


First Genome Sequence of the Microcolonial Black Fungus Saxispiralis lemnorum MUM 23.14: Insights into the Unique Genomic Traits of the Aeminiaceae Family



Abstract
:1. Introduction
2. Materials and Methods
2.1. Fungal Strain Isolation, DNA Extraction, and Whole-Genome Sequencing
2.2. Genome Assembly, Annotation, and Functional Analysis
2.3. Comparative Genomic Analysis
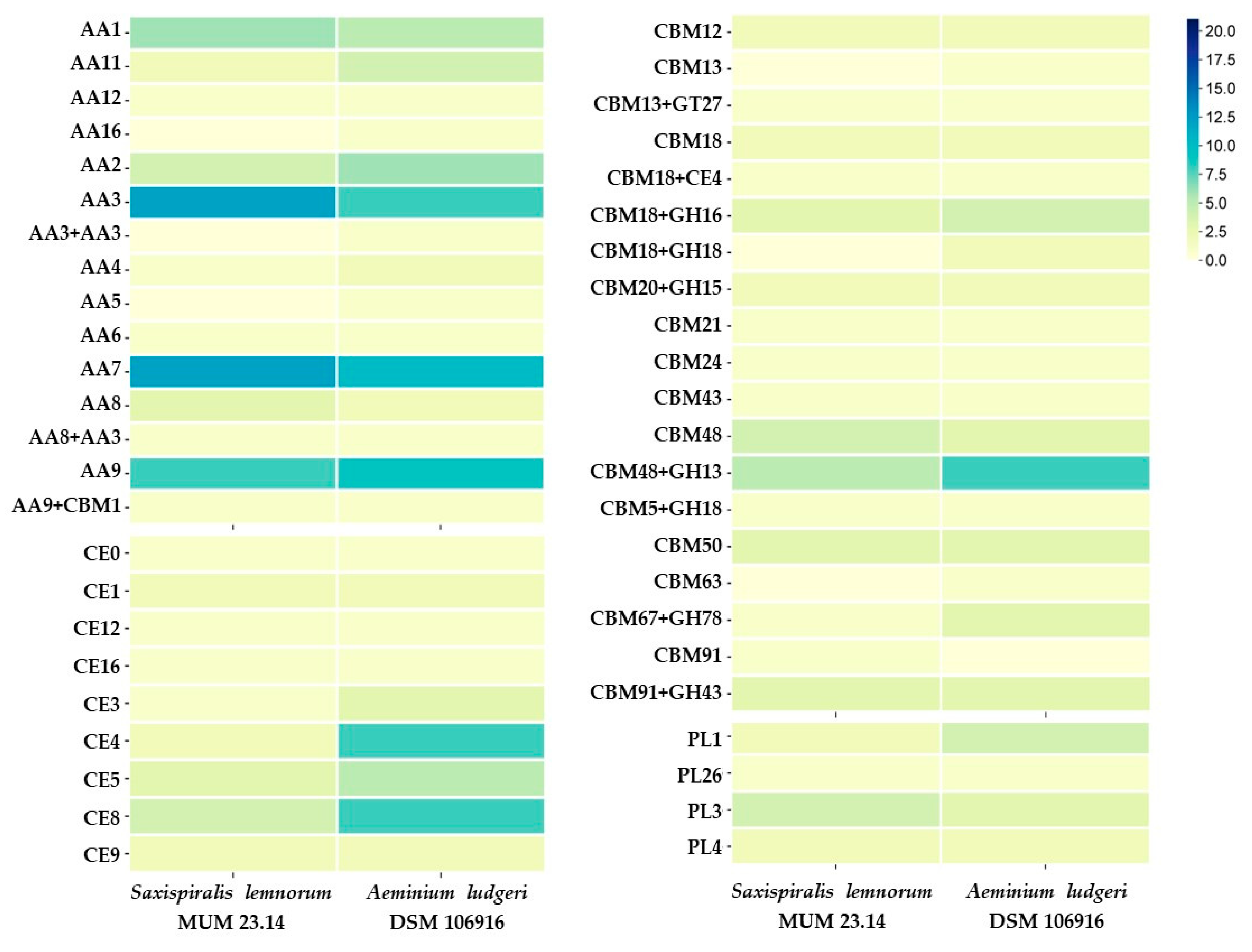
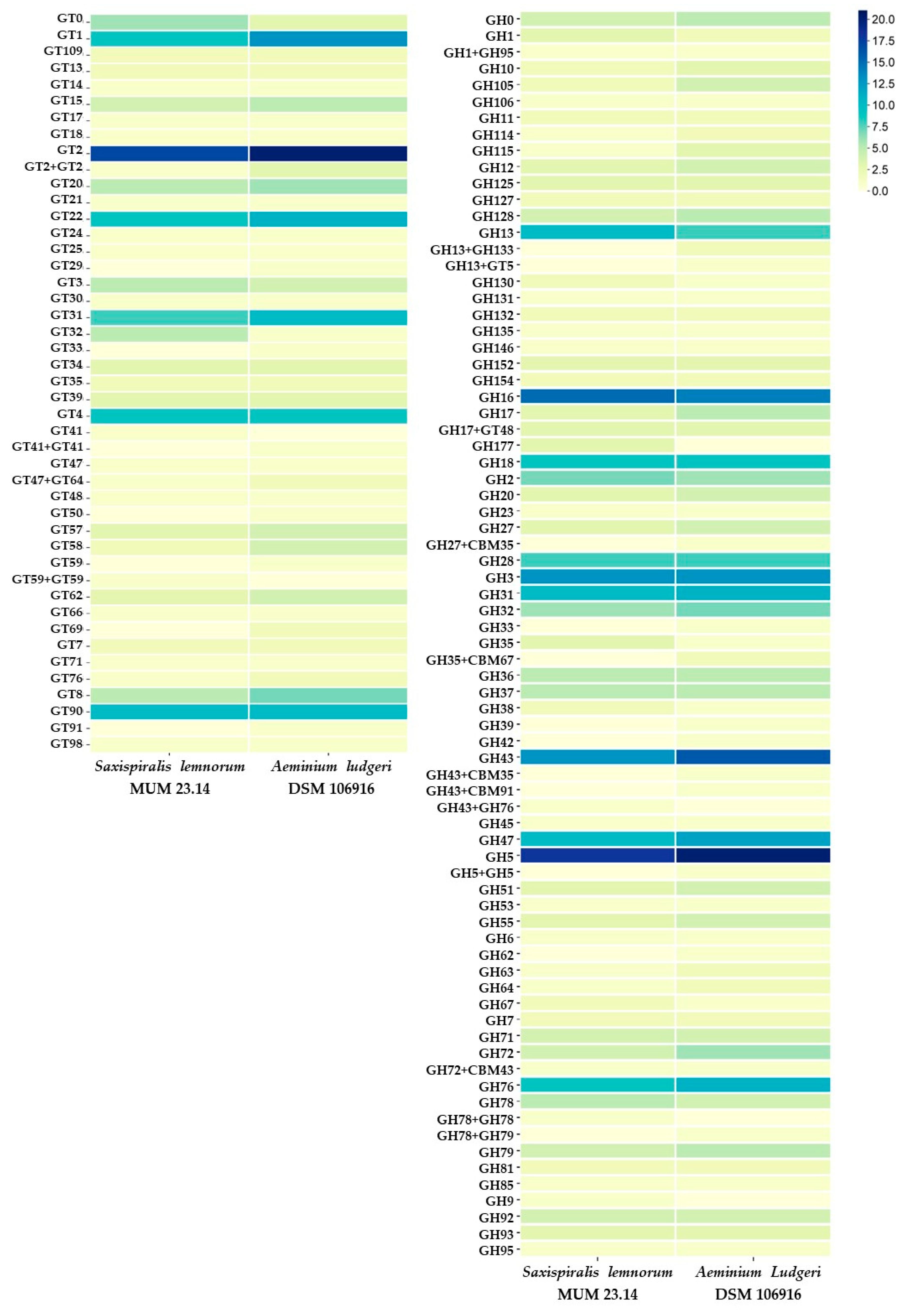
3. Results and Discussion
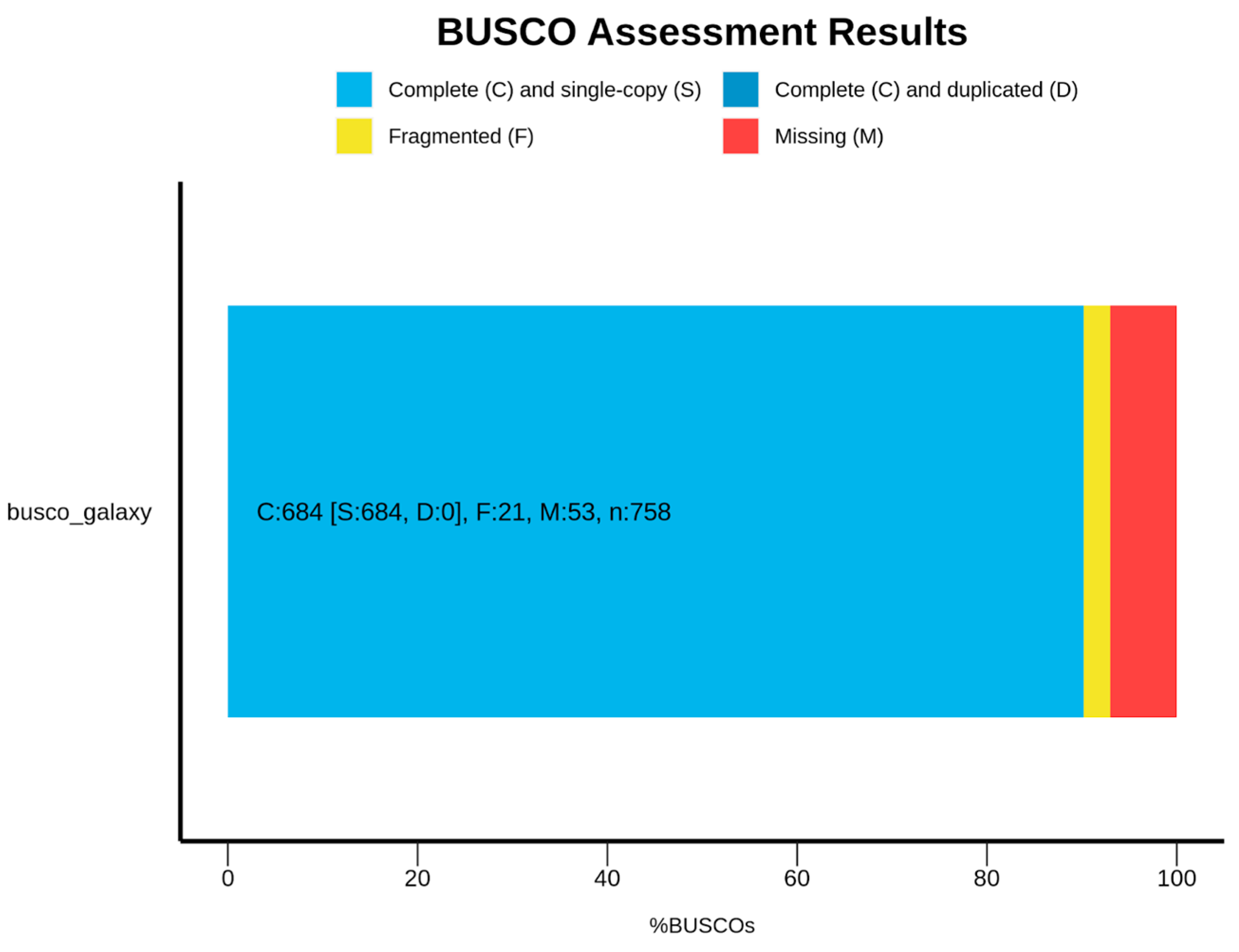
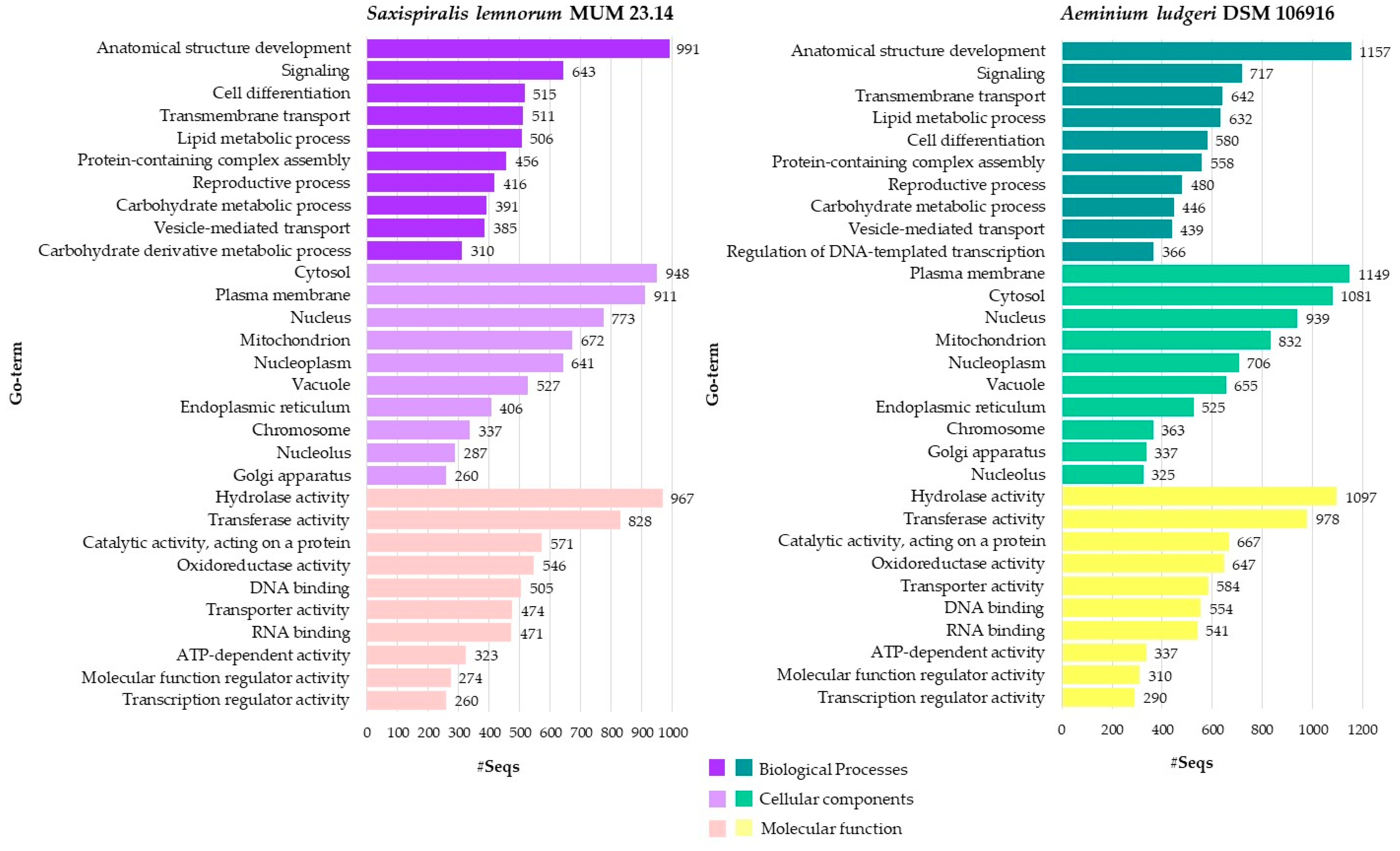
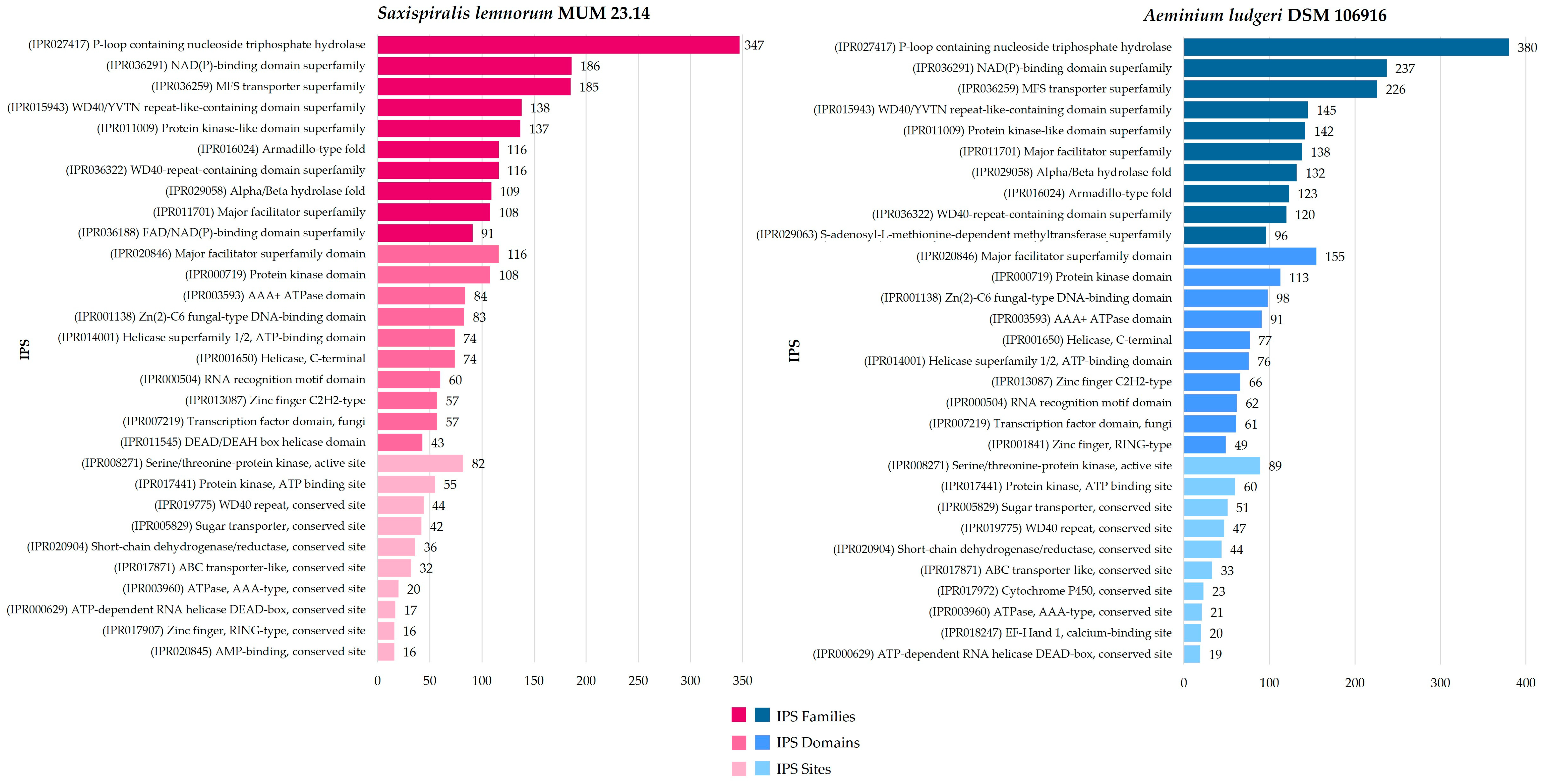
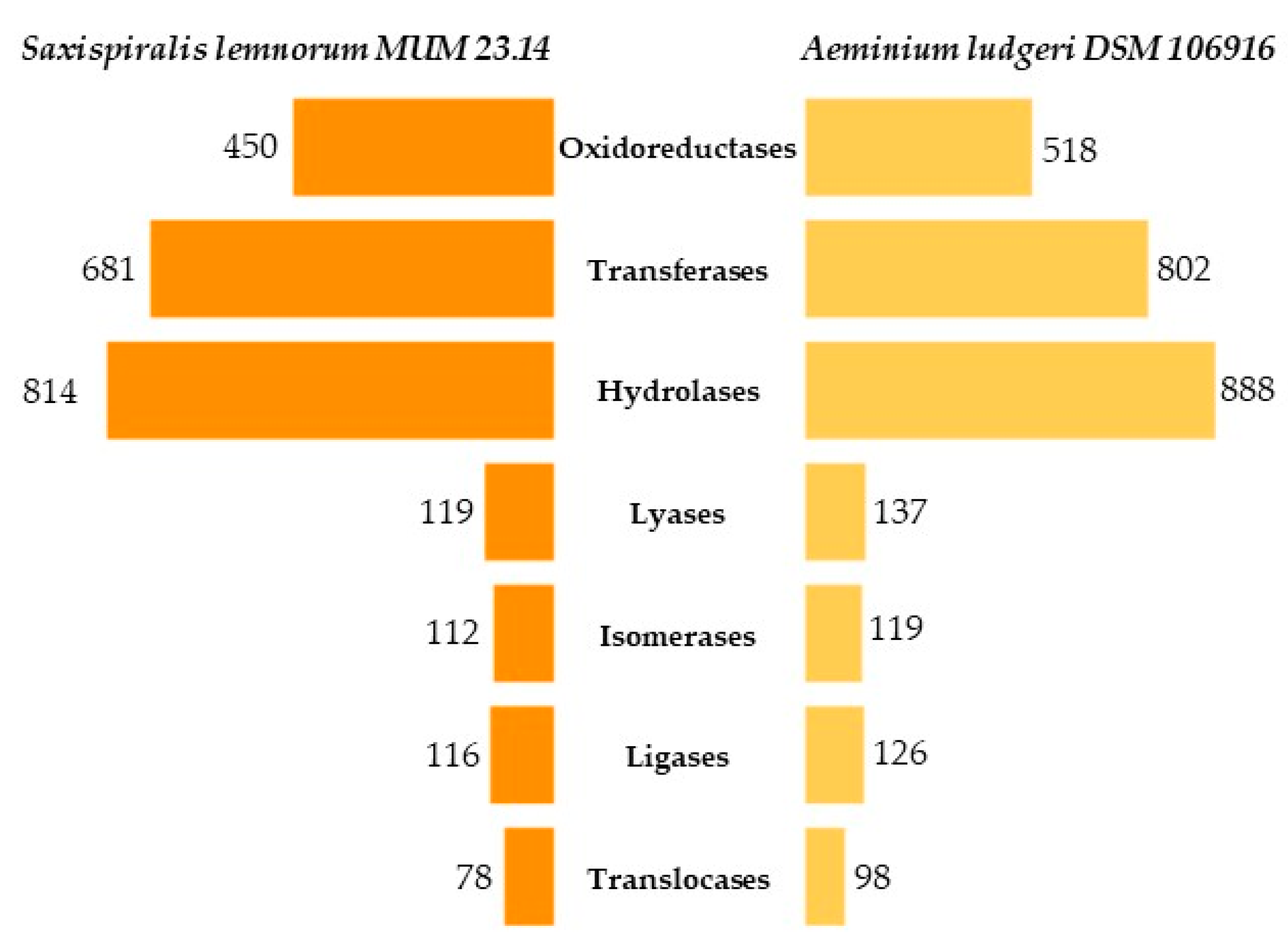
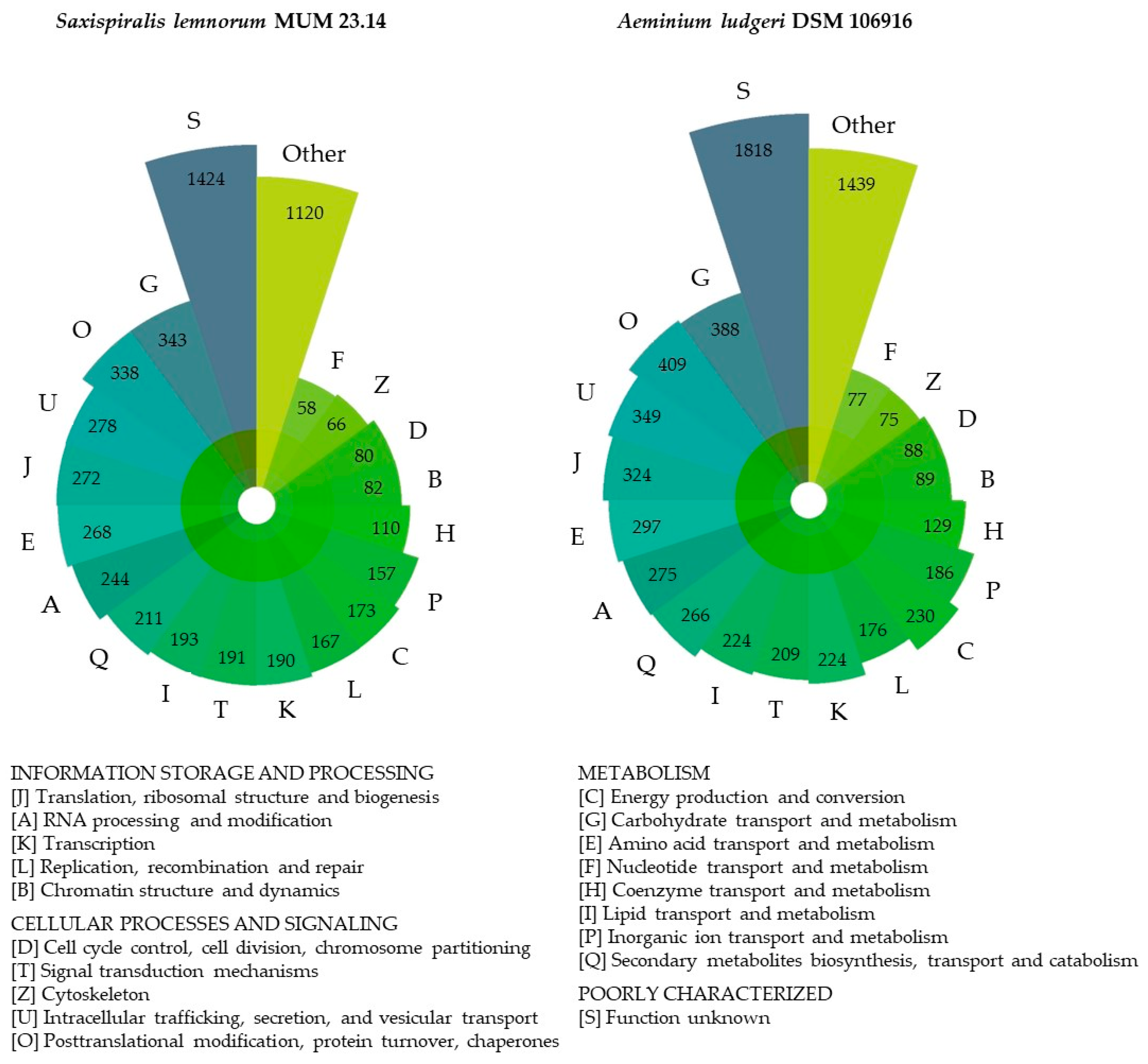
3.1. Saxispiralis lemnorum MUM 23.14 Genome Assembly, Annotation, and Funtional Characterization
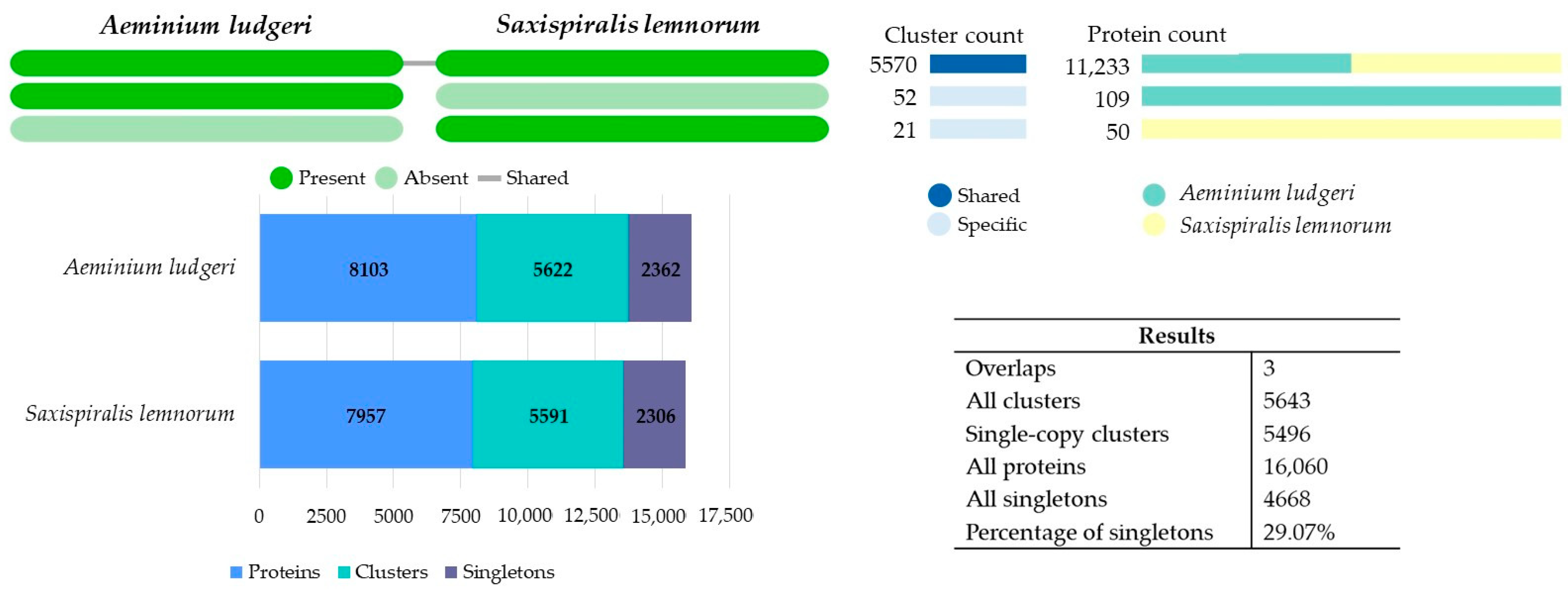
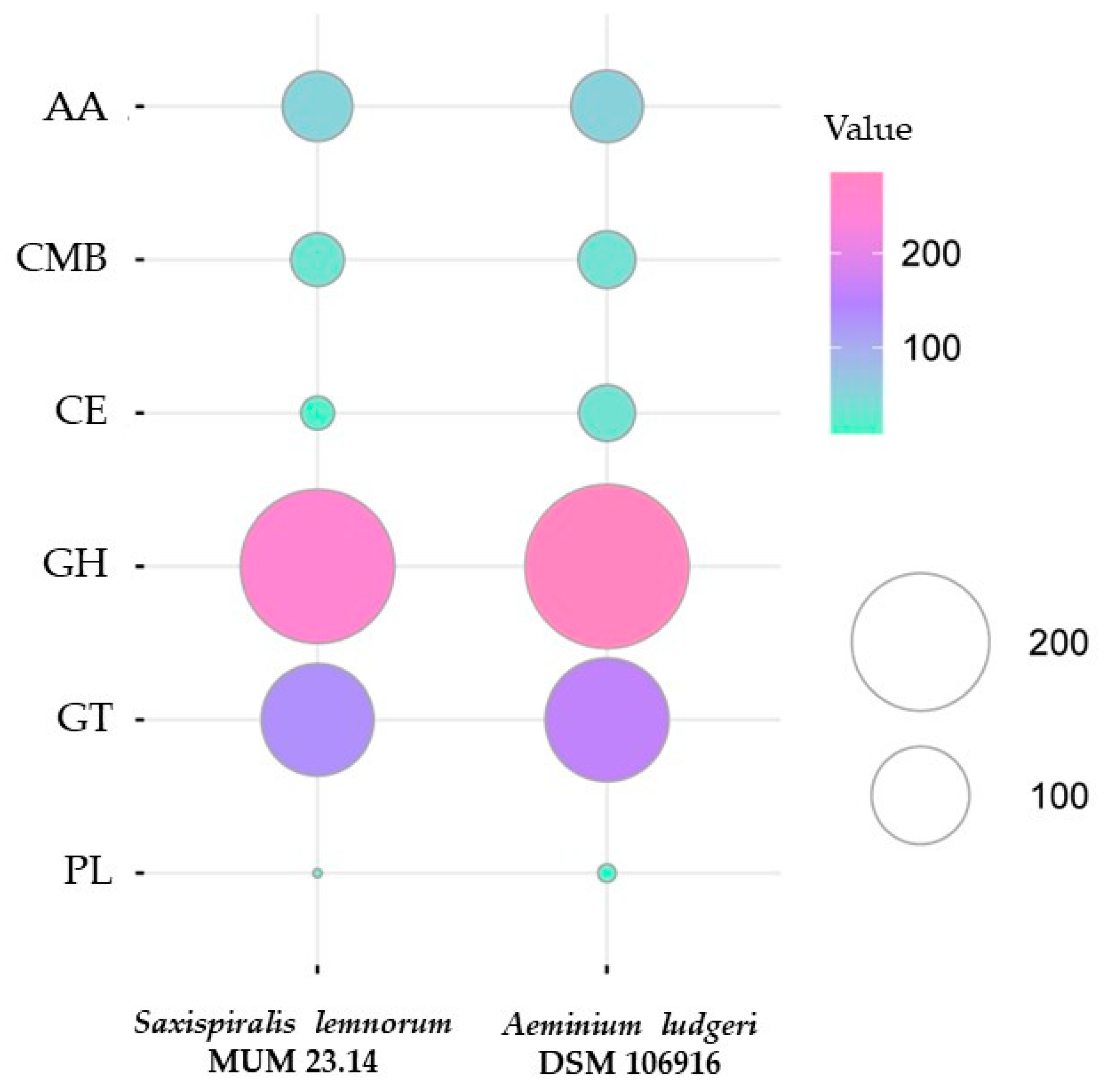
3.2. Aeminiaceae Comparative Genomic Analysis
3.2.1. Comparative KEGG Analysis and Stress-Related Genes
3.2.2. Comparative OrthoVenn3 Analysis
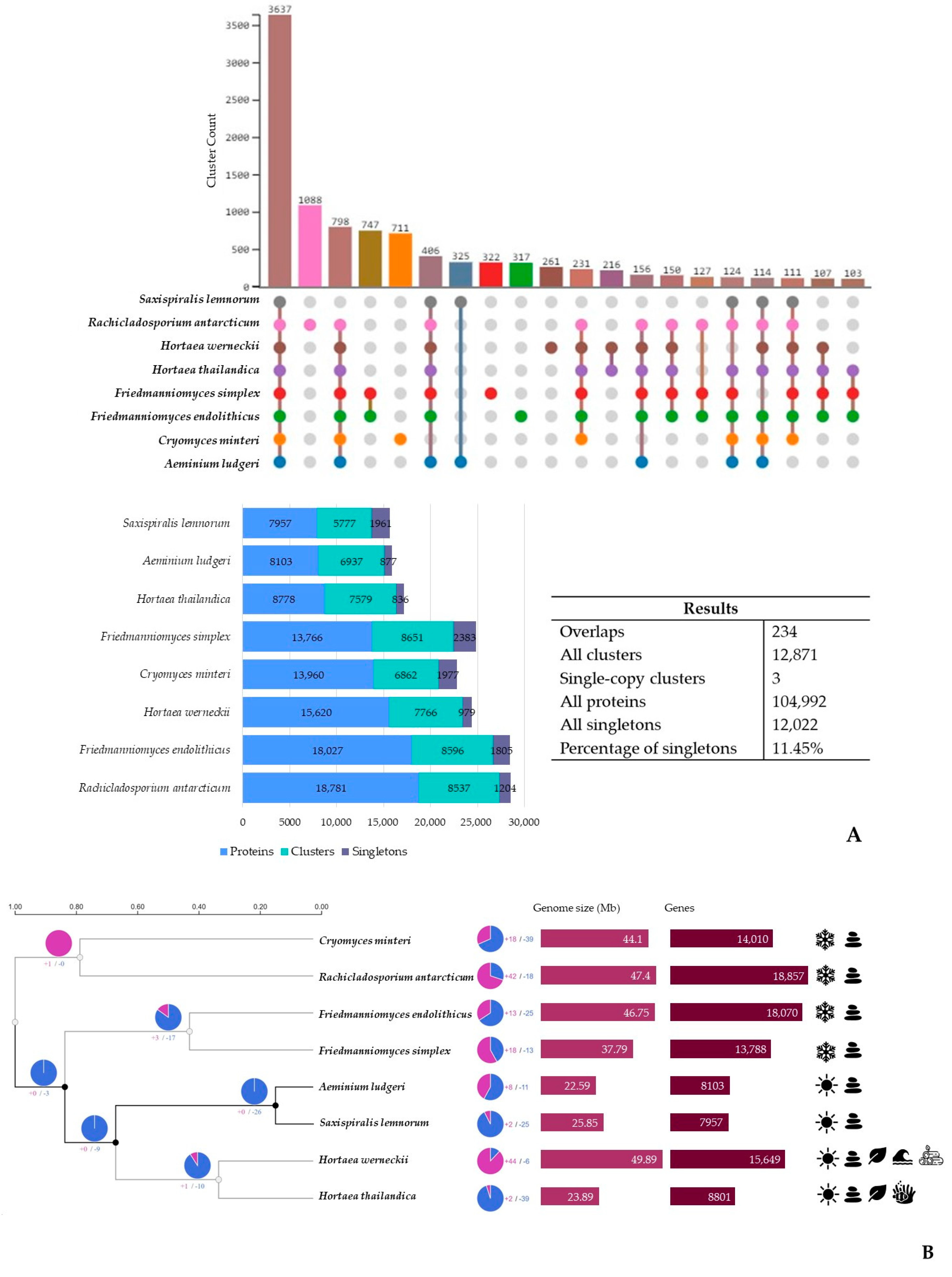
3.3. Additional Poikilotolerant Black Fungal Species Comparative Genomic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway | Protein | E.C. | S. lemnorum | A. ludgeri |
|---|---|---|---|---|
| Nitrogen metabolism | Nitrate Reductase (NAD(P)H) | 1.7.1.1; 1.7.1.2; 1.7.1.3 | ✓ | ✓ |
| Nitrite Reductase (NAD(P)H) | 1.7.1.4 | ✓ | ✓ | |
| Sulfur metabolism | Sulfate Adenylyltransferase | 2.7.7.4 | ✓ | ✓ |
| Bifunctional Enzyme CysN/CysC | 2.7.7.4; 2.7.1.25 | ✓ | ✓ | |
| Phosphoadenosine Phosphosulfate Reductase | 1.8.4.8; 1.8.4.10 | ✓ | ✓ | |
| Sulfite Reductase (NADPH) Flavoprotein Alpha-Component | 1.8.1.2 | ✓ | ✓ | |
| Terpenoid backbone biosynthesis | Prenylcysteine Oxidase/ Farnesylcysteine Lyase | 1.8.3.5; 1.8.3.6 | ✓ | ✓ |
| NAD+-Dependent Farnesol Dehydrogenase | 1.1.1.354 | - | ✓ | |
