
New Dimensions in Microbial Ecology—Functional Genes in Studies to Unravel the Biodiversity and Role of Functional Microbial Groups in the Environment


Abstract
:
1. Introduction
2. The Start: 16S rRNA Genes for Studies in the Environment
3. Progress and Limitations of Sequence Studies of 16S rRNA Genes and Metagenomes
4. Functional Gene Studies
5. Photosynthesis and Anoxygenic Phototrophic Bacteria
5.1. The Phylogeny of the fmoA Gene in Green Sulfur Bacteria
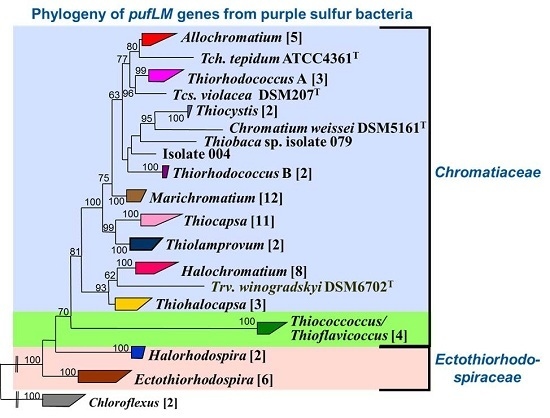
5.2. The Phylogeny of the pufLM Genes in Purple Sulfur Bacteria
5.3. Molecular Ecology and Species Recognition of Phototrophic Sulfur Bacteria in the Environment
5.4. Environmental Communities of Green Sulfur Bacteria
5.5. Environmental Communities of Phototrophic Purple Bacteria
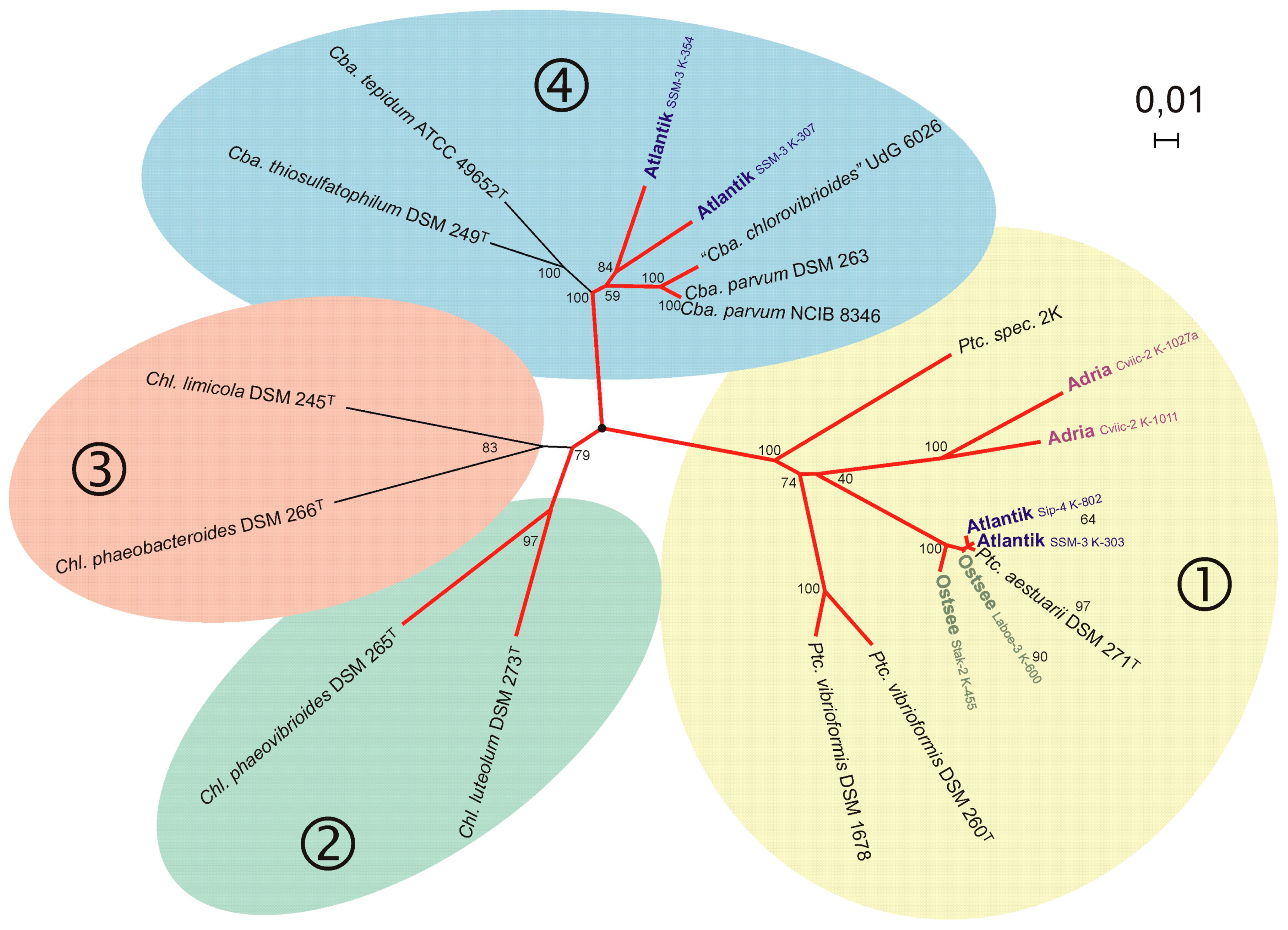
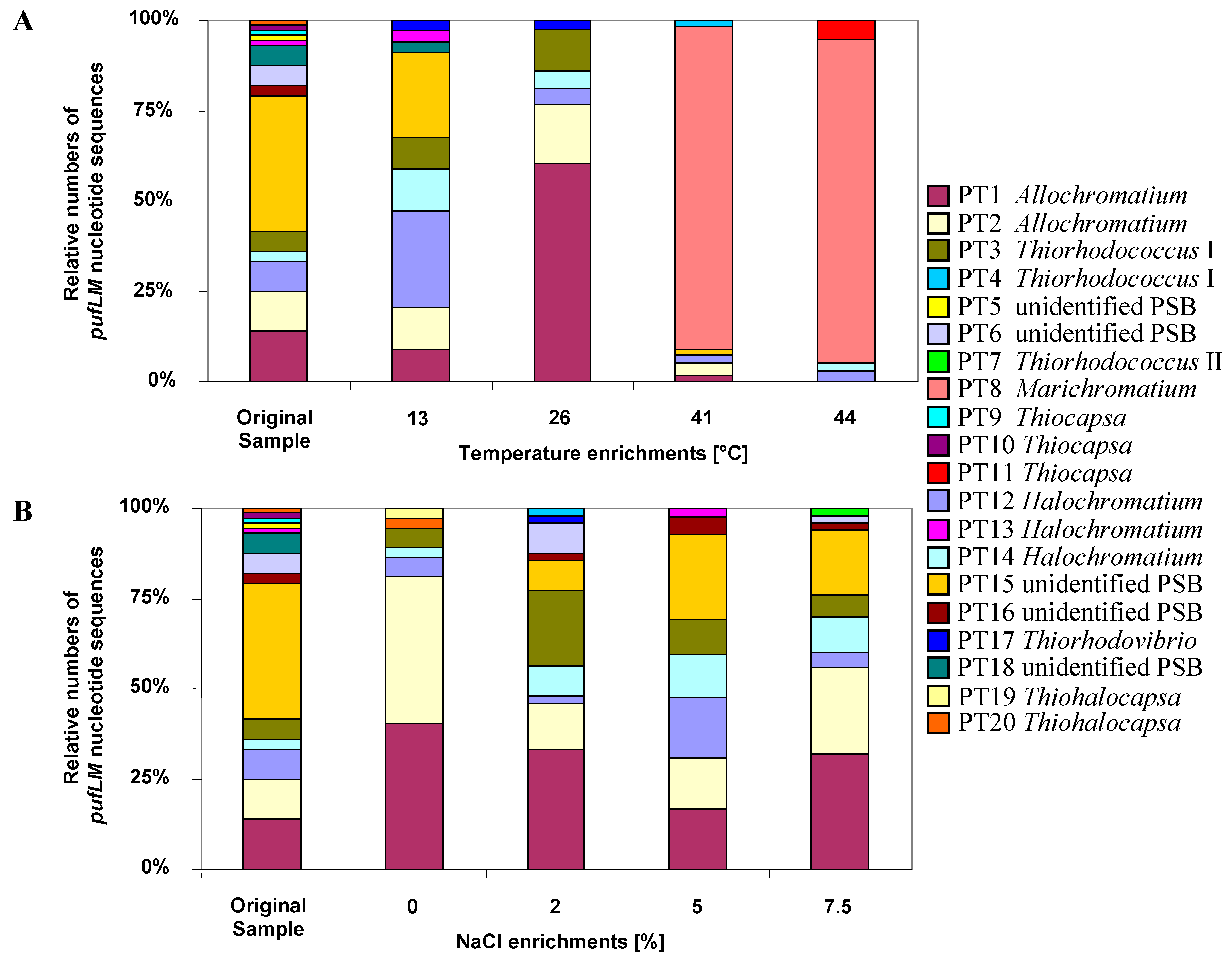
5.6. Baltic Sea Coastal Lagoon
5.7. Salt Lakes of the Salar de Atacama
5.8. Biodiversity of the bchY Gene
6. Oxidation and Reduction of Sulfur Compounds
6.1. Adenosine-5′-Phosphosulfate Reductase (APS Reductase), aprA
6.2. Dissimilatory Sulfite Reductase, dsrAB
6.3. Sulfate Thioesterase, soxB
7. Denitrification
7.1. Dissimilatory Nitrate Reductases, narH, narG and napA
7.2. Dissimilatory Nitrite Reductases, nirS and nirK
8. Nitrification—Oxidation of Ammonia—amoA
8.1. Aerobic Ammonia-Oxidizing Bacteria (AOB)
8.2. Ammonia-Oxidizing Archaea (AOA)
8.3. Anaerobic Ammonia-Oxidizing Bacteria (Anammox)
9. Oxidation of Methane—Methane Monooxygenase pmoA
10. Biosynthetic Pathways of Deep Sea Hot Vent Microorganisms
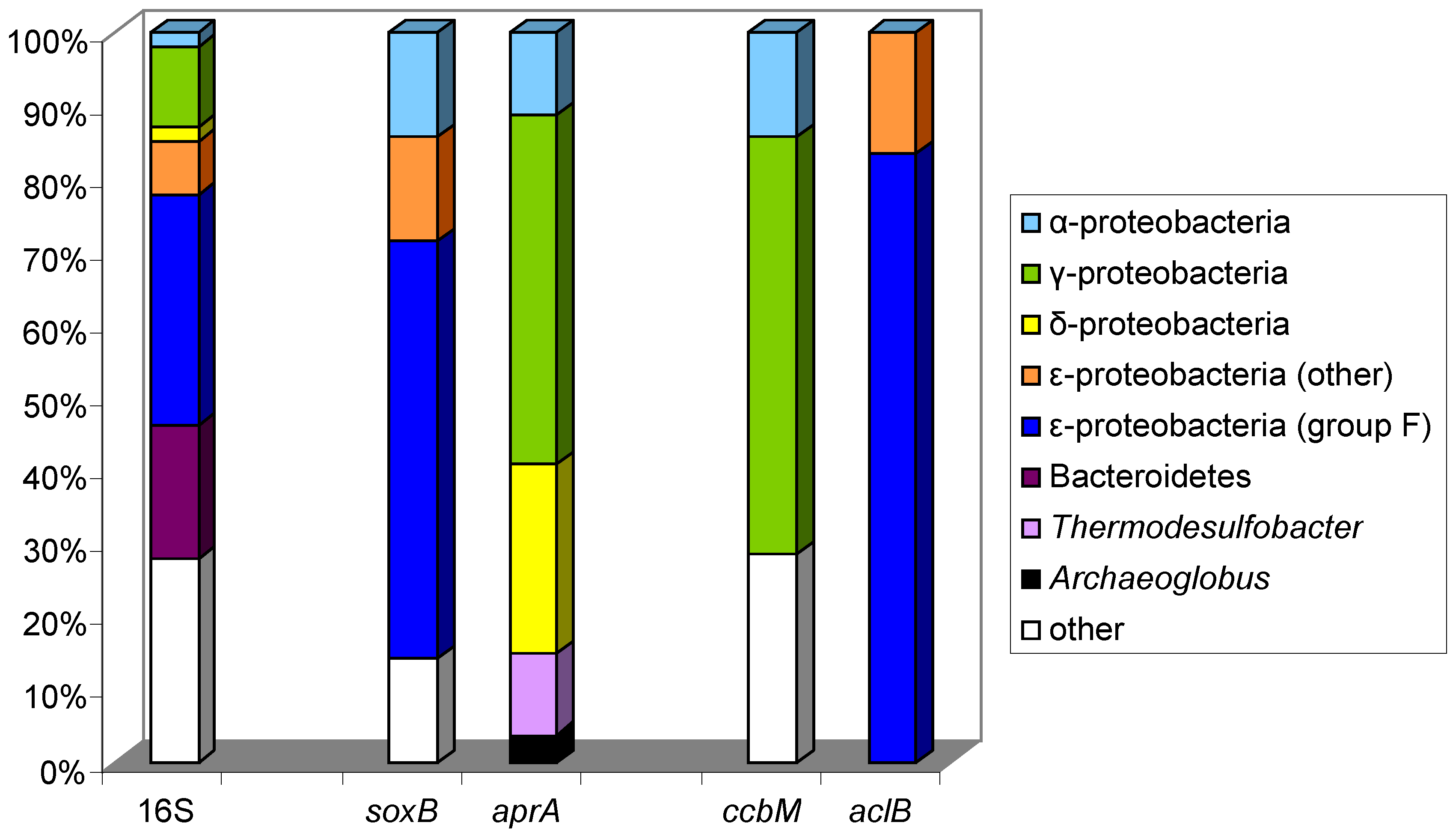
10.1. The Logatchev Hydrothermal Vent Field
10.2. Lamellibrachia Anaximandri
10.3. Rimicaris Exoculata
11. Conclusions
11.1. Functional Diversity—The Competition of Pathways in Nature
11.2. Environmental Conditions Determine Microbial Community Structure
11.3. The Challenges of Functional Diversity Studies in the Habitat
11.4. Selective Enrichment of Functional Groups
Acknowledgments
References
