
Whole-Genome Sequencing-Based Screening of MRSA in Patients and Healthcare Workers in Public Hospitals in Benin
 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Material and Methods
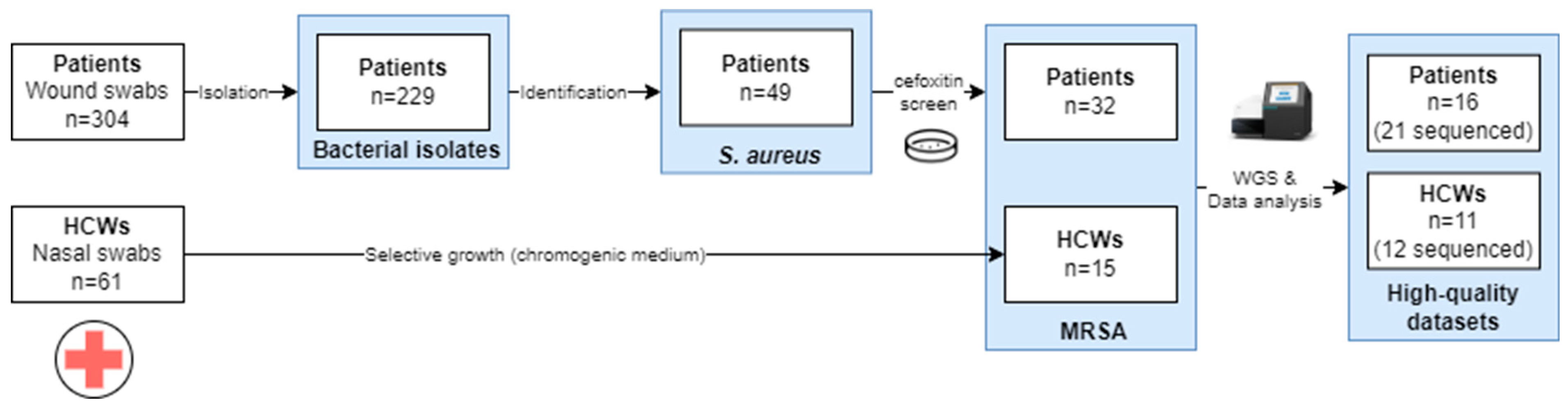
2.1. Study Design
2.2. Sample Collection
2.3. Bacterial Identification
2.4. Whole-Genome Sequencing
2.5. Quality Control and Preprocessing
2.6. Isolate Characterization
2.7. Phylogenomic Investigation
2.8. Ethics Approval and Consent to Participate
3. Results
3.1. Sample Collection and Species Identification
3.2. WGS Data Analysis
3.2.1. Preprocessing and Data Quality Evaluation
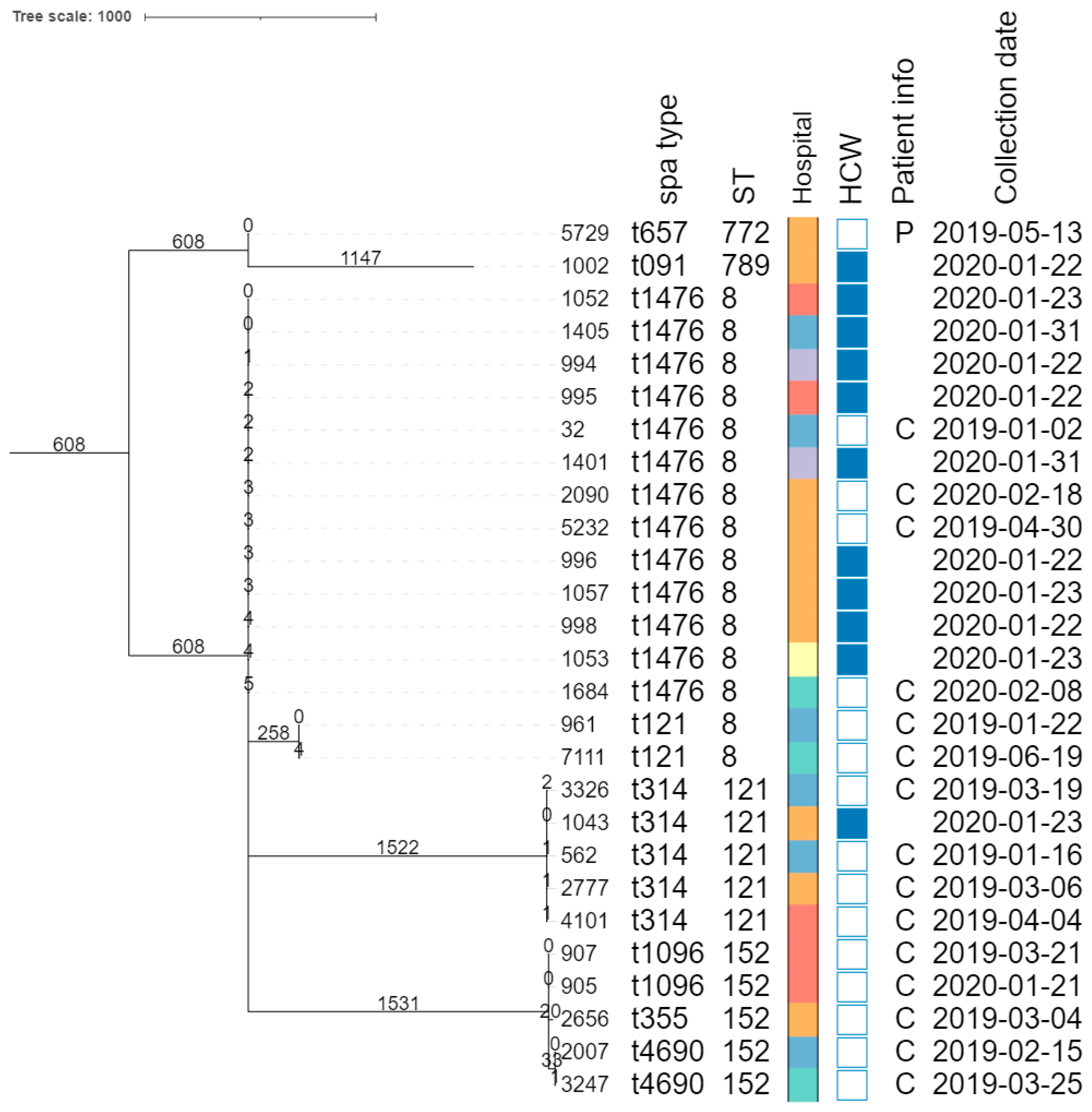
3.2.2. Isolate Typing
3.2.3. AMR Prediction
3.2.4. Virulence Gene Detection
