
Molecular Evolutionary Analyses of the Spike Protein Gene and Spike Protein in the SARS-CoV-2 Omicron Subvariants
 ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strain Selection
2.2. Time-Scaled Phylogenetic Analysis
2.3. Statistical Analysis
2.4. Similarity Plot (SimPlot) Analysis
2.5. Genome Network Analysis
2.6. Homology Modeling
2.7. Conformational Epitope Analysis
2.8. Selective Pressure Analysis
2.9. Visualizing Epitopes on 3D Structure Model of SARS-CoV-2 Spike Protein
3. Results
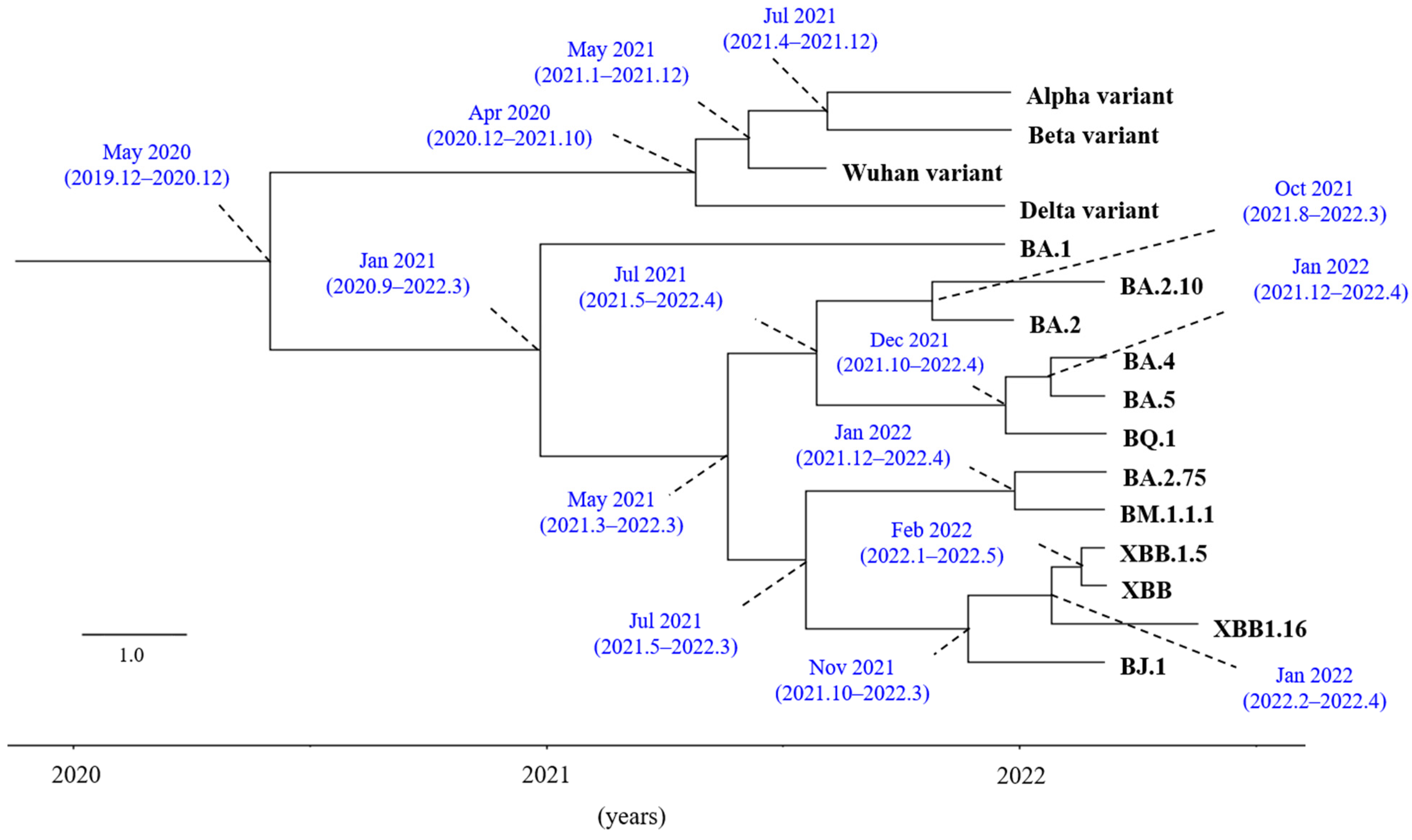
3.1. Time-Scaled Phylogeny of S Gene in the Various SARS-CoV-2 Variants/Subvariants
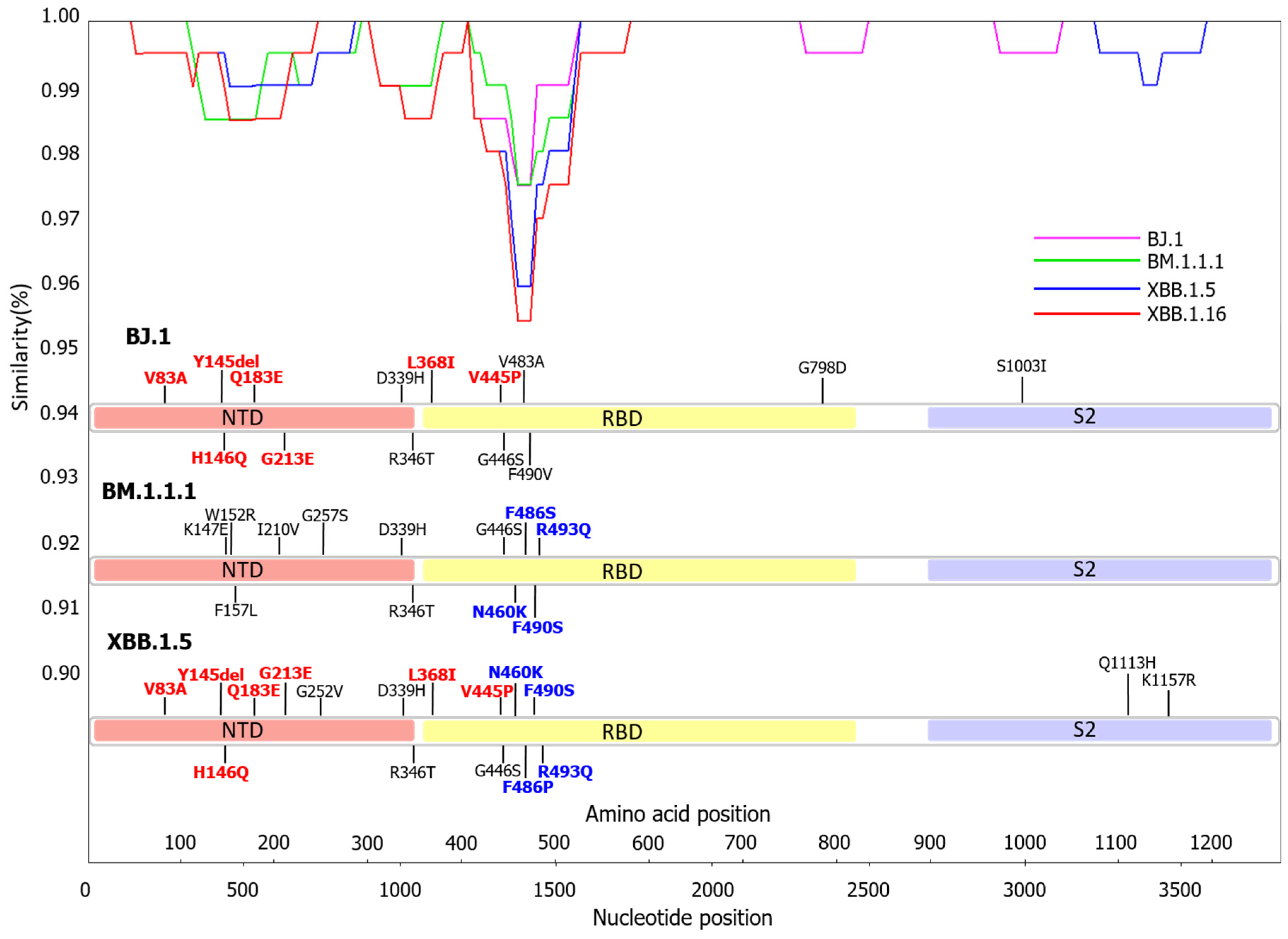
3.2. Similarity Plot Analysis of Omicron Subvariants BA.2, BJ.1, BM.1.1.1, XBB.1.5, and XBB.1.16
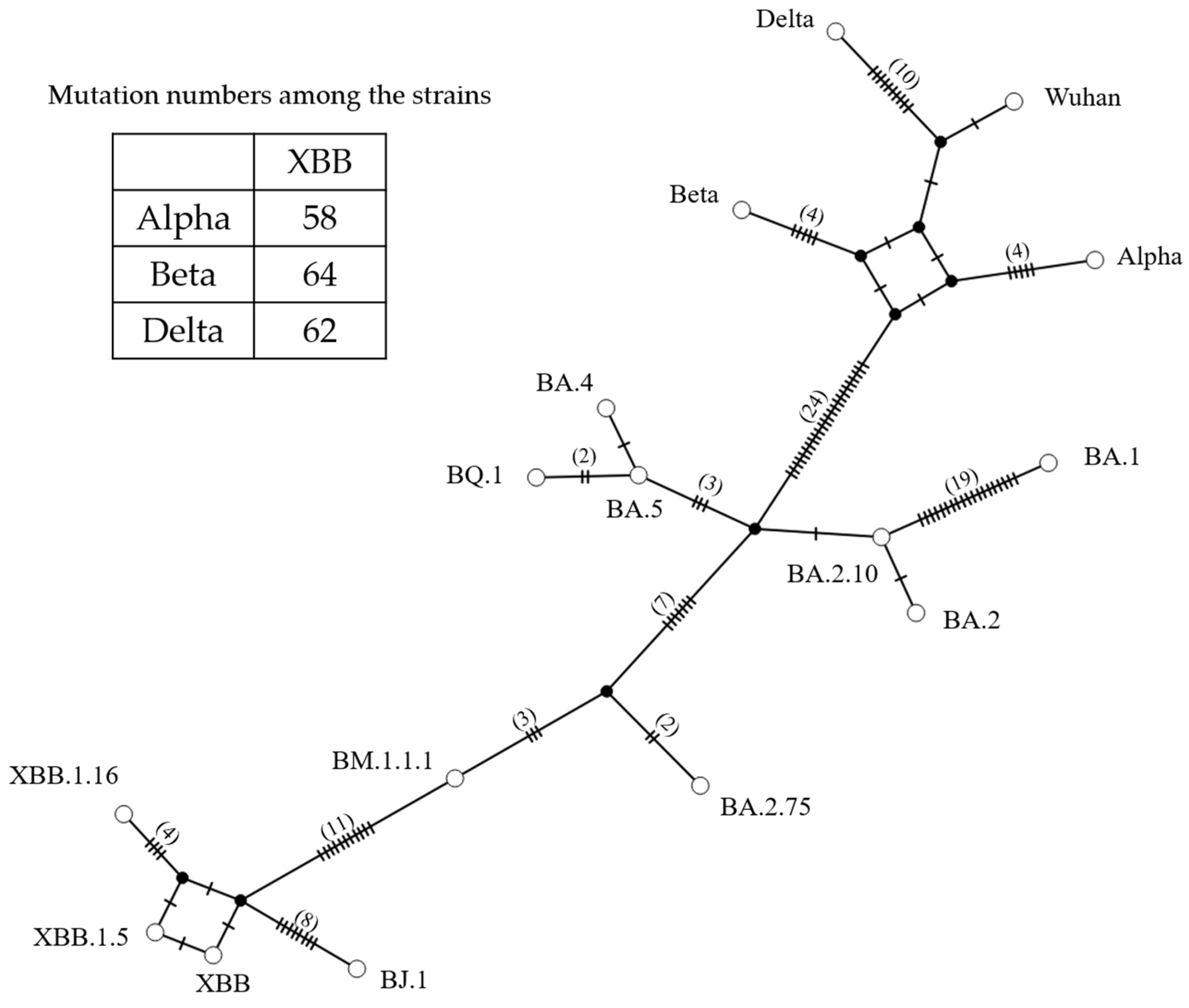
3.3. Genome Network Analysis Based on the S Gene Sequences
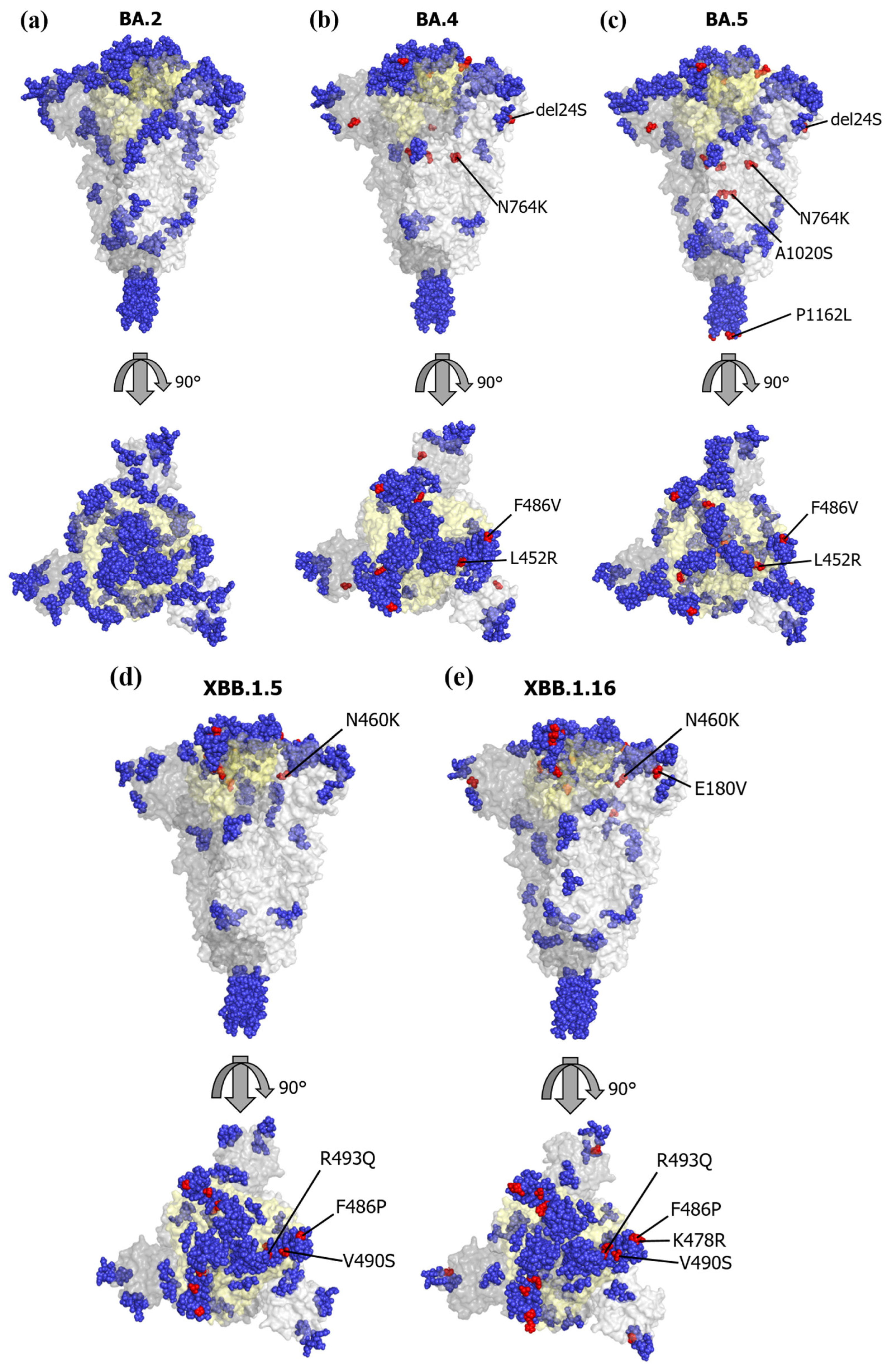
3.4. Mapping of the Conformational Epitopes and Amino Acid Mutations
3.5. Selective Pressure Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- WHO Coronavirus (COVID-19) Dashboard, Tracking SARS-CoV-2. Available online: https://www.who.int/activities/tracking-SARS-CoV-2-variants (accessed on 19 June 2023).
- Statistics and Research Coronavirus Pandemic (COVID-19). Available online: https://ourworldindata.org/coronavirus (accessed on 19 June 2023).
- González-Candelas, F.; Shaw, M.A.; Phan, T.; Kulkarni-Kale, U.; Paraskevis, D.; Luciani, F.; Kimura, H.; Sironi, M. One year into the pandemic: Short-term evolution of SARS-CoV-2 and emergence of new lineages. Infect. Genet. Evol. 2021, 92, 104869. [Google Scholar] [CrossRef]
- Genomic Epidemiology of Novel Coronavirus—Global Subsampling. Available online: https://nextstrain.org/ncov/gisaid/global/6m (accessed on 19 June 2023).