- Woese, C.R.; Fox, G.E. Phylogenetic structure of the prokaryotic domain: The primary kingdoms. Proc. Nat. Acad. Sci. USA 1977, 74, 5088–5090. [Google Scholar] [CrossRef] [PubMed]
- Woese, C.R. Bacterial evolution. Microbiol. Rev. 1987, 51, 221–271. [Google Scholar] [PubMed]
- Maidak, B.L.; Cole, J.R.; Parker, C.T.; Garrity, G.M.; Larsen, N.; Li, B.; Lilburn, T.G.; McCaughey, M.J.; Olsen, G.J.; Overbeek, R.; et al. A new version of the RDP (ribosomal database project). Nucleic Acids Res. 2009, 27, 171–173. [Google Scholar] [CrossRef]
- Muyzer, G.; DeWaal, E.C.; Uitterlinden, A.G. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl. Environ. Microbiol. 1993, 59, 695–700. [Google Scholar] [PubMed]
- Muyzer, G.; Smalla, K. Application of denaturing gradient gel electrophoresis (DGGE) and temperature gradient electrophoresis (TGGE) in microbial ecology. Antonie Leeuwenhoek 1998, 73, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Weitzel, B. Prokaryotische Aktivität und Diversität an Marinen Bio-Geo-Schnittstellen in der Tiefsee unter Besonderer Berücksichtigung der Prozesse der Methanoxidation. Ph.D. Thesis, University Kiel, Kiel, Germany, 2004. [Google Scholar]
- Marchesi, J.R.; Sato, T.; Weightman, A.J.; Martin, T.A.; Fry, J.C.; Hiom, S.J.; Wade, W.G. Design and evaluation of useful bacterium-specific PCR primers that amplify genes coding for bacterial 16S rRNA. Appl. Environ. Microbiol. 1998, 64, 795–799. [Google Scholar] [PubMed]
- Petri, R.; Imhoff, J.F. The relationship of nitrate reducing bacteria on the basis of narH gene sequences and the congruent phylogeny of narH and 16S rDNA based on pure culture studies and analyses of environmental DNA. Syst. Appl. Microbiol. 2000, 23, 47–57. [Google Scholar] [CrossRef]
- Tank, M.; Blümel, M.; Imhoff, J.F. Communities of purple sulfur bacteria in a Baltic Sea coastal lagoon analyzed by pufLM gene libraries and the impact of temperature and NaCl concentration in experimental enrichment cultures. FEMS Microbiol. Ecol. 2011, 78, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Schlegel, H.G. (Ed.) Anreicherungskultur und Mutantenauslese; Gustav Fischer: Stuttgart, Germany, 1965; p. 506.
