG-HD-NG-HD-NN-HD-HD-NS-NI-NG-NG | 1 | UA1381 |
| TALE20E | NI-NG-NI-NN-NG-HD-NS-NS-NN-NG-HD-NS-HD-NN-HD-NN-HD-NI-NS-NG | 1 | UA1235 |
| TALE21A | NI-NG-HD-NG-HD-N*-NG-NG-HD-HD-NG-NG-N*-NG-HD-NG-NG-NG-HD-NG-NG | 1 | CFBP1851 |
| TALE22A | NI-NG-HD-NG-NG-NG-HD-HD-NG-NG-HD-NG-HD-HD-NG-NG-HD-NG-NG-HD-NG-NG | 1 | UA1235 |
| TALE22B | NI-NG-HD-NG-NG-NG-HD-HD-NG-NG-NG-NG-HD-HD-NG-NG-HD-NG-NG-HD-NG-NG | 4 | UA306, UA318, UA522, UA681 |
| TALE22C | NI-NG-HD-NG-NG-NG-HD-HD-NG-NG-NG-NN-HD-NN-NG-NG-HD-NG-NG-HD-NG-NG | 2 | UA531, UA1061 |
| TALE22D | NI-NG-NI-NG-NI-NN-NG-HD-NN-HD-NN-NG-HD-NG-HD-NN-HD-NG-HD-NN-NG-NG | 1 | UA1061 |
| Target Gene ID a | Annotations a | EBE Prediction Quality c | Retained After Second Filter? e | Responsible for Activation (Ref) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| T13A (NC b) | T14E (NC b) | T14C (C4 b) | T15B (C3 b) | T20C (C1 b) | T22D (C1 b) | T22B (C2 b) | T22C (C2 b) | |||||
| Common DUGs | Manes.04G033900 | Dof Domain, Zinc Finger Protein | E | E | Yes | TAL22 [8] | ||||||
| Manes.06G123400 | Bidirectional Sugar Transporter Sweet10 | VG | E | Yes | TAL20 [8] | |||||||
| Manes.17G096100 | No Data | VG | VG | No | ||||||||
| Manes.15G026800 | Membrane-Associated Kinase Regulator | E | Yes | TAL14 [8] | ||||||||
| Manes.10G007900 | Abscisic Acid Receptor PYL4 | G | F | Yes | ||||||||
| Manes.11G151300 | Serine Carboxypeptidase S10 | F | G | Yes | TAL14 [8] | |||||||
| DUGs for UA681 | Manes.15G041200 | 12s Seed Storage Protein | VG | No | ||||||||
| Manes.18G060400 | Rho GTPase-Activating Protein Ren1 | E | Yes | |||||||||
| Manes.13G045100 | Clavata3/ESR (CLE)-Related Protein | E | Yes | TAL22 [8] | ||||||||
| Manes.S045100 | Class IV Chitinase | E | Yes | |||||||||
| Manes.04G109300 | Oxidoreductase, 2OG-Fe (II) Oxygenase Family Protein | F | VG | No | ||||||||
| Manes.06G159100 | Ring Finger Domain (ZF-Ring_2)/Wall-Associated Receptor Kinase C-Terminal | F | G | No | ||||||||
| Manes.09G134100 | mlo Protein | E | No | |||||||||
| Manes.14G044700 | Protein Containing an AP2 Domain | F | E | Yes | TAL15 [8] | |||||||
| Manes.04G096600 | Xyloglucan:Xyloglucosyl Transferase | VG d | No | |||||||||
| Manes.17G038600 | No Data | E | No | |||||||||
| Manes.05G118500 | No Data | G | F | F | No | |||||||
| Manes.02G120700 | Protein Containing a Tetratricopeptide Repeat (TPR_16) | G | No | |||||||||
| Manes.12G151000 | No Data | VG | Yes | |||||||||
| Manes.09G105800 | Protein Containing a Myb-Like DNA-Binding Domain and a Myb-CC Type Transfactor, LHEQLE Motif | G | Yes | |||||||||
| Manes.18G074600 | No Data | G | No | |||||||||
| Manes.S013800 | Basic Secretory Protein Family/Peptidase of Plants and Bacteria | G d | No | |||||||||
| Manes.06G057400 | Equilibrative Nucleoside Transporter | G | No | |||||||||
| Manes.07G135200 | Abscisic Acid Receptor PYL4 | G | Yes | |||||||||
| Manes.04G053400 | Galactolipase/Phospholipase A (1) | F | G | No | ||||||||
| Manes.08G036300 | Calcium-Dependent Protein Kinase | G | Yes | |||||||||
| DUGs for UA1061 | Manes.07G122400 | No Data | VG | Yes | ||||||||
| Manes.08G141800 | Ammonium Transporter 1 | E | Yes | |||||||||
| Manes.11G008300 | Member Of ‘GDXG’ Family of Lipolytic Enzymes | VG | Yes | |||||||||
| Manes.10G066600 | 2-Hydroxyisoflavanone Dehydratase | E | Yes | |||||||||
| Manes.16G117500 | No Data | VG | Yes | |||||||||
| Manes.13G136600 | Coniferyl-Alcohol Glucosyltransferase | E | No | |||||||||
| Manes.04G118200 | Protein of Unknown Function (Duf642) | G d | Yes | |||||||||
| Manes.15G090000 | Homeobox-Leucine Zipper Protein ATHB-52 | G | Yes | TAL14 [8] | ||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zárate-Chaves, C.A.; Osorio-Rodríguez, D.; Mora, R.E.; Pérez-Quintero, Á.L.; Dereeper, A.; Restrepo, S.; López, C.E.; Szurek, B.; Bernal, A. TAL Effector Repertoires of Strains of Xanthomonas phaseoli pv. manihotis in Commercial Cassava Crops Reveal High Diversity at the Country Scale. Microorganisms 2021, 9, 315. https://doi.org/10.3390/microorganisms9020315
Zárate-Chaves CA, Osorio-Rodríguez D, Mora RE, Pérez-Quintero ÁL, Dereeper A, Restrepo S, López CE, Szurek B, Bernal A. TAL Effector Repertoires of Strains of Xanthomonas phaseoli pv. manihotis in Commercial Cassava Crops Reveal High Diversity at the Country Scale. Microorganisms. 2021; 9(2):315. https://doi.org/10.3390/microorganisms9020315
Chicago/Turabian StyleZárate-Chaves, Carlos A., Daniela Osorio-Rodríguez, Rubén E. Mora, Álvaro L. Pérez-Quintero, Alexis Dereeper, Silvia Restrepo, Camilo E. López, Boris Szurek, and Adriana Bernal. 2021. "TAL Effector Repertoires of Strains of Xanthomonas phaseoli pv. manihotis in Commercial Cassava Crops Reveal High Diversity at the Country Scale" Microorganisms 9, no. 2: 315. https://doi.org/10.3390/microorganisms9020315