- Accorsi, E.K.; Britton, A.; Fleming-Dutra, K.E.; Smith, Z.R.; Shang, N.; Derado, G.; Miller, J.; Schrag, S.J.; Verani, J.R. Association Between 3 Doses of mRNA COVID-19 Vaccine and Symptomatic Infection Caused by the SARS-CoV-2 Omicron and Delta Variants. JAMA 2022, 327, 639–651. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.G.; Natarajan, K.; Irving, S.A.; Rowley, E.A.; Griggs, E.P.; Gaglani, M.; Klein, N.P.; Grannis, S.J.; DeSilva, M.B.; Stenehjem, E.; et al. Effectiveness of a Third Dose of mRNA Vaccines Against COVID-19-Associated Emergency Department and Urgent Care Encounters and Hospitalizations Among Adults During Periods of Delta and Omicron Variant Predominance—VISION Network, 10 States, August 2021–January 2022. MMWR Morb. Mortal Wkly. Rep. 2022, 71, 139–145. [Google Scholar] [PubMed]
- Andrews, N.; Stowe, J.; Kirsebom, F.; Toffa, S.; Rickeard, T.; Gallagher, E.; Gower, C.; Kall, M.; Groves, N.; O’Connell, A.M.; et al. COVID-19 Vaccine Effectiveness against the Omicron (B.1.1.529) Variant. N. Engl. J. Med. 2022, 386, 1532–1546. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Jian, F.; Wang, J.; Yu, Y.; Song, W.; Yisimayi, A.; An, R.; Chen, X.; Zhang, N.; Wang, Y.; et al. Imprinted SARS-CoV-2 humoral immunity induces convergent Omicron RBD evolution. Nature 2023, 614, 521–529. [Google Scholar] [CrossRef]
- Sharma, O.; Sultan, A.A.; Ding, H.; Triggle, C.R. A Review of the Progress and Challenges of Developing a Vaccine for COVID-19. Front. Immunol. 2020, 11, 585354. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, J.; Jian, F.; Xiao, T.; Song, W.; Yisimayi, A.; Huang, W.; Li, Q.; Wang, P.; An, R.; et al. Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature 2022, 602, 657–663. [Google Scholar] [CrossRef]
- Khoury, D.S.; Cromer, D.; Reynaldi, A.; Schlub, T.E.; Wheatley, A.K.; Juno, J.A.; Subbarao, K.; Kent, S.J.; Triccas, J.A.; Davenport, M.P. Neutralizing antibody levels are highly predictive of immune protection from symptomatic SARS-CoV-2 infection. Nat. Med. 2021, 27, 1205–1211. [Google Scholar] [CrossRef] [PubMed]
- Addetia, A.; Crawford, K.H.; Dingens, A.; Zhu, H.; Roychoudhury, P.; Huang, M.L.; Jerome, K.R.; Bloom, J.D.; Greninger, A.L. Neutralizing antibodies correlate with protection from SARS-CoV-2 in humans during a fishery vessel outbreak with high attack rate. J. Clin. Microbiol. 2020, 58, e02107–e02120. [Google Scholar] [CrossRef] [PubMed]