| NADP+-Dependent Farnesol Dehydrogenase | 1.1.1.216 | ✓ | - | |
| Carotenoid biosynthesis | Phytoene Desaturase (3,4-didehydrolycopene-forming) | 1.3.99.30 | ✓ | ✓ |
| 15-cis-Phytoene Synthase/ Lycopene beta-Cyclase | 2.5.1.32; 5.5.1.19 | - | ✓ | |
| Torulene Dioxygenase | 1.13.11.59 | - | ✓ | |
| beta-apo-4′-Carotenal Oxygenase | 1.2.1.82 | ✓ | ✓ | |
| DOPA-melanin pathway | Tyrosinase | 1.14.18.1 | ✓ | ✓ |
| Cytoplasmic Aspartate Aminotransferase | 2.6.1.1 | ✓ | ✓ | |
| Tyrosine Aminotransferase | 2.6.1.5 | ✓ | ✓ | |
| L-tyrosine degradation pathway | Histidinol-Phosphate Aminotransferase | 2.6.1.9 | ✓ | ✓ |
| Aromatic-Amino-Acid Transaminase | 2.6.1.57 | ✓ | ✓ | |
| 4-Hydroxyphenylpyruvate Dioxygenase | 1.13.11.27 | ✓ | ✓ | |
| Starch and sucrose metabolism | Glycogen Phosphorylase | 2.4.1.1 | ✓ | ✓ |
| UTP-A-Glucose-1-Phosphate Uridylyltransferase | 2.7.7.9 | ✓ | ✓ | |
| Trehalose 6-Phosphate Synthase | 2.4.1.15; 2.4.1.347 | ✓ | ✓ | |
| Trehalose 6-Phosphate Phosphatase | 3.1.3.12 | ✓ | ✓ |
| Stress Response | Gene | S. lemnorum | A. ludgeri |
|---|---|---|---|
| Oxidative and Heat Stress | Cell surface superoxide dismutase [Cu-Zn] 6 | - | LOCUSTAG 000040; LOCUSTAG 000737 |
| Catalase A | - | LOCUSTAG 000980 | |
| Catalase-1 | NODE_958 | - | |
| Peroxisomal Catalase | NODE_9; NODE_139 | - | |
| Probable Ras-related protein Rab7 | - | LOCUSTAG 001245 | |
| Ras-related protein ced-10 | NODE_1791 | - | |
| Ras-related protein Rab-2A | NODE_15 | - | |
| Ras-related protein Rab-8A | NODE_38 | - | |
| Ras-related protein RSR1 | NODE_75 | - | |
| Superoxide dismutase [Mn], mitochondrial | NODE_4; NODE_1087 | LOCUSTAG 001431; LOCUSTAG 006179 | |
| Cell surface Cu-only superoxide dismutase ARB | NODE_1080 | LOCUSTAG 003114 | |
| Mitogen-activated protein kinase Hog1 | NODE_87 | LOCUSTAG 006209 | |