- Gilbert, W.; Maxam, A. The nucleotide sequence of the lac operator. Proc. Natl. Acad. Sci. USA 1973, 70, 3581–3584. [Google Scholar] [CrossRef] [PubMed]
- Sanger, F.; Coulson, A.R. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J. Mol. Biol. 1975, 94, 441–448. [Google Scholar] [CrossRef]
- Sogin, M.L.; Morrison, H.G.; Huber, J.A.; Welch, D.M.; Huse, S.M.; Neal, P.R.; Arrieta, J.M.; Herndl, G.J. Microbial diversity in deep sea and thze underexplored “rare biosphere”. Proc. Natl. Acad. Sci. USA 2006, 103, 12115–12120. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.; Kelso, J. High-throughput DNA sequencing—Concepts and limitations. Bioessays 2010, 32, 524–536. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.A.; Dupont, C.L. Microbial metagenomics: Beyond the genome. Annu. Rev. Mar. Sci. 2011, 3, 347–371. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, R.; Adams, M.; White, O.; Clayton, R.; Kirkness, E.; Kerlavage, A.; Bult, C.; Tomb, J.; Dougherty, B.; Merrick, J.; et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 1995, 269, 496–512. [Google Scholar] [CrossRef] [PubMed]
- Venter, J.C.; Remington, K.; Heidelberg, J.F.; Halpern, A.L.; Rusch, D.; Eisen, J.A.; Wu, D.; Paulsen, I.; Nelson, K.E.; Nelson, W.; et al. Environmental genome shotgun sequencing of the Sargasso Sea. Science 2004, 304, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Henckel, T.; Friedrich, M.; Conrad, R. Molecular analyses of the methane-oxidizing microbial community in rice field soil by targeting the genes of the 16S rRNA, particulate methane monooxygenase, and methanol dehydrogenase. Appl. Environ. Microbiol. 1999, 65, 1980–1990. [Google Scholar] [PubMed]
- Müller, A.L.; Kjeldsen, K.U.; Rattei, T.; Pester, M.; Loy, A. Phylogenetic and environmental diversity of DsrAB-type dissimilatory (bi)sulfite reductases. ISME J. 2015, 9, 1152–1165. [Google Scholar] [CrossRef] [PubMed]
- Meyer, B.; Kuever, J. Molecular analysis of the diversity of sulfate-reducing and sulfur-oxidizing prokaryotes in the environment, using aprA as functional marker gene. Appl. Environ. Microbiol. 2007, 73, 7664–7679. [Google Scholar] [CrossRef] [PubMed]
- Meyer, B.; Imhoff, J.F.; Kuever, J. Molecular analysis of the distribution and phylogeny of soxB gene among sulfur-oxidizing bacteria—Evolution of the Sox sulfur oxidation enzyme system. Environ. Microbiol. 2007, 9, 2957–2977. [Google Scholar] [CrossRef] [PubMed]
- Loy, A.; Duller, S.; Baranyi, C.; Mussmann, M.; Ott, J.; Sharon, I.; Beja, O.; Paslier, D.L.; Dahl, C.; Wagner, M. Reverse dissimilatory sulfite reductase as phylogenetic marker for a subgroup of sulfur-oxidizing prokaryotes. Environ. Microbiol. 2009, 11, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Prosser, J.I.; Nicol, G.W. Relative contributions of archaea and bacteria to aerobic ammonia oxidation in the environment. Environ. Microbiol. 2008, 10, 2931–2941. [Google Scholar] [CrossRef] [PubMed]
- Knief, C. Diversity and habitat preferences of cultivated and uncultivated aerobic methanotrophic bacteria evaluated based on pmoA as molecular marker. Front. Microbiol. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Peck, H.D. Symposium on metabolism of inorganic compounds. V. Comparative metabolism in organic sulfur compounds in microorganisms. Bact. Rev. 1962, 26, 67–94. [Google Scholar] [PubMed]
- Holmes, A.J.; Costello, A.; Lidstrom, M.E.; Murrell, J.C. Evidence that particulate methane monooxygenase and ammonia monooxygenase maybe evolutionary related. FEMS Microbiol. Lett. 1995, 132, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Pfennig, N. Phototrophic green and purple bacteria: A comparative systematic survey. Ann. Rev. Microbiol. 1977, 31, 275–290. [Google Scholar] [CrossRef] [PubMed]
- Imhoff, J.F. Taxonomy and physiology of phototrophic purple bacteria and green sulfur bacteria. In Anoxygenic Photosynthetic Bacteria; Blankenship, R.E., Madigan, M.T., Bauer, C.E., Eds.; Kluwer Academic Publ.: Dordrecht, The Netherlands, 1995; pp. 1–15. [Google Scholar]
- Imhoff, J.F.; Hiraishi, A.; Süling, J. Anoxygenic phototrophic purple bacteria. In Bergey’s Manual of Systematic Bacteriology, 2nd ed.; Brenner, D.J., Krieg, N.R., Staley, J.T., Eds.; Springer Verlag: New York, NY, USA, 2005; Volume 2, pp. 119–132. [Google Scholar]
- Imhoff, J.F.; Hiraishi, A. Aerobic bacteria containing bacteriochlorophyll and belonging to the alphaproteobacteria. In Bergey’s Manual of Systematic Bacteriology, 2nd ed.; Brenner, D.J., Krieg, N.R., Staley, J.T., Eds.; Springer Verlag: New York, NY, USA, 2005; Volume 2, pp. 133–136. [Google Scholar]
- Yurkov, V.V.; Beatty, J.T. Aerobic anoxygenic phototrophic bacteria. Microbiol. Mol. Biol. Rev. 1998, 62, 695–724. [Google Scholar] [PubMed]
- Imhoff, J.F. True marine and halophilic anoxygenic phototrophic bacteria. Arch. Microbiol. 2001, 176, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Imhoff, J.F. The family Chlorobiaceae. In The Prokaryotes. Other Major Lineages of Bacteria and Archaea, 4th ed.; DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer Verlag: Berlin, Germany; Heidelberg, Germany, 2014; pp. 501–514. [Google Scholar]
- Madigan, M.T.; Ormerod, J.G. Taxonomy, physiology and ecology of heliobacteria. In Anoxygenic Photosynthetic Bacteria; Blankenship, R.E., Madigan, M.T., Bauer, C.E., Eds.; Kluwer Academic Publ.: Dordrecht, The Netherlands, 1995; pp. 17–30. [Google Scholar]
- Pierson, B.K.; Castenholz, R.W. Taxonomy and physiology of filamentous anoxygenic phototrophs. In Anoxygenic Photosynthetic Bacteria; Blankenship, R.E., Madigan, M.T., Bauer, C.E., Eds.; Kluwer Academic Publ.: Dordrecht, The Netherlands, 1995; pp. 31–47. [Google Scholar]
- Bryant, D.A.; Costas, A.M.G.; Maresca, J.A.; Chew, A.G.M.; Klatt, C.G.; Bateson, M.M.; Tallon, L.J.; Hostetler, J.; Nelson, W.C.; Heidelberg, J.F.; et al. Candidatus Chloracidobacterium thermophilum: An aerobic phototrophic acidobacterium. Science 2007, 317, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Tank, M.; Bryant, D.A. Chloracidobacterium thermophilum gen. nov., sp. nov.: An anoxygenic microaerophilic chlorophotoheterotrophic acidobacterium. Int. J. Syst. Evol. Microbiol. 2015, 65, 1426–1430. [Google Scholar] [CrossRef] [PubMed]
- Imhoff, J.F. The phototrophic Alphaproteobacteria. In The Prokaryotes. A Handbook on the Biology of Bacteria, 3rd ed.; Dworkin, M., Falkow, S., Rosenberg, E., Schleifer, K.-H., Stackebrandt, E., Eds.; Springer Verlag: New York, NY, USA, 2006; Volume 5, pp. 41–64. [Google Scholar]
- Imhoff, J.F. The phototrophic Betaproteobacteria. In The Prokaryotes. A Handbook on the Biology of Bacteria, 3rd ed.; Dworkin, M., Falkow, S., Rosenberg, E., Schleifer, K.-H., Stackebrandt, E., Eds.; Springer Verlag: New York, NY, USA, 2006; Volume 5, pp. 593–601. [Google Scholar]
- Imhoff, J.F. The Chromatiaceae. In The Prokaryotes. A Handbook on the Biology of Bacteria, 3rd ed.; Dworkin, M., Falkow, S., Rosenberg, E., Schleifer, K.-H., Stackebrandt, E., Eds.; Springer Verlag: New York, NY, USA, 2006; Volume 6, pp. 846–873. [Google Scholar]
- Imhoff, J.F. The Family Ectothiorhodospiraceae. In The Prokaryotes. A Handbook on the Biology of Bacteria, 3rd ed.; Dworkin, M., Falkow, S., Rosenberg, E., Schleifer, K.-H., Stackebrandt, E., Eds.; Springer Verlag: New York, NY, USA, 2006; Volume 6, pp. 874–886. [Google Scholar]
- Shimada, K. Aerobic anoxygenic phototrophic bacteria. In Anoxygenic Photosynthetic Bacteria; Blankenship, R.E., Madigan, M.T., Bauer, C.E., Eds.; Kluwer Academic Publ.: Dordrecht, The Netherlands, 1995; pp. 105–122. [Google Scholar]
- Yurkov, V.V. Aerobic phototrophic bacteria. In The Prokaryotes. A Handbook on the Biology of Bacteria, 3rd ed.; Dworkin, M., Falkow, S., Rosenberg, E., Schleifer, K.-H., Stackebrandt, E., Eds.; Springer Verlag: New York, NY, USA, 2006; Volume 6, pp. 562–584. [Google Scholar]