- Tuekprakhon, A.; Nutalai, R.; Dijokaite-Guraliuc, A.; Zhou, D.; Ginn, H.M.; Selvaraj, M.; Liu, C.; Mentzer, A.J.; Supasa, P.; Duyvesteyn, H.M.E.; et al. Antibody escape of SARS-CoV-2 Omicron BA.4 and BA.5 from vaccine and BA.1 serum. Cell 2022, 185, 2422–2433.e13. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchene, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Nert, K.D.; De Maio, N.; et al. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2019, 15, e1006650. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Kanda, Y. Investigation of the freely available easy-to-use software ‘EZR’ for medical statistics. Bone Marrow Transpl. 2013, 48, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, L.B.; Foster, C.; Rawlinson, W.; Tedla, N.; Bull, R.A. Evolution of the SARS-CoV-2 omicron variants BA.1 to BA.5: Implications for immune escape and transmission. Rev. Med. Virol. 2022, 32, e2381. [Google Scholar] [CrossRef]
- Tegally, H.; Moir, M.; Everatt, J.; Giovanetti, M.; Scheepers, C.; Wilkinson, E.; Subramoney, K.; Makatini, Z.; Moyo, S.; Amoako, D.G.; et al. Emergence of SARS-CoV-2 Omicron lineages BA.4 and BA.5 in South Africa. Nat. Med. 2022, 28, 1785–1790. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.C.; Shirk, P.; Lambrou, A.S.; Hassell, N.; Zheng, X.Y.; Payne, A.B.; Ali, A.R.; Batra, D.; Caravas, J.; Chau, R.; et al. Genomic Surveillance for SARS-CoV-2 Variants: Circulation of Omicron Lineages—United States, January 2022–May 2023. MMWR Morb. Mortal Wkly. Rep. 2023, 72, 651–656. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef] [PubMed]
- Shashkova, T.I.; Umerenkov, D.; Salnikov, M.; Strashnov, P.V.; Konstantinova, A.V.; Lebed, I.; Shcherbinin, D.N.; Asatryan, M.N.; Kardymon, O.L.; Ivanisenko, N.V. SEMA: Antigen B-cell conformational epitope prediction using deep transfer learning. Front. Immunol. 2022, 13, 960985. [Google Scholar] [CrossRef] [PubMed]