| Superoxide dismutase [Cu-Zn] | NODE_9; NODE_48; NODE_86 | LOCUSTAG 004510 | |
| WSC domain-containing protein ARB 07870 | NODE_7 | - | |
| Heat shock 70 | - | LOCUSTAG 000326 | |
| Heat shock 70 protein 2 | NODE_3 | LOCUSTAG 000548 | |
| Heat shock protein 78, mitochondrial | NODE_261 | LOCUSTAG 000712 | |
| Heat shock protein 9/12 | - | LOCUSTAG 003049 | |
| 30 kDa heat shock protein | NODE_27 | LOCUSTAG 003145 | |
| Heat shock protein 90 | NODE_96 | LOCUSTAG 003389 | |
| Heat shock protein 70-like protein lhs1 | - | LOCUSTAG 003932 | |
| Heat shock protein hsp98 | NODE_39 | LOCUSTAG 003953 | |
| Heat shock protein hsp88 | - | LOCUSTAG 004025 | |
| Heat shock protein sti1-like protein | - | LOCUSTAG 005642 | |
| Heat shock protein sti1 homolog | NODE_72 | - | |
| 10 kDa heat shock protein, mitochondrial | - | LOCUSTAG 006313 | |
| Heat shock protein 60 | NODE_101 | LOCUSTAG 006680 | |
| Heat shock factor protein | - | LOCUSTAG 006684 | |
| Heat shock factor protein 2 | NODE_101 | - | |
| Heat shock factor binding protein 1 | - | LOCUSTAG 006956 | |
| Domain-containing protein ARB | NODE_7 | - | |
| Acidic Stress | Calcium-transporting ATPase 1 | - | LOCUSTAG 003279; LOCUSTAG 006615 |
| Calcium-transporting ATPase 2 | NODE_4 | LOCUSTAG 000733 | |
| Calcium-transporting ATPase 3 | - | LOCUSTAG 006658 | |
| Calcium-transporting ATPase pmrA | NODE_99 | - | |
| Osmotic Stress | alpha-1,2-mannosyltransferase | NODE_10; NODE_35; NODE_46 | LOCUSTAG 003717 |
| alpha-1,3-mannosyltransferase | - | LOCUSTAG 007040; LOCUSTAG 007816 | |
| alpha-1,3/1,6-mannosyltransferase alg-2 | NODE_103 | - | |
| alpha-1,6-mannosyltransferase | - | LOCUSTAG 008081; LOCUSTAG 000265; LOCUSTAG 003765 | |
| Initiation-specific alpha-1,6-mannosyltransferase | NODE_2; NODE_11 | - | |
| Probable alpha-1,6-mannosyltransferase MNN10 | NODE_4 | - | |