- Yutin, N.; Suzuki, M.T.; Teeling, H.; Weber, M.; Venter, J.C.; Rusch, D.B.; Beja, O. Assessing diversity and biogeography of aerobic anoxygenic phototrophic bacteria in surface waters of the Atlantic and Pacific Oceans using the Global Ocean Sampling expedition metagenomes. Environ. Microbiol. 2007, 9, 1464–1475. [Google Scholar] [CrossRef] [PubMed]
- Beja, O.; Suzuki, M.T.; Heidelberg, J.F.; Nelson, W.C.; Preston, C.M.; Hamada, T.; Eisen, J.A.; Fraser, C.M.; DeLong, E.F. Unsuspected diversity among marine aerobic anoxygenic phototrophs. Nature 2002, 415, 630–633. [Google Scholar] [CrossRef] [PubMed]
- Ranchou-Peyruse, A.; Herbert, R.; Caumette, P.; Guyoneaud, R. Comparison of cultivation-dependent and molecular methods for studying the diversity of anoxygenic purple phototrophs in sediments of an eutrophic brackish lagoon. Environ. Microbiol. 2006, 8, 1590–1599. [Google Scholar] [CrossRef] [PubMed]
- Alexander, B.; Andersen, J.H.; Cox, R.P.; Imhoff, J.F. Phylogeny of green sulfur bacteria on the basis of gene sequences of 16S rRNA and of the Fenna-Matthews-Olson protein. Arch. Microbiol. 2002, 178, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Tank, M.; Thiel, V.; Imhoff, J.F. Phylogenetic relationship of phototrophic purple sulfur bacteria according to pufL and pufM genes. Int. Microbiol. 2009, 12, 175–185. [Google Scholar] [PubMed]
- Imhoff, J.F. Reassignment of the genus Ectothiorhodospira Pelsh 1936 to a new family Ectothiorhodospiraceae fam. nov., and emended description of the Chromatiaceae Bavendamm 1924. Int. J. Syst. Bacteriol. 1984, 34, 338–339. [Google Scholar] [CrossRef]
- Imhoff, J.F.; Trüper, H.G.; Pfennig, N. Rearrangement of the species and genera of the phototrophic “Purple Nonsulfur Bacteria”. Int. J. Syst. Bacteriol. 1984, 34, 340–343. [Google Scholar] [CrossRef]
- Imhoff, J.F.; Süling, J. The phylogenetic relationship among Ectothiorhodospiraceae. A reevaluation of their taxonomy on the basis of 16S rDNA analyses. Arch. Microbiol. 1996, 165, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Guyoneaud, R.; Süling, J.; Petri, R.; Matheron, R.; Caumette, P.; Pfennig, N.; Imhoff, J.F. Taxonomic rearrangements of the genera Thiocapsa and Amoebobacter on the basis of 16S rDNA sequence analyses and description of Thiolamprovum gen. nov. Int. J. Syst. Bacteriol. 1998, 48, 957–964. [Google Scholar] [CrossRef] [PubMed]
- Imhoff, J.F.; Süling, J.; Petri, R. Phylogenetic relationships among the Chromatiaceae, their taxonomic reclassification and description of the new genera Allochromatium, Halochromatium, Isochromatium, Marichromatium, Thiococcus, Thiohalocapsa, and Thermochromatium. Int. J. Syst. Bacteriol. 1998, 48, 1129–1143. [Google Scholar] [CrossRef] [PubMed]
- Imhoff, J.F. Phylogenetic taxonomy of the family Chlorobiaceae on the basis of 16S rRNA and fmo (Fenna Matthews-Olson protein) gene sequences. Int. J. Syst. Evol. Microbiol. 2003, 53, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Fenna, R.E.; Matthews, B.W.; Olson, J.M.; Shaw, E.K. Structure of a bacteriochlorophyll protein from green photosynthetic bacterium Chlorobium limicola—Crystallographic evidence for a trimer. J. Mol. Biol. 1974, 84, 231–240. [Google Scholar] [CrossRef]
- Blankenship, R.E.; Olson, J.M.; Miller, M. Antenna complexes from green photosynthetic bacteria. In Anoxygenic Photosynthetic Bacteria; Blankenship, R.E., Madigan, M.T., Bauer, C.E., Eds.; Kluwer Academic Publ.: Dordrecht, The Netherlands, 1995; pp. 399–435. [Google Scholar]
- Alexander, B.; Imhoff, J.F. Communities of green sulfur bacteria in marine and saline habitats analyzed by gene sequences of 16S rRNA and Fenna-Matthews-Olson protein. Int. Microbiol. 2006, 9, 259–266. [Google Scholar] [PubMed]
- Imhoff, J.F.; Thiel, V. Phylogeny and taxonomy of Chlorobiaceae. Photosynth. Res. 2010, 104, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Imhoff, J.F. The anoxygenic phototrophic purple bacteria. In Bergey’s Manual of Systematic Bacteriology, 2nd ed.; Springer Verlag: New York, NY, USA, 2001; Volume 1, pp. 621–627. [Google Scholar]
- Stackebrandt, E.; Ebers, J. Taxonomic parameters revisited: Tarnished gold standards. Microbiol. Today 2006, 8, 6–9. [Google Scholar]
- Repeta, D.J.; Simpson, D.J.; Jørgensen, B.B.; Jannasch, H.W. Evidence for anoxygenic photosynthesis from the distribution of bacteriochlorophylls in the Black Sea. Nature 1989, 342, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Imhoff, J.F. Halophilic phototrophic bacteria. In Halophilic Bacteria; Rodriguez-Valera, F., Ed.; CRC Press: Boca Raton, FL, USA, 1988; Volume 1, pp. 85–108. [Google Scholar]
- Overmann, J.; Cypionka, H.; Pfennig, N. An extremely low-light-adapted phototrophic sulfur bacterium from the Black Sea. Limnol. Oceanogr. 1992, 37, 150–155. [Google Scholar] [CrossRef]
- Kumar, A.P.; Srinivas, T.N.R.; Sasikala, C.; Ramana, C.V.; Süling, J.; Imhoff, J.F. Prosthecochloris indica sp. nov., a novel green sulfur bacterium from a marine aquaculture pond of Kakinada, India. J. Gen. Appl. Microbiol. 2009, 55, 163–169. [Google Scholar] [CrossRef]
- Manske, A.K.; Glaeser, J.; Kuypers, M.M.M.; Overmann, J. Physiology and phylogeny of green sulfur bacteria forming a monospecific phototrophic assemblage at a depth of 100 m in the Black Sea. Appl. Environ. Microbiol. 2005, 71, 8049–8060. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, K.V.P.; Hiraishi, A.; Shimada, K.; Matsuura, K. Horizontal transfer of genes coding for the photosynthetic reaction centers of purple bacteria. J. Mol. Evol. 1977, 45, 131–136. [Google Scholar] [CrossRef]
- Achenbach, L.A.; Carey, J.; Madigan, M.T. Photosynthetic and phylogenetic primers for detection of anoxygenic phototrophs in natural environments. Appl. Environ. Microbiol. 2001, 67, 2922–2926. [Google Scholar] [CrossRef] [PubMed]
- Karr, E.A.; Sattley, W.M.; Jung, D.O.; Madigan, M.T.; Achenbach, L.A. Remarkable diversity of phototrophic purple bacteria in a permanently frozen Antarctic lake. Appl. Environ. Microbiol. 2003, 69, 4910–4914. [Google Scholar] [CrossRef] [PubMed]
- Voget, S.; Wemheuer, B.; Brinkhoff, T.; Vollmers, J.; Dietrich, S.; Giebel, H.-A.; Beardsley, C.; Sardemann, C.; Bakenhus, I.; Billerbeck, S.; et al. Adaptation of an abundant Roseobacter RCA organism to pelagic systems revealed by genomic and transcriptomic analyses. ISME J. 2015, 9, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Oz, A.; Sabehi, G.; Koblizek, M.; Massana, R.; Beja, O. Roseobacter-like bacteria in Red and Mediterranean Sea aerobic anoxygenic photosynthetic populations. Appl. Environ. Microbiol. 2005, 71, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Yutin, N.; Béjà, O.; Suzuki, M.T. The use of denaturing gradient gel electrophoresis with fully-degenerate pufM primers to monitor aerobic anoxygenic phototrophic assemblages. Limnol. Oceanogr. Methods 2008, 6, 427–440. [Google Scholar] [CrossRef]
- Mukkata, K.; Kantachote, D.; Wittayaweerasak, B.; Techkarnjanaruk, S.; Boonapatcharoen, N. Diversity of purple nonsulfur bacteria in shrimp ponds with varying mercury levels. Saudi J. Biol. Sci. 2015. [Google Scholar] [CrossRef]
- Waidner, L.A.; Kirchman, D.L. Diversity and distribution of ecotypes of the aerobic anoxygenic phototrophic gene pufM in the Delaware estuary. Appl. Environ. Microbiol. 2008, 74, 4012–4021. [Google Scholar] [CrossRef] [PubMed]
- Asao, M.; Pinkart, H.C.; Madigan, M.T. Diversity of extremophilic purple phototrophic bacteria in Soap Lake, a central Washington (USA) soda lake. Environ. Microbiol. 2011, 13, 2146–2157. [Google Scholar] [CrossRef] [PubMed]
- Thiel, V.; Tank, M.; Neulinger, S.C.; Gehrmann, L.; Dorador, C.; Imhoff, J.F. Unique communities of anoxygenic phototrophic bacteria in saline lakes of Salar de Atacama (Chile). Evidence for a new phylogenetic lineage of phototrophic Gammaproteobacteria from pufLM gene analyses. FEMS Microbiol. Ecol. 2010, 74, 510–522. [Google Scholar] [CrossRef] [PubMed]