- Kringelum, J.V.; Lundegaard, C.; Lund, O.; Nielsen, M. Reliable B cell epitope predictions: Impacts of method development and improved benchmarking. PLoS Comput. Biol. 2012, 8, e1002829. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Chen, Z.; Zhang, L.; Yan, D.; Mao, T.; Tang, K.; Qiu, T.; Cao, Z. SEPPA 3.0-enhanced spatial epitope prediction enabling glycoprotein antigens. Nucleic Acids Res. 2019, 47, W388–W394. [Google Scholar] [CrossRef] [PubMed]
- Ponomarenko, J.; Bui, H.H.; Li, W.; Fusseder, N.; Bourne, P.E.; Sette, A.; Peters, B. ElliPro: A new structure-based tool for the prediction of antibody epitopes. BMC Bioinform. 2008, 9, 514. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Akagawa, M.; Kimura, R.; Sada, M.; Shirai, T.; Okayama, K.; Hayashi, Y.; Kondo, M.; Takeda, M.; Ryo, A.; et al. Molecular evolutionary analyses of the fusion protein gene in human respirovirus 1. Virus Res. 2023, 333, 199142. [Google Scholar] [CrossRef]
- Pond, S.L.; Frost, S.D. Datamonkey: Rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 2005, 21, 2531–2533. [Google Scholar] [CrossRef]
- DeLano, W.L. The PyMOL Molecular Graphics System; DeLano Scientific: San Carlos, CA, USA, 2002. [Google Scholar]
- Yoshihara, K.; Minh, L.N.; Okada, T.; Toizumi, M.; Nguyen, H.A.; Vo, H.M.; Hashizume, M.; Dang, D.A.; Kimura, H.; Yoshida, L.M. Evolutionary dynamics of influenza B strains detected from paediatric acute respiratory infections in central Vietnam. Infect. Genet. Evol. 2020, 81, 104264. [Google Scholar] [CrossRef]
- Kobayashi, M.; Matsushima, Y.; Motoya, T.; Sakon, N.; Shigemoto, N.; Okamoto-Nakagawa, R.; Nishimura, K.; Yamashita, Y.; Kuroda, M.; Saruki, N.; et al. Molecular evolution of the capsid gene in human norovirus genogroup II. Sci. Rep. 2016, 6, 29400. [Google Scholar] [CrossRef]
- Chatterjee, S.; Bhattacharya, M.; Nag, S.; Dhama, K.; Chakraborty, C. A Detailed Overview of SARS-CoV-2 Omicron: Its Sub-Variants, Mutations and Pathophysiology, Clinical Characteristics, Immunological Landscape, Immune Escape, and Therapies. Viruses 2023, 15, 167. [Google Scholar] [CrossRef]