| 26S proteasome regulatory subunit RPN7 | - | LOCUSTAG 007830 | |
| Probable 26S proteasome regulatory subunit RPN7 | NODE_152 | - | |
| Adenosylhomocysteinase | NODE_9 | LOCUSTAG 001515 | |
| UV-A Stress | Deoxyribodipyrimidine photo-lyase | NODE_10; NODE_1685 | LOCUSTAG 001515; LOCUSTAG 006406 |
References
- Paiva, D.S.; Fernandes, L.; Trovão, J.; Mesquita, N.; Tiago, I.; Portugal, A. Uncovering the Fungal Diversity Colonizing Lime-stone Walls of a Forgotten Monument in the Central Region of Portugal by High-Throughput Sequencing and Culture-Based Methods. Appl. Sci. 2022, 12, 10650. [Google Scholar] [CrossRef]
- Paiva, D.S.; Trovão, J.; Fernandes, L.; Mesquita, N.; Tiago, I.; Portugal, A. Expanding the microcolonial black fungi Aeminiaceae family: Saxispiralis lemnorum gen. et sp. nov. (Mycosphaerellales), isolated from deteriorated limestone in the Lemos Pantheon, Portugal. J. Fungi 2023, 9, 916. [Google Scholar] [CrossRef]
- Trovão, J.; Tiago, I.; Soares, F.; Paiva, D.S.; Mesquita, N.; Coelho, C.; Catarino, L.; Gil, F.; Portugal, A. Description of Aeminiaceae fam. nov., Aeminium gen. nov. and Aeminium ludgeri sp. nov. (Capnodiales), isolated from a biodeteriorated art-piece in the old cathedral of Coimbra, Portugal. MycoKeys 2019, 45, 57–73. [Google Scholar] [CrossRef]
- Liu, B.; Fu, R.; Wu, B.; Liu, X.; Xiang, M. Rock-inhabiting fungi: Terminology, diversity, evolution, and adaptation mechanisms. Mycology 2021, 13, 1–31. [Google Scholar] [CrossRef]
- De Leo, F.; Marchetta, A.; Urzì, C. Black Fungi on Stone-Built Heritage: Current Knowledge and Future Outlook. Appl. Sci. 2022, 12, 3969. [Google Scholar] [CrossRef]
- Gorbushina, A.A.; Whitehead, K.; Dornieden, T.; Niesse, A.; Schulte, A.; Hedges, J.I. Black fungal colonies as units of survival: Hyphal mycosporines synthesized by rock-dwelling microcolonial fungi. Can. J. Bot. 2003, 81, 131–138. [Google Scholar] [CrossRef]