- Caumette, P.; Baulaigue, R.; Matheron, R. Characterization of Chromatium salexigens sp. nov., a halophilic Chromatiaceae isolated from Mediterranean salinas. Syst. Appl. Microbiol. 1988, 10, 284–292. [Google Scholar] [CrossRef]
- Caumette, P.; Baulaigue, R.; Matheron, R. Thiocapsa halophila sp. nov., a new halophilic phototrophic purple sulfur bacterium. Arch. Microbiol. 1991, 155, 170–176. [Google Scholar] [CrossRef]
- Caumette, P.; Imhoff, J.F.; Süling, J.; Matheron, R. Chromatium glycolicum sp. nov., a moderately halophilic purple sulfur bacterium that uses glycolate as substrate. Arch. Microbiol. 1997, 167, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Serrano, W.; Amann, R.; Fischer, U. A new moderately thermophilic and high sulfide tolerant biotype of Marichromatium gracile, isolated from tidal sediments of the German Wadden Sea: Marichromatium gracile biotype thermosulfidiphilum. Syst. Appl. Microbiol. 2009, 32, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Dorador, C.; Vila, I.; Witzel, K.-P.; Imhoff, J.F. Cyanobacterial diversity in Salar de Huasco, a high altitude saline wetland in Northern Chile, are highly similar to Antarctic cyanobacteria. FEMS Microbiol. Ecol. 2008, 64, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Dorador, C.; Meneses, D.; Urtuvia, V.; Demergasso, C.; Vila, I.; Witzel, K.-P.; Imhoff, J.F. Diversity of Bacteroidetes in high altitude saline evaporitic basins in northern Chile. J. Geophys. Res. Biogeosci. 2009, 114, 35–44. [Google Scholar] [CrossRef]
- Dorador, C.; Vila, I.; Witzel, K.-P.; Imhoff, J.F. Bacterial and archaeal diversity in high altitude wetlands of the Chilean Altiplano. Fundam. Appl. Limnol. 2013, 182, 135–159. [Google Scholar]
- Gorlenko, V.M.; Bryantseva, I.A.; Rabold, S.; Tourova, T.P.; Rubtsova, D.; Smirnova, E.; Thiel, V.; Imhoff, J.F. A novel alkaliphilic and halophilic purple sulfur bacterium Ectothiorhodospira variabilis from soda lakes. Int. J. Syst. Evol. Microbiol. 2009, 59, 658–664. [Google Scholar] [CrossRef] [PubMed]
- Bryantseva, I.A.; Gorlenko, V.M.; Kompantseva, E.I.; Imhoff, J.F. Thioalkalicoccus limnaeus gen. nov., sp. nov., a new alkaliphilic purple sulfur bacterium with bacteriochlorophyll b. Int. J. Syst. Evol. Microbiol. 2000, 50, 2157–2163. [Google Scholar] [CrossRef] [PubMed]
- Imhoff, J.F.; Pfennig, N. Thioflavicoccus mobilis gen. nov., sp. nov., a novel purple sulfur bacterium with bacteriochlorophyll b. Int. J. Syst. Evol. Microbiol. 2001, 51, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.A.M.; Stolz, J.F.; Pierson, B.K. Structure of a microbial mat at Great Sippewissett Marsh, Cape Cod, Massachusetts. FEMS Microbiol. Ecol. 1987, 45, 343–364. [Google Scholar] [CrossRef]
- Pfennig, N.; Lünsdorf, H.; Süling, J.; Imhoff, J.F. Rhodospira trueperi, gen. nov. and spec. nov., a new phototrophic Proteobacterium of the alpha-group. Arch. Microbiol. 1997, 168, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Yutin, N.; Suzuki, M.T.; Rosenberg, M.; Rotem, D.; Madigan, M.T.; Süling, J.; Imhoff, J.F.; Beja, O. BchY-based degenerate primers target all types of anoxygenic photosynthetic bacteria in a single PCR. Appl. Environ. Microbiol. 2009, 75, 7556–7559. [Google Scholar] [CrossRef] [PubMed]
- Chew, A.G.M.; Bryant, D.A. Chlorophyll biosynthesis in bacteria: The origins of structural and functional diversity. Annu. Rev. Microbiol. 2007, 61, 113–129. [Google Scholar] [CrossRef] [PubMed]
- Jörgensen, B.B. Mineralisation of organic matter in the seabed—The role of sulfate reduction. Nature 1982, 296, 643–645. [Google Scholar] [CrossRef]
- Jörgensen, B.B.; Bang, M.; Blackburn, T.H. Anaerobic mineralization in marine sediments from the Baltic Sea-North Sea transition. Mar. Ecol. Prog. Ser. 1990, 59, 39–54. [Google Scholar]
- Hipp, W.M.; Pott, A.S.; Thum-Schmitz, N.; Faath, I.; Dahl, C.; Trüper, H.G. Towards the phylogeny of APS reductases and sirohaem sulfite reductases in sulfate-reducing and sulfur-oxidizing prokaryotes. Microbiology 1997, 143, 2891–2902. [Google Scholar] [CrossRef] [PubMed]
- Frigaard, N.U.; Dahl, C. Sulfur metabolism in phototrophic sulfur bacteria. Adv. Microb. Physiol. 2009, 54, 103–200. [Google Scholar] [PubMed]
- Friedrich, C.G.; Rother, D.; Bradischewsky, F.; Quentmeier, A.; Fischer, J. Oxidation of reduced inorganic sulfur compounds by bacteria: Emergence of a common mechanism? Appl. Environ. Microbiol. 2001, 67, 2873–2882. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, C.G.; Bradischewsky, F.; Rother, D.; Quentmeier, A.; Fischer, J. Prokaryotic sulfur oxidation. Curr. Opin. Microbiol. 2005, 8, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Petri, R.; Podgorsek, L.; Imhoff, J.F. Phylogeny and distribution of the soxB gene among thiosulfate-oxidizing bacteria. FEMS Microbiol. Lett. 2001, 197, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Hügler, M.; Gärtner, A.; Imhoff, J.F. Functional genes as markers for sulfur cycling and CO2 fixation in microbial communities of hydrothermal vents of the Logatchev field. FEMS Microbiol. Ecol. 2010, 73, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Hügler, M.; Petersen, J.M.; Dubilier, N.; Imhoff, J.F.; Sievert, S.M. Pathways of carbon and energy metabolism of the epibiotic community associated with the deep-sea hydrothermal vent shrimp Rimicaris exoculata. PLoS ONE 2011, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Meyer, B.; Kuever, J. Phylogenetic diversity and spatial distribution of the microbial community associated with the Caribbean deep-water sponge Polymastia cf. corticata by 16S rRNA, aprA, and amoA gene analysis. Microb. Ecol. 2008, 56, 306–321. [Google Scholar] [CrossRef] [PubMed]
- Aoki, M.; Kakiuchi, R.; Yamaguchi, T.; Takai, K.; Inagaki, F.; Imachi, H. Phylogenetic diversity of aprA genes in subseafloor sediments on the northwestern Pacific margin off Japan. Microbes Environ. 2015, 30, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Lenk, S.; Arnds, J.; Zerjatke, K.; Musat, N.; Amann, R.; Mussmann, M. Novel groups of Gammaproteobacteria catalyse sulfur oxidation and carbon fixation in a coastal, intertidal sediment. Environ. Microbiol. 2011, 13, 758–774. [Google Scholar] [CrossRef] [PubMed]
- Blazejak, A.; Schippers, A. Real-time PCR quantification and diversity analysis of the functional genes aprA and dsrA of sulfate-reducing prokaryotes in marine sediments of the Peru continental margin and the Black Sea. Front. Microbiol. 2011, 2, 253. [Google Scholar] [CrossRef] [PubMed]
- Schedel, M.; Vanselow, M.; Trüper, H.G. Siroheme sulfite reductase from Chromatium vinosum. Purification and investigation of some of its molecular and catalytic properties. Arch. Microbiol. 1979, 121, 29–36. [Google Scholar] [CrossRef]
- Wagner, M.; Roger, A.J.; Flax, J.L.; Brusseau, G.A.; Stahl, D.A. Phylogeny of dissimilatory sulfite reductases supports an early origin of sulfate respiration. J. Bacteriol. 1998, 180, 2975–2982. [Google Scholar] [PubMed]
- Minz, D.; Flax, J.L.; Green, S.J.; Muyzer, G.; Cohen, Y.; Wagner, M.; Rittmann, B.E.; Stahl, D.A. Diversity of sulfate-reducing bacteria in oxic and anoxic regions of a microbial mat characterized by comparative analysis of dissimilatory sulfite reductase genes. Appl. Environ. Microbiol. 1999, 65, 4666–4671. [Google Scholar] [PubMed]
- Muyzer, G.; Stams, A.J.M. The ecology and biotechnology of sulfate-reducing bacteria. Nat. Rev. 2008, 6, 441–454. [Google Scholar]
- Dhillon, A.; Teske, A.; Dillon, J.; Stahl, D.A.; Sogin, M.L. Molecular characterization of sulfate-reducing bacteria in the Guaymas Basin. Appl. Environ. Microbiol. 2003, 69, 2765–2772. [Google Scholar] [CrossRef] [PubMed]