- Scarpa, F.; Sanna, D.; Azzena, I.; Casu, M.; Cossu, P.; Fiori, P.L.; Benvenuto, D.; Imperia, E.; Giovanetti, M.; Ceccarelli, G.; et al. Genome-based comparison between the recombinant SARS-CoV-2 XBB and its parental lineages. J. Med. Virol. 2023, 95, e28625. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhi, H.; Teng, Y. The outbreak of SARS-CoV-2 Omicron lineages, immune escape, and vaccine effectivity. J. Med. Virol. 2023, 95, e28138. [Google Scholar] [CrossRef]
- Ju, B.; Fan, Q.; Liu, C.; Shen, S.; Wang, M.; Guo, H.; Zhou, B.; Ge, X.; Zhang, Z. Omicron BQ.1.1 and XBB.1 unprecedentedly escape broadly neutralizing antibodies elicited by prototype vaccination. Cell Rep. 2023, 42, 112532. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.C.; O’Toole, A.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- Gobeil, S.M.; Janowska, K.; McDowell, S.; Mansouri, K.; Parks, R.; Stalls, V.; Kopp, M.F.; Manne, K.; Li, D.; Wiehe, K.; et al. Effect of natural mutations of SARS-CoV-2 on spike structure, conformation, and antigenicity. Science 2021, 373, 6555. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Zhao, S.; Liu, Q.; Zhang, X.; Sha, T.; Su, Y.; Zhao, W.; Bao, Y.; Xue, Y.; Chen, H. Ongoing Positive Selection Drives the Evolution of SARS-CoV-2 Genomes. Genom. Proteom. Bioinform. 2022, 20, 1214–1223. [Google Scholar] [CrossRef] [PubMed]
- Lon, J.R.; Bai, Y.; Zhong, B.; Cai, F.; Du, H. Prediction and evolution of B cell epitopes of surface protein in SARS-CoV-2. Virol. J. 2020, 17, 165. [Google Scholar] [CrossRef] [PubMed]
- Gattinger, P.; Niespodziana, K.; Stiasny, K.; Sahanic, S.; Tulaeva, I.; Borochova, K.; Dorofeeva, Y.; Schlederer, T.; Sonnweber, T.; Hofer, G.; et al. Neutralization of SARS-CoV-2 requires antibodies against conformational receptor-binding domain epitopes. Allergy 2022, 77, 230–242. [Google Scholar] [CrossRef]
- Yi, Z.; Ling, Y.; Zhang, X.; Chen, J.; Hu, K.; Wang, Y.; Song, W.; Ying, T.; Zhang, R.; Lu, H.; et al. Functional mapping of B-cell linear epitopes of SARS-CoV-2 in COVID-19 convalescent population. Emerg. Microbes Infect. 2020, 9, 1988–1996. [Google Scholar] [CrossRef]