- Butinar, L.; Sonjak, S.; Zalar, P.; Plemenitaš, A.; Gunde-Cimerman, N. Melanized halophilic fungi are eukaryotic members of microbial communities in hypersaline waters of solar salterns. Bot. Mar. 2005, 48, 73–79. [Google Scholar] [CrossRef]
- Selbmann, L.; De Hoog, G.S.; Mazzaglia, A.; Friedmann, E.I.; Onofri, S. Fungi at the edge of life: Cryptoendolithic black fungi from Antarctic desert. Stud. Mycol. 2005, 51, 1–32. [Google Scholar]
- Gorbushina, A.A.; Kotlova, E.R.; Sherstneva, O.A. Cellular responses of microcolonial rock fungi to long-term desiccation and subsequent rehydration. Stud. Mycol. 2008, 61, 91–97. [Google Scholar] [CrossRef]
- Zakharova, K.; Tesei, D.; Marzban, G.; Dijksterhuis, J.; Wyatt, T.; Sterflinger, K. Microcolonial fungi on rocks: A life in constant drought? Mycopathologia 2013, 175, 537–547. [Google Scholar] [CrossRef]
- Selbmann, L.; Zucconi, L.; Isola, D.; Onofri, S. Rock black fungi: Excellence in the extremes, from the Antarctic to space. Curr. Genet. 2015, 61, 335–345. [Google Scholar] [CrossRef]
- Tesei, D. Black Fungi Research: Out-of-This-World Implications. Encyclopedia 2022, 2, 212–229. [Google Scholar] [CrossRef]
- Onofri, S.; Zucconi, L.; Isola, D.; Selbmann, L. Rock-inhabiting fungi and their role in deterioration of stone monuments in the Mediterranean area. Plant Biosyst. 2014, 148, 384–391. [Google Scholar] [CrossRef]
- Sterflinger, K. Fungi: Their role in deterioration of cultural heritage. Fungal Biol. Rev. 2010, 24, 47–55. [Google Scholar] [CrossRef]
- Sterflinger, K.; Piñar, G. Microbial deterioration of cultural heritage and works of art—Tilting at windmills? Appl. Microbiol. Biotechnol. 2013, 97, 9637–9646. [Google Scholar] [CrossRef]