- Widdel, F. Anaerober Abbau von Fettsäuren und Benzoesäure Durch neu Isolierte Arten Sulfatreduzierender Bakterien. Ph.D. Thesis, University of Göttingen, Göttingen, Germany, 1980. [Google Scholar]
- Gosh, W.; Dam, B. Biochemistry and molecular biology of lithotrophic sulfur oxidation by taxonomically and ecologically diverse bacteria and archaea. FEMS Microbiol. Rev. 2009, 33, 999–1043. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, L.H.; Bryant, D.A.; Frigaard, N.U. Mechanisms and evolution of oxidative sulfur metabolism in green sulfur bacteria. Front. Microbiol. 2011, 2, 116–130. [Google Scholar] [CrossRef] [PubMed]
- Headd, B.; Engel, A.S. Evidence for niche partitioning revealed by the distribution of sulfur oxidation genes collected from areas of a terrestrial sulfide spring with differing geochemical conditions. Appl. Environ. Microbiol. 2013, 79, 1171–1182. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Jiang, H.; Dong, H.; Wu, G.; Hou, W.; Zhao, W.; Sun, Y.; Lai, Z. Abundance and diversity of sulfur-oxidizing bacteria along a salinity gradient in four Qinghai-Tibetan lakes, China. Geomicrobiol. J. 2013, 30, 851–860. [Google Scholar] [CrossRef]
- Tourova, T.P.; Slobodova, N.V.; Bumazhkin, B.K.; Kolganova, T.V.; Muyzer, G.; Sorokin, D.Y. Analysis of community composition of sulfur-oxidizing bacteria in hypersaline and soda lakes using soxB as a functional marker. FEMS Microbiol. Ecol. 2013, 84, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Imhoff, J.F.; Hashwa, F.; Trüper, H.G. Isolation of extremely halophilic phototrophic bacteria from the alkaline Wadi Natrun, Egypt. Arch. Hydrobiol. 1978, 84, 381–388. [Google Scholar]
- Imhoff, J.F.; Sahl, H.G.; Soliman, G.S.H.; Trüper, H.G. The Wadi Natrun: Chemical composition and microbial mass developments in alkaline brines of eutrophic desert lakes. Geomicrobiol. J. 1979, 1, 219–234. [Google Scholar] [CrossRef]
- Imhoff, J.F.; Trüper, H.G. Ectothiorhodospira halochloris sp. nov., a new extremely halophilic phototrophic bacterium containing bacteriochlorophyll b. Arch. Microbiol. 1977, 114, 115–121. [Google Scholar] [CrossRef]
- Imhoff, J.F.; Trüper, H.G. Ectothiorhodospira abdelmalekii sp. nov., a new halophilic and alkaliphilic phototrophic bacterium. Zent. Bakteriol. Mikrobiol. Hyg. 1981, 2, 228–234. [Google Scholar]
- Petri, R. Diversität Nitratreduzierender Bakteriengemeinschaften in den Sedimenten der Ostsee und Untersuchungen zur Phylogenie der Respiratorischen Nitratreduktase. Ph.D. Thesis, University Kiel, Kiel, Germany, 2000; p. 148. [Google Scholar]
- Gregory, L.G.; Bond, P.L.; Richardson, D.J.; Spiro, S. Characterization of a nitrate-respiring bacterial community using the nitrate reductase gene (narG) as a functional marker. Microbiology 2003, 149, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Vetriani, C.; Voordeckers, J.W.; Crespo-Medina, M.; O’Brien, C.E.; Giovanelli, D.; Lutz, R.A. Deep-sea hydrothermal vent Epsilonproteobacteria encode a conserved and widespread nitrate reduction pathway (Nap). ISME J. 2014, 8, 1510–1521. [Google Scholar] [CrossRef] [PubMed]
- Zumft, W.G. Cell biology and molecular basis of denitrification. Microbiol. Mol. Biol. Rev. 1997, 61, 533–616. [Google Scholar] [PubMed]
- Braker, G.; Zhou, J.; Wu, L.; Devol, A.H.; Tiedje, J.M. Nitrite reductase genes (nirK and nirS) as functional markers to investigate diversity of denitrifying bacteria in Pacific Northwest marine sediment communities. Appl. Environ. Microbiol. 2000, 66, 2096–2104. [Google Scholar] [CrossRef] [PubMed]
- Braker, G.; Fesefeldt, A.; Witzel, K.-P. Development of PCR primer systems for amplification of nitrite reductase genes (nirK and nirS) to detect denitrifying bacteria in environmental samples. Appl. Environ. Microbiol. 1998, 64, 3769–3775. [Google Scholar] [PubMed]
- Huang, S.; Chen, C.; Yang, X.; Wu, Q.; Zhang, R. Distribution of typical denitrifying functional genes and diversity of nirS-encoding bacterial community related to environmental characteristics of river sediments. Biogeosciences 2011, 8, 3041–3051. [Google Scholar] [CrossRef]
- Junier, P.; Kim, O.-S.; Witzel, K.-P.; Imhoff, J.F.; Hadas, O. Habitat-partitioning of denitrifying bacterial communities carrying nirS/nirK genes in the stratified water column of Lake Kinneret, Israel. Aquat. Microb. Ecol. 2008, 51, 129–140. [Google Scholar] [CrossRef]
- Castro-Gonzalez, M.; Braker, G.; Farias, L.; Ulloa, O. Communities of nirS-type denitrifiers in the water column of the oxygen-minimum zone in the eastern South Pacific. Environ. Microbiol. 2005, 7, 1298–1306. [Google Scholar] [CrossRef] [PubMed]
- Oakley, B.B.; Francis, C.A.; Roberts, K.J.; Fuchsman, C.A.; Srinivasan, S.; Staley, J.T. Analysis of nitrite reductase (nirK and nirS) genes and cultivation reveal depauperate community of denitrifying bacteria in the Black Sea suboxic zone. Environ. Microbiol. 2007, 9, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Casciotti, K.L.; Ward, B.B. Dissimilatory nitrite reductase genes from autotrophic ammonia-oxidizing bacteria. Appl. Environ. Microbiol. 2001, 67, 2213–2221. [Google Scholar] [CrossRef] [PubMed]
- Rotthauwe, J.-H.; Witzel, K.-P.; Liesack, W. The ammonia monooxygenase structural gene amoA as a functional marker: Molecular fine-scale analysis of natural ammonia-oxidizing populations. Appl. Environ. Microbiol. 1997, 63, 4704–4712. [Google Scholar] [PubMed]
- Junier, P.; Kim, O.-S.; Junier, T.; Imhoff, J.F.; Ahn, T.-S.; Witzel, K.-P. Community analysis in betaproteobacterial ammonia-oxidizing bacteria using the amoCAB operon. Appl. Microbiol. Biotechnol. 2009, 83, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Francis, C.A.; Roberts, K.J.; Beman, J.M.; Santoro, A.E.; Oakley, B.B. Ubiquity and diversity of ammonia-oxidizing archaea in water columns and sediments of the ocean. Proc. Natl. Acad. Sci. USA 2005, 102, 14683–14688. [Google Scholar] [CrossRef] [PubMed]
- Lund, M.B.; Smith, J.M.; Francis, C.A. Diversity, abundance and expression of nitrite reductase (nirK)-like genes in marine thaumarchaea. ISME J. 2012, 6, 1966–1977. [Google Scholar] [CrossRef] [PubMed]
- Tavormina, P.L.; Ussler, W.; Orphan, V.J. Planktonic and sediment-associated aerobic methanotrophs in two seep systems along the North American margin. Appl. Environ. Microbiol. 2008, 74, 3985–3995. [Google Scholar] [CrossRef] [PubMed]
- Casciotti, K.L.; Ward, B.B. Phylogenetic analysis of nitric oxide reductase gene homologues from aerobic ammonia-oxidizing bacteria. FEMS Microbiol. Ecol. 2005, 52, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Garbeva, P.; Baggs, E.M.; Prosser, J.I. Phylogeny of nitrite reductase (nirK) and nitric oxide reductase (norB) genes from Nitrosopsira species isolated from soil. FEMS Microbiol. Lett. 2007, 266, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.E.; Boehm, A.B.; Francis, C.A. Denitrifier community composition along a nitrate and salinity gradient in a coastal aquifer. Appl. Environ. Microbiol. 2006, 72, 2102–2109. [Google Scholar] [CrossRef] [PubMed]
- Treusch, A.H.; Leininger, S.; Kletzin, A.; Schuster, S.C.; Klenk, H.P.; Schleper, C. Novel genes for nitrite reductase and Amo-related proteins indicate a role of uncultivated mesophilic crenarchaeota in nitrogen cycling. Environ. Microbiol. 2005, 7, 1985–1995. [Google Scholar] [CrossRef] [PubMed]
- Könneke, M.; Bernhard, A.E.; de la Torre, J.R.; Walker, C.B.; Waterbury, J.B.; Stahl, D.A. Isolation of autotrophic ammonia-oxidizing marine archaeon. Nature 2005, 437, 543–546. [Google Scholar] [CrossRef] [PubMed]
- Purkhold, U.; Pommerening-Röser, A.; Juretschko, S.; Schmid, M.C.; Koops, H.-P.; Wagner, M. Phylogeny of all recognized species of ammonia oxidizers based on comparative 16S rRNA and amoA sequence analysis: Implications for molecular diversity studies. Appl. Environ. Microbiol. 2000, 66, 5368–5382. [Google Scholar] [CrossRef] [PubMed]