- Wang, Q.; Iketani, S.; Li, Z.; Liu, L.; Guo, Y.; Huang, Y.; Bowen, A.D.; Liu, M.; Wang, M.; Yu, J.; et al. Alarming antibody evasion properties of rising SARS-CoV-2 BQ and XBB subvariants. Cell 2023, 186, 279–286.e8. [Google Scholar] [CrossRef]
- Tamura, T.; Ito, J.; Uriu, K.; Zahradnik, J.; Kida, I.; Anraku, Y.; Nasser, H.; Shofa, M.; Oda, Y.; Lytras, S.; et al. Virological characteristics of the SARS-CoV-2 XBB variant derived from recombination of two Omicron subvariants. Nat. Commun. 2023, 14, 2800. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BA.2 | BA.4 | BA.5 | |||
| Position | Predicted epitopes | Position | Predicted epitopes | Position | Predicted epitopes |
| 109–114 | TLDSKT | 108–114 | TTLDSKT | 109–114 | TLDSKT |
| 144–154 | YYHKNNKSWME | 142–152 | DVYYHKNNKSW | 145–152 | YHKNNKSW |
| 160–169 | YSSANNCTFE | 164–167 | NNCT | 160–167 | YSSANNCT |
| 177–185 | MDLEGKQGN | 182–185 | KQGN | 182–185 | KQGN |
| 436–452 | WNSNKLDSKVGGNYNYL | 437–450 | NSNKLDSKVGGNYN | 443–450 | SKVGGNYN |
| 458–460 | KSN | 455–460 | LFRKSN | 457–460 | RKSN |
| 474–488 | QAGNKPCNGVAGFNC | 468–494 | ISTEIYQAGNKPCNGVAGVNCYFPLQS | 472–489 | IYQAGNKPCNGVAG VNCY |
| 498–506 | RPTYGVGHQ | 497–506 | FRPTYGVGHQ | 496–506 | GFRPTYGVGHQ |
| 703–705 | NSV | 703–705 | NSV | 703–705 | NSV |
| 834–842 | IKQYGDCLG | 836–841 | QYGDCL | 836–841 | QYGDCL |
| 890–897 | AGAALQIP | 890–895 | AGAALQ | 890–897 | AGAALQIP |
| 1140–1162 | PLQPELDSFKEELDKYFKNHTSP | 1140–1162 | PLQPELDSFKEELDKYFKNHTSP | 1140–1162 | PLQPELDSFKEELDKYFKNHTSL |
| BA.4 | XBB.1.5 | XBB.1.16 | |||
| Position | Predicted epitopes | Position | Predicted epitopes | Position | Predicted epitopes |
| 142–152 | DVYYHKNNKSW | 146–151 | QKNNKS | 147–151 | KNNKS |
| 164–167 | NNCT | 164–166 | NNC | 162–166 | SANNC |
| 182–185 | KQGN | 182–185 | KEGN | 182–185 | KEGN |
| 437–450 | NSNKLDSKVGGNYN | 436–452 | WNSNKLDSKPSGNYNYL | 436–452 | WNSNKLDSKPSGNYNYL |