- Ametrano, C.G.; Grewe, F.; Crous, P.W.; Goodwin, S.B.; Liang, C.; Selbmann, L.; Lumbsch, H.T.; Leavitt, S.D.; Muggia, L. Genome-scale data resolve ancestral rock-inhabiting lifestyle in Dothideomycetes (Ascomycota). IMA Fungus 2019, 10, 19. [Google Scholar] [CrossRef]
- Sinha, S.; Flibotte, S.; Neira, M.; Formby, S.; Plemenitaš, A.; Cimerman, N.G.; Lenassi, M.; Gostinčar, C.; Stajich, J.E.; Nislow, C. Insight into the Recent Genome Duplication of the Halophilic Yeast Hortaea werneckii: Combining an Improved Genome with Gene Expression and Chromatin Structure. G3 2017, 7, 2015–2022. [Google Scholar] [CrossRef]
- Jalili, V.; Afgan, E.; Gu, Q.; Clements, D.; Blankenberg, D.; Goecks, J.; Taylor, J.; Nekrutenko, A. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2020 update. Nucleic Acids Res. 2020, 48, W395–W402. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. MetaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef]
- Alonge, M.; Soyk, S.; Ramakrishnan, S.; Wang, X.; Goodwin, S.; Sedlazeck, F.J.; Lippman, Z.B.; Schatz, M.C. RaGOO: Fast and accurate reference-guided scaffolding of draft genomes. Genome Biol. 2019, 20, 224. [Google Scholar] [CrossRef]
- Trovão, J.; Tiago, I.; Soares, F.; Paiva, D.S.; Mesquita, N.; Coelho, C.; Catarino, L.; Gil, F.; Portugal, A. High-Quality Draft Genome Sequence of the Microcolonial Black Fungus Aeminium ludgeri DSM 106916. Microbiol. Resour. Announc. 2019, 8, e00202-19. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Kriventseva, E.V.; Kuznetsov, D.; Tegenfeldt, F.; Manni, M.; Dias, R.; Simão, F.A.; Zdobnov, E.M. OrthoDB v10: Sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Res. 2019, 47, D807–D811. [Google Scholar] [CrossRef]
- Seemann, T. Barrnap 0.7: Rapid Ribosomal RNA Prediction. 2013. Available online: https://github.com/tseemann/barrnap (accessed on 21 August 2023).