- Norton, J.M.; Alzerreca, J.J.; Suwa, Y.; Klotz, M.G. Diversity of ammonia monooxygenase operon in autotrophic ammonia-oxidizing bacteria. Arch. Microbiol. 2002, 177, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Junier, P.; Molina, V.; Dorador, C.; Hadas, O.; Kim, O.-S.; Junier, T.; Witzel, K.-P.; Imhoff, J.F. Phylogenetic and functional markers to study ammonia-oxidising microorganisms (AOM) in the environment. Appl. Microbiol. Biotechnol. 2010, 85, 425–440. [Google Scholar] [CrossRef] [PubMed]
- Purkhold, U.; Wagner, M.; Timmermann, G.; Pommerening-Röser, A.; Koops, H.P. 16S rRNA and amoA-based phylogeny of 12 novel betaproteobacterial ammonia-oxidizing isolates: Extension of the dataset and proposal of a new lineage within the nitrosomonads. Int. J. Syst. Evol. Microbiol. 2003, 53, 1485–1494. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, H.; Takai, K.; Inagaki, F.; Yamato, Y.; Suzuki, M.; Nealson, K.H.; Horikoshi, K. Bacterial community shift along a subsurface geothermal water stream in a Japanese gold mine. Extremophiles 2005, 9, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Carini, S.A.; Joye, S.B. Nitrification in Mono Lake, California: Activity and community composition during contrasting hydrological regimes. Limnol. Oceanogr. 2008, 53, 2546–2557. [Google Scholar] [CrossRef]
- De la Torre, J.R.; Walker, C.B.; Ingalls, A.E.; Könneke, M.; Stahl, D.A. Cultivation of a thermophilic ammonia oxidizing archaeon synthesizing crenarchaeol. Environ. Microbiol. 2008, 10, 810–818. [Google Scholar] [CrossRef] [PubMed]
- Hatzenpichler, R.; Lebedeva, E.V.; Spieck, E.; Stoecker, K.; Richter, A.; Daims, H.; Wagner, M. A moderately thermophilic ammonia-oxidizing crenarchaeote from a hot spring. Proc. Natl. Acad. Sci. USA 2008, 105, 2134–2139. [Google Scholar] [CrossRef] [PubMed]
- Brochier-Armanet, C.; Boussau, B.; Bribaldo, S. Mesophilic crenarchaeota: Proposal for a third archaeal phylum, the Thaumarchaeota. Nat. Rev. Microbiol. 2008, 6, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Erguder, T.H.; Boon, N.; Wittebolle, L.; Marzorati, M.; Verstraete, W. Environmental factors shaping the ecological niches of ammonia-oxidizing archaea. FEMS Microbiol. Rev. 2009, 33, 855–869. [Google Scholar] [CrossRef] [PubMed]
- Stahl, D.A.; de la Torre, J.R. Physiology and diversity of ammonia-oxidizing archaea. Annu. Rev. Microbiol. 2012, 66, 83–101. [Google Scholar] [CrossRef] [PubMed]
- Dodsworth, J.A.; Hungate, B.; de la Torre, J.R.; Jiang, H.; Hedlund, B.P. Measuring nitrification, denitrification, and related biomarkers in terrestrial geothermal ecosystems. Methods Enzymol. 2011, 486, 172–203. [Google Scholar]
- Pester, M.; Raqttei, T.; Flechl, S.; Grönkröft, A.; Richter, A.; Overmann, J.; Reinhold-Hurek, B.; Loy, A.; Wagner, M. amoA-based consensus phylogeny of ammonia-oxidizing archaea and deep sequencing of amoA genes from soils of four different geographic regions. Environ. Microbiol. 2012, 14, 525–539. [Google Scholar] [CrossRef] [PubMed]
- Reigstad, L.J.; Richter, A.; Daims, H.; Urich, T.; Schwark, L.; Schleper, C. Nitrification in terrestrial hot springs of Iceland and Kamchatka. FEMS Microbiol. Ecol. 2008, 64, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Dong, H.; Yu, B.; Ly, G.; Deng, S.; Berzins, N.; Dai, M. Diversity and abundance of ammonia-oxidizing archaea and bacteria in Qinghai Lake, Northwestern China. Geomicrobiol. J. 2009, 26, 199–211. [Google Scholar] [CrossRef]
- Dorador, C.; Vila, I.; Remosellez, F.; Witzel, K.-P.; Imhoff, J.F. Unique clusters of Archaea in Salar des Huasco, an athalassohaline evaporitic basin of the Chilean Altiplano. FEMS Microbiol. Ecol. 2010, 73, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Tamegai, H.; Aoki, R.; Arakawa, S.; Kato, C. Molecular analysis of the nitrogen cycle in deep-sea microorganisms from the Nankai Trough: Genes for nitrification and denitrification from deep-sea environmental DNA. Extremophiles 2007, 11, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Bayer, K.; Schmitt, S.; Hentschel, U. Physiology, phylogeny and in situ evidence for bacterial and archaeal nitrifiers in the marine sponge Aplysina aerophoba. Environ. Microbiol. 2008, 10, 2942–2955. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Hou, L.; Newell, S.; Liu, M.; Zhou, J.; Zhao, H.; You, L.; Cheng, X. Community dynamics and activity of ammonia-oxidizing prokaryotes in intertidal sediments of the Zangtze estuary. Appl. Environ. Microbiol. 2014, 80, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Sintes, E.; Bergauer, K.; de Corte, D.; Yokokawa, T.; Herndl, G.J. Archaeal amoA gene diversity points to distinct biogeography of ammonia-oxidizing Crenarchaeota in the ocean. Environ. Microbiol. 2013, 15, 1647–1658. [Google Scholar] [CrossRef] [PubMed]
- Sintes, E.; DeCorte, D.; Haberleitner, E.; Herndl, G.J. Geographic distribution of archaeal ammonia oxidizing ecotypes in the Atlantic Ocean. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Strous, M.; Fuerst, J.A.; Kramer, E.H.M.; Logeman, S.; Muyzer, G.; van de Pas-Schoonen, K.T.; Webb, R.; Kuebeb, J.G.; Jetten, M.S.M. Missing lithotroph identified as new planctomycete. Nature 1999, 400, 446–449. [Google Scholar] [PubMed]
- Kartal, B.; Koleva, M.; Arsov, R.; van der Star, W.; Jetten, M.S.; Strous, M. Adaptation of a freshwater anammox population to high salinity wastewater. J. Biotechnol. 2006, 126, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Strous, M.; Pelletier, E.; Mangenot, S.; Rattei, T.; Lehner, A.; Taylor, M.W.; Horn, M.; Daims, H.; Bartol-Mavel, D.; Wincker, P.; et al. Deciphering the evolution and metabolism of an anammox bacterium from a community genome. Nature 2006, 440, 790–794. [Google Scholar] [CrossRef] [PubMed]
- Lam, P.; Lavik, G.; Jensen, M.M.; van den Vossenberg, J.; Schmid, M.; Woebken, D.; Gutierrez, D.; Amann, R.; Jetten, M.S.M.; Kuypers, M.M.M. Revising the nitrogen cycle in the Peruvian oxygen minimum zone. Proc. Natl. Acad. Sci. USA 2009, 106, 4752–4757. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Hong, Y.; Klotz, M.G.; Gu, J.D. A comparison of primer sets for detecting 16S rRNA and hydrazine oxidoreductase genes of anaerobic ammonium-oxidizing bacteria in marine sediments. Appl. Microbiol. Biotechnol. 2010, 86, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Harhangi, H.R.; LeRoy, M.; van Alen, T.; Hu, B.; Groen, J.; Kartal, B.; Tringe, S.G.; Quan, Z.-X.; Jetten, M.S.M.; Op den Camp, H.J.M. Hydrazine synthase, a unique phylomarker with which to study the presence and biodiversity of anammox bacteria. Appl. Environ. Microbiol. 2012, 78, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Whittenbury, R.; Phillips, K.C.; Wilkinson, J.F. Enrichment, isolation and some properties of methane utilizing bacteria. J. Gen. Microbiol. 1970, 61, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Brusseau, G.A.; Bulygina, E.S.; Hanson, R.S. Phylogenetic analysis and development of probes for differentiating methylotrophic bacteria. Appl. Environ. Microbiol. 1994, 60, 626–636. [Google Scholar] [PubMed]
- Reeburgh, W.S. Oceanic methane biogeochemistry. Chem. Rev. 2007, 107, 486–513. [Google Scholar] [CrossRef] [PubMed]
- Conrad, R. Microbial ecology of methanogens and metanotrophs. Adv. Agron. 2007, 96, 1–63. [Google Scholar]
- Dumont, M.G.; Lüke, C.; Deng, Y.; Frenzel, P. Classification of pmoA amplicon pyrosequences using BLAST and the lowest common ancestor method in MEGAN. Front. Microbiol. 2014, 5. [Google Scholar] [CrossRef]
- Costello, A.M.; Lidstrom, M.E. Molecular characterization of functional and phylogenetic genes from natural populations of methanotrophs in lake sediments. Appl. Environ. Microbiol. 1999, 65, 5066–5074. [Google Scholar] [PubMed]