| 455–460 | LFRKSN | 455–460 | LFRKSK | 455–460 | LFRKSK |
| 468–494 | ISTEIYQAGNKPCNGVAGVNCYFPLQS | 472–494 | IYQAGNKPCNGVAGP NCYSPLQS | 469–494 | STEIYQAGNRPCNGVAGP NCYSPLQS |
| 497–506 | FRPTYGVGHQ | 498–506 | RPTYGVGHQ | 496–506 | GFRPTYGVGHQ |
| 703–705 | NSV | 703–705 | NSV | 703–705 | NSV |
| 836–841 | QYGDCL | 836–843 | QYGDCLGD | 834–841 | IKQYGDCL |
| 890–895 | AGAALQ | 890–895 | AGAALQ | 890–894 | AGAAL |
| 1140–1162 | PLQPELDSFKEELDKYFKNHTSP | 1140–1162 | PLQPELDSFKEELDKYFKNHTSP | 1140–1162 | PLQPELDSFKEELDKYFKNHTSP |
| BA.5 | XBB.1.5 | XBB.1.16 | |||
| Position | Predicted epitopes | Position | Predicted epitopes | Position | Predicted epitopes |
| 142–152 | YHKNNKSW | 146–151 | QKNNKS | 147–151 | KNNKS |
| 160–167 | YSSANNCT | 164–166 | NNC | 162–166 | SANNC |
| 182–185 | KQGN | 182–185 | KEGN | 182–185 | KEGN |
| 443–450 | SKVGGNYN | 436–452 | WNSNKLDSKPSGNYNYL | 436–452 | WNSNKLDSKPSGNYNYL |
| 457–460 | RKSN | 455–460 | LFRKSK | 455–460 | LFRKSK |
| 472–489 | IYQAGNKPCNGVAGV NCY | 472–494 | IYQAGNKPCNGVAGP NCYSPLQS | 469–494 | STEIYQAGNRPCNGVAGP NCYSPLQS |
| 496–506 | GFRPTYGVGHQ | 498–506 | RPTYGVGHQ | 496–506 | GFRPTYGVGHQ |
| 703–705 | NSV | 703–705 | NSV | 703–705 | NSV |
| 836–841 | QYGDCL | 836–843 | QYGDCLGD | 834–841 | IKQYGDCL |
| 890–897 | AGAALQIP | 890–895 | AGAALQ | 890–894 | AGAAL |
| 1140–1162 | PLQPELDSFKEELDKYFKNHTSL | 1140–1162 | PLQPELDSFKEELDKYFKNHTSP | 1140–1162 | PLQPELDSFKEELDKYFKNHTSP |
| BJ.1 | BM.1.1.1 | ||
| Position | Predicted epitopes | Position | Predicted epitopes |
| 147–151 | KNNKS | 146–152 | HENNKSR |
| 182–185 | KEGN | 182–185 | KQGN |
| 437–452 | NSNKLDSKPSGNYNYL | 439–452 | NKLDSKVSGNYNYL |
| 455–460 | LFRKSN | 457–460 | RKSK |
| 474–489 | QAGNKPCNGAAGFNCY | 472–489 | IYQAGNKPCNGVAGSNCY |
| 491–506 | PLRSYGFRPTYGVGHQ | 498–506 | RPTYGVGHQ |
| 703–705 | NSV | 703–705 | NSV |
| 836–842 | QYGDCLG | 834–841 | IKQYGDCL |
| 890–896 | AGAALQI | 890–896 | AGAALQI |
| 1140–1162 | PLQPELDSFKEELDKYFKNHTSP | 1140–1162 | PLQPELDSFKEELDKYFKNHTSP |
| XBB.1.5 | XBB.1.16 | ||