- Laslett, D.; Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef]
- Stanke, M.; Morgenstern, B. AUGUSTUS: A web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005, 33, W465–W467. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. EggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Blum, M.; Chang, H.Y.; Chuguransky, S.; Grego, T.; Kandasaamy, S.; Mitchell, A.; Nuka, G.; Paysan-Lafosse, T.; Qureshi, M.; Raj, S.; et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 2021, 49, D344–D354. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef]
- Aramaki, T.; Blanc-Mathieu, R.; Endo, H.; Ohkubo, K.; Kanehisa, M.; Goto, S.; Ogata, H. KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 2020, 36, 2251–2252. [Google Scholar] [CrossRef]
- Zheng, J.; Ge, Q.; Yan, Y.; Zhang, X.; Huang, L.; Yin, Y. dbCAN3: Automated carbohydrate-active enzyme and substrate annotation. Nucleic Acids Res. 2023, 51, W115–W121. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; van Wezel, G.P.; Medema, M.H.; Weber, T. AntiSMASH 6.0: Improving Cluster Detection and Comparison Capabilities. Nucleic Acids Res. 2021, 49, W29–W35. [Google Scholar] [CrossRef]
- Sun, J.; Lu, F.; Luo, Y.; Bie, L.; Xu, L.; Wang, Y. OrthoVenn3: An integrated platform for exploring and visualizing orthologous data across genomes. Nucleic Acids Res. 2003, 51, W397–W403. [Google Scholar] [CrossRef]
- Li, L.; Stoeckert, C.J.; Roos, D.S. OrthoMCL: Identification of Ortholog Groups for Eukaryotic Genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef]
- Mendes, F.K.; Vanderpool, D.; Fulton, B.; Hahn, M.W. CAFE 5 models’ variation in evolutionary rates among gene families. Bioinformatics 2020, 36, 5516–5518. [Google Scholar] [CrossRef] [PubMed]
- Coleine, C.; Masonjones, S.; Sterflinger, K.; Onofri, S.; Selbmann, L.; Stajich, J.E. Peculiar genomic traits in the stress-adapted cryptoendolithic Antarctic fungus Friedmanniomyces endolithicus. Fungal Biol. 2020, 124, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Medema, M.H.; Kottmann, R.; Yilmaz, P.; Cummings, M.; Biggins, J.B.; Blin, K.; de Bruijn, I.; Chooi, Y.H.; Claesen, J.; Coates, R.C.; et al. Minimum Information about a Biosynthetic Gene cluster. Nat. Chem. Biol. 2015, 11, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Kautsar, S.A.; Blin, K.; Shaw, S.; Navarro-Muñoz, J.C.; Terlouw, B.R.; van der Hooft, J.J.J.; van Santen, J.A.; Tracanna, V.; Suarez Duran, H.G.; Pascal Andreu, V.; et al. MIBiG 2.0: A repository for biosynthetic gene clusters of known function. Nucleic Acids Res. 2020, 48, D454–D458. [Google Scholar] [CrossRef]
- Yang, S.L.; Chung, K.R. Transcriptional regulation of elsinochrome phytotoxin biosynthesis by an EfSTE12 activator in the citrus scab pathogen Elsinoë fawcettii. Fungal Biol. 2010, 114, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, M.M.; Moreno, L.F.; Stielow, B.J.; Muszewska, A.; Hainaut, M.; Gonzaga, L.; Abouelleil, A.; Patané, J.S.L.; Priest, M.; Souza, R.; et al. Exploring the genomic diversity of black yeasts and relatives (Chaetothyriales, Ascomycota). Stud. Mycol. 2017, 86, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Stajich, J.E. Fungal Genomes and Insights into the Evolution of the Kingdom. Microbiol. Spectr. 2017, 5. [Google Scholar] [CrossRef]
- Sterflinger, K.; Lopandic, K.; Pandey, R.V.; Blasi, B.; Kriegner, A. Nothing Special in the Specialist? Draft Genome Sequence of Cryomyces antarcticus, the Most Extremophilic Fungus from Antarctica. PLoS ONE 2014, 9, e109908. [Google Scholar] [CrossRef]
- Raina, M.; Ibba, M. tRNAs as regulators of biological processes. Front. Genet. 2014, 5, 171. [Google Scholar] [CrossRef]
- Pao, S.S.; Paulsen, I.T.; Saier, M.H. Major Facilitator Superfamily. Microbiol. Mol. Biol. Rev. 1998, 62, 1–34. [Google Scholar] [CrossRef]
- Martínez, A.T.; Speranza, M.; Ruiz-Dueñas, F.J.; Ferreira, P.; Camarero, S.; Guillén, F.; Martínez, M.J.; Gutiérrez, A.; del Río, J.C. Biodegradation of lignocellulosics: Microbial, chemical, and enzymatic aspects of the fungal attack of lignin. Int. Microbiol. 2005, 8, 195–204. [Google Scholar] [PubMed]
- Baldrian, P.; Valášková, V. Degradation of cellulose by basidiomycetous fungi. FEMS Microbiol. Rev. 2008, 32, 501–521. [Google Scholar] [CrossRef] [PubMed]
- Kirtzel, J.; Siegel, D.; Krause, K.; Kothe, E. Stone-Eating Fungi: Mechanisms in Bioweathering and the Potential Role of Laccases in Black Slate Degradation with the Basidiomycete Schizophyllum commune. In Advances in Applied Microbiology; Sariaslani, S., Gadd, G.M., Eds.; Academic Press: Cambridge, MA, USA, 2017; pp. 83–101. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for Glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef] [PubMed]
- De Vries, R.P.; Riley, R.; Wiebenga, A.; Aguilar-Osorio, G.; Amillis, S.; Uchima, C.A.; Anderluh, G.; Asadollahi, M.; Askin, M.; Barry, K.; et al. Comparative genomics reveals high biological diversity and specific adaptations in the industrially and medically important fungal genus Aspergillus. Genome Biol. 2017, 18, 28. [Google Scholar] [CrossRef] [PubMed]
- Rokas, A.; Mead, M.E.; Steenwyk, J.L.; Raja, H.A.; Oberlies, N.H. Biosynthetic gene clusters and the evolution of fungal chemodiversity. Nat. Prod. Rep. 2020, 37, 868–878. [Google Scholar] [CrossRef] [PubMed]
- Hoffmeister, D.; Keller, N.P. Natural products of filamentous fungi: Enzymes, genes, and their regulation. Nat. Prod. Rep. 2007, 24, 393–416. [Google Scholar] [CrossRef]
- Gluck-Thaler, E.; Haridas, S.; Binder, M.; Grigoriev, I.V.; Crous, P.W.; Spatafora, J.W.; Bushley, K.; Slot, J.C. The Architecture of Metabolism Maximizes Biosynthetic Diversity in the Largest Class of Fungi. Mol. Biol. Evol. 2020, 37, 2838–2856. [Google Scholar] [CrossRef]
- Waites, M.J.; Morgan, N.L.; Rockey, J.S.; Higton, G. Industrial Microbiology: An Introduction; Wiley: Hoboken, NJ, USA, 2001. [Google Scholar]
- Dadachova, E.; Bryan, R.A.; Huang, X.; Moadel, T.; Schweitzer, A.D.; Aisen, P.; Nosanchuk, J.D.; Casadevall, A. Ionizing Radiation Changes the Electronic Properties of Melanin and Enhances the Growth of Melanized Fungi. PLoS ONE 2007, 2, e457. [Google Scholar] [CrossRef]
- Revankar, S.G.; Sutton, D.A. Melanized Fungi in Human Disease. Clin. Microbiol. Rev. 2012, 25, 720. [Google Scholar] [CrossRef]
- Gostinčar, C.; Muggia, L.; Grube, M. Polyextremotolerant black fungi: Oligotrophism, adaptive potential, and a link to lichen symbioses. Front. Microbiol. 2012, 3, 390. [Google Scholar] [CrossRef]
- Naranjo-Ortiz, M.A.; Gabaldón, T. Fungal evolution: Major ecological adaptations and evolutionary transitions. Biol. Rev. 2019, 94, 1443–1476. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Jiang, Z.; Liu, H.; Zhou, D.; Sanchez-Silva, M. Microbiologically induced deterioration of concrete—A Review. Braz. J. Microbiol. 2014, 44, 1001–1007. [Google Scholar] [CrossRef] [PubMed]