- Rastogi, G.; Ranade, D.R.; Yeole, T.Y.; Gupta, A.K.; Patole, M.S.; Shouche, Y.S. Novel methanotroph diversity evidenced by molecular characterization of particulate methane monooxygenase A (pmoA) genes in a biogas reactor. Microbiol. Res. 2009, 164, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Tavormina, P.L.; Ussler, W.; Joy, S.B.; Harrison, B.K.; Orphan, V.J. Distribution of putative aerobic methanotrophs in diverse marine environments. ISME J. 2010, 4, 700–710. [Google Scholar] [CrossRef] [PubMed]
- Jannasch, H.W.; Wirsen, C.O. Chemosynthetic primary production at East Pacific sea floor spreading centers. BioScience 1979, 29, 592–598. [Google Scholar] [CrossRef]
- Jannasch, H.W.; Taylor, C.D. Deep-sea microbiology. Annu. Rev. Microbiol. 1984, 38, 487–514. [Google Scholar] [CrossRef] [PubMed]
- Jannasch, H.W. Microbial interactions with hydrothermal fluids. In Seafloor Hydrothermal Systems: Physical, Chemical, Biological, and Geological Interactions; Humphris, S.E., Zierenberg, R.A., Mullineaud, L.S., Thompsen, R.E., Eds.; Geophysical Monograph, American Geophysical Union: Washington, DC, USA, 1995; Volume 91, pp. 273–296. [Google Scholar]
- Petersen, S.; Kuhn, K.; Kuhn, T.; Augustin, N.; Hekinian, R.; Franz, L.; Borowski, C. The geological setting of the ultramafic-hosted Logatchev hydrothermal field (14°14′50″ N, Mid-Atlantic Ridge) and its influence on massive sulfide formation. Lithos 2009, 112, 40–56. [Google Scholar] [CrossRef]
- Campbell, B.C.; Engel, A.S.; Porter, M.L.; Takai, K. The versatile epsilon-proteobacteria: Key players in sulfidic habitats. Nat. Rev. Microbiol. 2006, 4, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Takai, K.; Campbell, B.J.; Cary, S.C.; Suzuki, M.; Oida, H.; Nunoura, T.; Hirayama, H.; Nakagawa, S.; Suzuki, Y.; Inagaki, F.; et al. Enzymatic and genetic characterization of carbon and energy metabolisms by deep-sea hydrothermal chemolithoautotrophic isolates of Epsilonproteobacteria. Appl. Environ. Microbiol. 2005, 71, 7310–7320. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Takai, K.; Inagaki, F.; Hirayama, H.; Nunoura, T.; Horikoshi, K.; Sako, Y. Distribution, phylogenetic diversity and physiological characteristics of epsilonproteobacteria in a deep-sea hydrothermal field. Environ. Microbiol. 2005, 7, 1619–1632. [Google Scholar] [CrossRef] [PubMed]
- Thiel, V.; Hügler, M.; Blümel, M.; Baumann, H.I.; Gärtner, A.; Schmaljohann, R.; Strauss, H.; Garbe-Schönberg, D.; Petersen, S.; Cowart, D.A.; et al. Widespread occurrence of two carbon fixation pathways in tubeworm endosymbionts: Lessons from hydrothermal vent associated tubeworms from the Mediterranean Sea. Front. Microbiol. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Markert, S.; Arndt, C.; Felbeck, H.; Becher, D.; Sievert, S.M.; Hügler, M.; Albrecht, D.; Robidart, J.; Bench, S.; Feldman, R.A.; et al. Physiological proteomics of the uncultured endosymbiont of Riftia pachyptila. Science 2007, 315, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Goffredi, S.K. Indigenous ectosymbiotic bacteria associated with diverse hydrothermal vent invertebrates. Environ. Microbiol. Rep. 2010, 2, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Könneke, M.; Schubert, D.M.; Brown, P.C.; Hügler, M.; Standfest, S.; Schwander, T.; Schada von Borzyskowski, L.; Erb, T.J.; Stahl, D.A.; Berg, I.A. Ammonia-oxidizing archaea use the most energy-efficient aerobic pathway for CO2 fixation. Proc. Natl. Acad. Sci. USA 2014, 111, 8239–8244. [Google Scholar] [CrossRef] [PubMed]
- Pfennig, N. Eine vollsynthetische Nährlösung zur selektiven Anreicherung einiger Schwefelpurpurbakterien. Naturwissenschaften 1961, 48, 136–137. [Google Scholar] [CrossRef]
- Pfennig, N. Anreicherungskulturen für rote und grüne Schwefelbakterien. Zentralbl. Bakt. Parasitenk Abt. 1 1965, 179–189 (Suppl. S1), 503–505. [Google Scholar]
- Pfennig, N.; Trüper, H.G. Isolation of members of the families Chromatiaceae and Chlorobiaceae. In The Prokaryotes, 1st ed.; Starr, M.P., Stolp, H., Trüper, H.G., Balows, A., Schlegel, H.G., Eds.; Springer: New York, NY, USA, 1981; pp. 279–289. [Google Scholar]
- Giovannoni, S.J.; Stingl, U. The importance of culturing bacterioplankton in the omics age. Nat. Rev. Microbiol. 2007, 5, 820–826. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Biogeochemical Fucnction | Gene Product | Primer Name | Primer Sequence (5'-3') | Reference |
|---|---|---|---|---|---|
| pufLM | photosynthesis | photosynthetic reaction center | pufL67F | TTCGACTTYTGGRTNGGNCC | [48] |
| pufM781R | CCAKSGTCCAGCGCCAGAANA | [48] | |||
| fmoA | photosynthesis | bacteriochlorophyll-a-protein | F-Start-fmo-modif | ATT ATG GCT CTN TTC GGC | [75] |
| r-889-FMO | CCGACCATNCCGTGRTG | [47] | |||
| bchlY | photosynthesis | chlorophyllide reductase | bchY-fwd | CCNCARACNATGTGYCCNGCNTTYGG | [19] |
| bchY-rev | GGRTCNRCNGGRAANATYTCNCCC | [19] | |||
| soxB | “sulfur” oxidation | sulfate thioesterase | soxB432F | GAYGGNGGNGAYACNTGG | [96] |
| soxB1446B | CATGTCNCCNCCRTGYTG | [96] | |||
| aprA | “sulfur” oxidation | APS reductase | AprA-1-FW | TGGCAGATCATGATYMATGG | [20] |
| sulfur reduction | AprA-5-rv | GCGCCAACYGGRCCTTA | [20] | ||
| dsrAB | “sulfur” oxidation | dissimilatory disulfite reductase | rDSR1Fa | AARGGNTAYTGGAARG | [22] |
| sulfur reduction | rDSR1Fb | TTYGGNTAYTGGAARG | [22] | ||
| rDSR1Fc | ATGGGNTAYTGGAARG | [22] | |||
| rDSR4Ra | CCRAARCAIGCNCCRCA | [22] | |||
| rDSR4Rb | GGRWARCAIGCNCCRCA | [22] | |||
| narH | nitrate reduction | dissimilatory nitrate reductase | narH50F | AARTGYATCGGYTGCCA | [8] |
| denitrification | narH1040B | GTNCGRTYTCNGG | [8] | ||
| narH403F | GGNCCNAACTGGGNGA | [8] | |||
| narH403B | TCNTCCCAGTTNGGNCC | [8] | |||
| nirS | nitrate reduction | dissimilatory nitrate reductase | nirS1F | CCTAYTGGCCGCCRCART | [123] |
| denitrification | nirS6R | CGTTGAACTTRCCGGT | [123] | ||
| nirK | nitrate reduction | dissimilatory nitrate reductase | nirK1F | GGMATGGTKCCSTGGCA | [123] |
| denitrification | nirK5R | GCCTCGATCAGRTTRTGG | [123] | ||
| Cunir3 | CGTCTAYCAYTCCGCVCC | [128] | |||
| Cunir4 | GCCTCGATCAGRTTRTGG | [128] | |||
| amoA | ammonia oxidation | ammonia monooxygenase | amoA-1F | GGGGTTTCTACTGGTGGT | [129] |
| amoA-2R | CCCCTCKGSAAAGCCTTCTTC | [129] | |||
| amoCAB | ammonia oxidation | ammonia monooxygenase | amoC58f | CTAYGACATGTCRCTGTGG | [130] |
| amoB1179r | CCAAARCGRCTTTCCGG | [130] | |||
| amoA | ammonia oxidation | archaeal | Arch-amoAF | STAATGGTCTGGCTTAGACG | [131] |
| ammonia monooxygenase | Arch-amoAR | GCGGCCATCCATCTGTATGT | [131] | ||
| anirKa | archaeal | anirKa-58F | ACBYTATTCGGAAGYACATACACA | [132] | |
| dissimilatory nitrate reductase | anirKa-579R | GYMATTCCGTACATKCCGGA | [132] | ||
| anirKb | archaeal | anirKb-61F | CTATTCGGARGTWCTTTYACTGC | [132] | |
| dissimilatory nitrate reductase | anirKa-555R | ACGTGTTGGTCCATTGCTGC | [132] | ||
| pmoAC | methane oxidation | methane monooxygenase | pmoC617f | ACACCTTCTGGTTCATGG | [133] |
| pmoA682r | GAASGCNGAGAAGAASGC | [133] | |||
| napA | nitrate reduction | periplasmic nitrate reductase | NapV16F | GCNCCNTGYMGNTTYTGYGG | [120] |
| NapV17R | RTGYTGRTTRAANCCCATNGTCCA | [120] |
© 2016 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Imhoff, J.F. New Dimensions in Microbial Ecology—Functional Genes in Studies to Unravel the Biodiversity and Role of Functional Microbial Groups in the Environment. Microorganisms 2016, 4, 19. https://doi.org/10.3390/microorganisms4020019
Imhoff JF. New Dimensions in Microbial Ecology—Functional Genes in Studies to Unravel the Biodiversity and Role of Functional Microbial Groups in the Environment. Microorganisms. 2016; 4(2):19. https://doi.org/10.3390/microorganisms4020019
Chicago/Turabian StyleImhoff, Johannes F. 2016. "New Dimensions in Microbial Ecology—Functional Genes in Studies to Unravel the Biodiversity and Role of Functional Microbial Groups in the Environment" Microorganisms 4, no. 2: 19. https://doi.org/10.3390/microorganisms4020019