| Position | Predicted epitopes | Position | Predicted epitopes |
| 146–151 | QKNNKS | 147–151 | KNNKS |
| 182–185 | KEGN | 182–185 | KEGN |
| 436–452 | WNSNKLDSKPSGNYNYL | 436–452 | WNSNKLDSKPSGNYNYL |
| 455–460 | LFRKSK | 455–460 | LFRKSK |
| 472–494 | IYQAGNKPCNGVAGPNCYSPLQS | 469–494 | STEIYQAGNRPCNGVAGPNCYSPLQS |
| 498–506 | RPTYGVGHQ | 496–506 | GFRPTYGVGHQ |
| 703–705 | NSV | 703–705 | NSV |
| 836–843 | QYGDCLGD | 834–841 | IKQYGDCL |
| 890–895 | AGAALQ | 890–894 | AGAAL |
| 1140–1162 | PLQPELDSFKEELDKYFKNHTSP | 1140–1162 | PLQPELDSFKEELDKYFKNHTSP |
| Amino Acid Changes | |
| Val3Gly | |
| Ala83Val | |
| His146Gln | |
| Gln180Glu | |
| Glu183Val | |
| Gly213Glu | |
| Gly252Val | |
| RBD | Asp339His |
| Arg346Thr | |
| Leu368Ile | |
| Asn417Lys | |
| Lys440Asn | |
| Val445Pro, Ala | |
| Gly446Ser | |
| Leu452Arg | |
| Asn460Lys, Ser | |
| Lys478Arg | |
| Phe486Val, Pro, Ser | |
| Phe490Ser | |
| Arg493Gln | |
| Pro521Thr, Gln | |
| Asn658Ser |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagasawa, N.; Kimura, R.; Akagawa, M.; Shirai, T.; Sada, M.; Okayama, K.; Sato-Fujimoto, Y.; Saito, M.; Kondo, M.; Katayama, K.; et al. Molecular Evolutionary Analyses of the Spike Protein Gene and Spike Protein in the SARS-CoV-2 Omicron Subvariants. Microorganisms 2023, 11, 2336. https://doi.org/10.3390/microorganisms11092336
Nagasawa N, Kimura R, Akagawa M, Shirai T, Sada M, Okayama K, Sato-Fujimoto Y, Saito M, Kondo M, Katayama K, et al. Molecular Evolutionary Analyses of the Spike Protein Gene and Spike Protein in the SARS-CoV-2 Omicron Subvariants. Microorganisms. 2023; 11(9):2336. https://doi.org/10.3390/microorganisms11092336
Chicago/Turabian StyleNagasawa, Norika, Ryusuke Kimura, Mao Akagawa, Tatsuya Shirai, Mitsuru Sada, Kaori Okayama, Yuka Sato-Fujimoto, Makoto Saito, Mayumi Kondo, Kazuhiko Katayama, and et al. 2023. "Molecular Evolutionary Analyses of the Spike Protein Gene and Spike Protein in the SARS-CoV-2 Omicron Subvariants" Microorganisms 11, no. 9: 2336. https://doi.org/10.3390/microorganisms11092336