- Bindschedler, S.; Cailleau, G.; Verrecchia, E. Role of Fungi in the Biomineralization of Calcite. Minerals 2016, 6, 41. [Google Scholar] [CrossRef]
- Coccato, A.; Moens, L.; Vandenabeele, P. On the stability of mediaeval inorganic pigments: A literature review of the effect of climate, material selection, biological activity, analysis and conservation treatments. Herit. Sci. 2017, 5, 12. [Google Scholar] [CrossRef]
- Singh, S.M.; Singh, P.N.; Singh, S.K.; Sharma, P.K. Pigment, fatty acid and extracellular enzyme analysis of a fungal strain Thelebolus microsporus from Larsemann Hills, Antarctica. Polar Rec. 2014, 50, 31–36. [Google Scholar] [CrossRef]
- Trochine, A.; Turchetti, B.; Vaz, A.B.M.; Brandao, L.; Rosa, L.H.; Buzzini, P.; Rosa, C.; Libkind, D. Description of Dioszegia patagonica sp. nov., a novel carotenogenic yeast isolated from cold environments. Int. J. Syst. Evol. Microbiol. 2017, 67, 4332–4339. [Google Scholar] [CrossRef] [PubMed]
- Flieger, K.; Knabe, N.; Toepel, J. Development of an Improved Carotenoid Extraction Method to Characterize the Carotenoid Composition under Oxidative Stress and Cold Temperature in the Rock Inhabiting Fungus Knufia petricola A95. J. Fungi 2018, 4, 124. [Google Scholar] [CrossRef]
- Nai, C. Rock-Inhabiting Fungi Studied with the Aid of the Model Black Fungus Knufia Petricola A95 and Other Related Strains. Ph.D. Thesis, Freie Universität Berlin, Berlin, Germany, 2014. [Google Scholar]
- Li, X.Q.; Guo, B.L.; Cai, W.Y.; Zhang, J.M.; Huang, H.Q.; Zhan, P.; Xi, L.Y.; Vicente, V.A.; Stielow, B.; Sun, J.F.; et al. The Role of Melanin Pathways in Extremotolerance and Virulence of Fonsecaea Revealed by De Novo Assembly Transcriptomics Using Illumina Paired-End Sequencing. Stud. Mycol. 2016, 83, 1–18. [Google Scholar] [CrossRef]
- Jacobson, E.S. Pathogenic Roles for Fungal Melanins. Clin. Microbiol. Rev. 2000, 13, 708–717. [Google Scholar] [CrossRef]
- Langfelder, K.; Streibel, M.; Jahn, B.; Haase, G.; Brakhage, A.A. Biosynthesis of fungal melanins and their importance for human pathogenic fungi. Fungal Genet. Biol. 2003, 38, 143–158. [Google Scholar] [CrossRef]
- Cordero, R.J.B.; Vij, R.; Casadevall, A. Microbial melanins for radioprotection and bioremediation. Microb. Biotechnol. 2017, 10, 1186–1190. [Google Scholar] [CrossRef] [PubMed]
- Sterflinger, K. Temperature and NaCl- tolerance of rock-inhabiting meristematic fungi. Antonie Van Leeuwenhoek 1998, 74, 271–281. [Google Scholar] [CrossRef]
- Robinson, C.H. Cold adaptation in Arctic and Antarctic fungi. New Phytol. 2001, 151, 341–353. [Google Scholar] [CrossRef]
- Abad, A.; Victoria Fernández-Molina, J.; Bikandi, J.; Ramírez, A.; Margareto, J.; Sendino, J.; Luis Hernando, F.; Pontón, J.; Garaizar, J.; Rementeria, A. What makes Aspergillus fumigatus a successful pathogen? Genes and molecules involved in invasive aspergillosis. Rev. Iberoam. Micol. 2010, 27, 155–182. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Bermúdez, P.; Srebotnik, E.; Hammel, K.E. Chlorination and Cleavage of Lignin Structures by Fungal Chloroperoxidases. Appl. Environ. Microbiol. 2003, 69, 5015–5018. [Google Scholar] [CrossRef] [PubMed]
- Tesei, D.; Marzban, G.; Zakharova, K.; Isola, D.; Selbmann, L.; Sterflinger, K. Alteration of Protein Patterns in Black Rock-Inhabiting Fungi as a Response to Different Temperatures. Fungal Biol. 2012, 116, 932–940. [Google Scholar] [CrossRef]
- Pacelli, C.; Bryan, R.A.; Onofri, S.; Selbmann, L.; Zucconi, L.; Shuryak, I.; Dadachova, E. Survival and Redox Activity of Friedmanniomyces endolithicus, an Antarctic Endemic Black Meristematic Fungus, after Gamma Rays Exposure. Fungal Biol. 2018, 122, 1222–1227. [Google Scholar] [CrossRef]
- Biswas, S.; Dijck, P.V.; Datta, A. Environmental Sensing and Signal Transduction Pathways Regulating Morphopathogenic Determinants of Candida albicans. Microbiol. Mol. Biol. Rev. 2007, 71, 348–376. [Google Scholar] [CrossRef]
- Wang, Q.; Szaniszlo, P.J. WdStuAp, an APSES Transcription Factor, Is a Regulator of Yeast-Hyphal Transitions in Wangiella (Exophiala) dermatitidis. Eukaryot. Cell 2007, 6, 1595–1605. [Google Scholar] [CrossRef]
- Connolly, L.A.; Riccombeni, A.; Grózer, Z.; Holland, L.M.; Lynch, D.B.; Andes, D.R.; Gácser, A.; Butler, G. The APSES transcription factor Efg1 is a global regulator that controls morphogenesis and biofilm formation in Candida parapsilosis. Mol. Microbiol. 2013, 90, 36–53. [Google Scholar] [CrossRef]
- Sun, L.; Xiao, L.; Xiao, B.; Wang, W.; Pan, C.; Wang, S.; Lian, B. Differences in the gene expressive quantities of carbonic anhydrase and cysteine synthase in the weathering of potassium-bearing minerals by Aspergillus niger. Sci. China Earth Sci. 2013, 56, 2135–2140. [Google Scholar] [CrossRef]
- Tekaia, F. Inferring Orthologs: Open Questions and Perspectives. Genom. Insights 2016, 9, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Gostinčar, C.; Gunde-Cimerman, N. Overview of Oxidative Stress Response Genes in Selected Halophilic Fungi. Genes 2018, 9, 143. [Google Scholar] [CrossRef] [PubMed]


 Antarctic climate;
Antarctic climate;  moderate climate;
moderate climate;  rock;
rock;  plant;
plant;  water;
water;  coral;
coral;  wood).
Antarctic climate; moderate climate; rock; plant; water; coral; wood).
wood).
Antarctic climate; moderate climate; rock; plant; water; coral; wood).
| Data | S. lemnorum MUM 23.14 | A. ludgeri DSM 106916 |
|---|---|---|
| Size | 25,845,107 bp | 22,585,551 bp |
| GC content | 57.49% | 58.57% |
| Scaffold number | 1414 | 228 |
| Scaffold N50 | 152,083 bp | 181,365 bp |
| Genes | 7957 | 8103 |
| rRNAs | 3 | 2 |
| tRNAs | 41 | 23 |
| CAZome | 488 | 581 |
| BGCs | 6 | 11 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paiva, D.S.; Fernandes, L.; Portugal, A.; Trovão, J. First Genome Sequence of the Microcolonial Black Fungus Saxispiralis lemnorum MUM 23.14: Insights into the Unique Genomic Traits of the Aeminiaceae Family. Microorganisms 2024, 12, 104. https://doi.org/10.3390/microorganisms12010104
Paiva DS, Fernandes L, Portugal A, Trovão J. First Genome Sequence of the Microcolonial Black Fungus Saxispiralis lemnorum MUM 23.14: Insights into the Unique Genomic Traits of the Aeminiaceae Family. Microorganisms. 2024; 12(1):104. https://doi.org/10.3390/microorganisms12010104
Chicago/Turabian StylePaiva, Diana S., Luís Fernandes, António Portugal, and João Trovão. 2024. "First Genome Sequence of the Microcolonial Black Fungus Saxispiralis lemnorum MUM 23.14: Insights into the Unique Genomic Traits of the Aeminiaceae Family" Microorganisms 12, no. 1: 104. https://doi.org/10.3390/microorganisms12010104