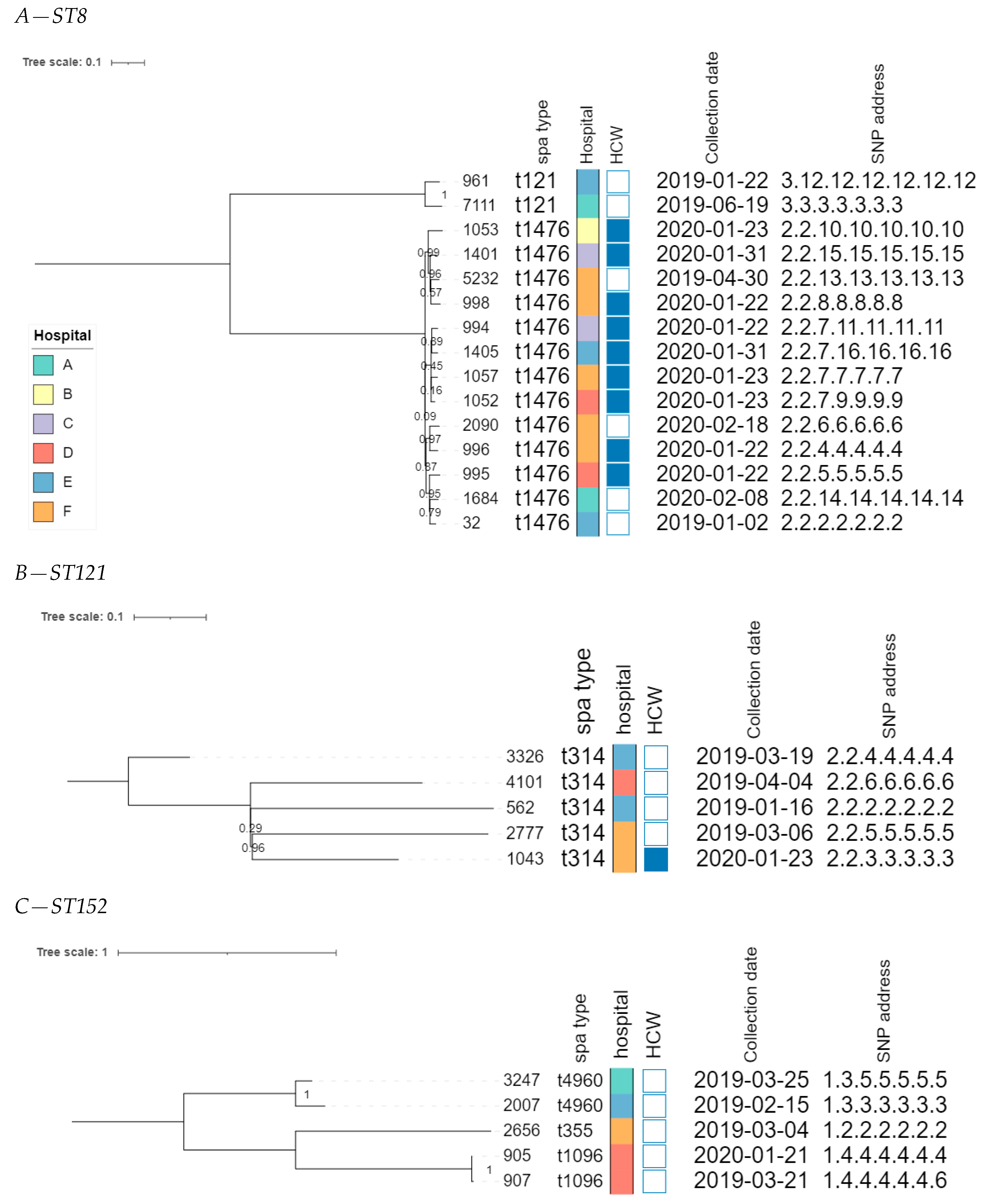
3.2.5. Phylogenomic Investigation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kondo, S.; Phokhaphan, P.; Tongsima, S.; Ngamphiw, C.; Phornsiricharoenphant, W.; Ruangchai, W.; Disratthakit, A.; Tingpej, P.; Mahasirimongkol, S.; Lulitanond, A.; et al. Molecular characterization of methicillin-resistant Staphylococcus aureus genotype ST764-SCCmec type II in Thailand. Sci. Rep. 2022, 12, 2085. [Google Scholar] [CrossRef] [PubMed]
- Laxminarayan, R.; Duse, A.; Wattal, C.; Zaidi, A.K.M.; Wertheim, H.F.L.; Sumpradit, N.; Vlieghe, E.; Hara, G.L.; Gould, I.M.; Goossens, H.; et al. Antibiotic resistance-the need for global solutions. Lancet Infect. Dis. 2013, 13, 1057–1098. [Google Scholar] [CrossRef] [Green Version]
- Hiramatsu, K.; Ito, T.; Tsubakishita, S.; Sasaki, T.; Takeuchi, F.; Morimoto, Y.; Katayama, Y.; Matsuo, M.; Kuwahara-Arai, K.; Hishinuma, T.; et al. Genomic basis for methicillin resistance in Staphylococcus aureus. Infect. Chemother. 2013, 45, 117–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Achek, R.; El-Adawy, H.; Hotzel, H.; Hendam, A.; Tomaso, H.; Ehricht, R.; Neubauer, H.; Nabi, I.; Hamdi, T.M.; Monecke, S. Molecular characterization of staphylococcus aureus isolated from human and food samples in northern algeria. Pathogens 2021, 10, 1276. [Google Scholar] [CrossRef] [PubMed]
- Allegranzi, B.; Nejad, S.B.; Combescure, C.; Graafmans, W.; Attar, H.; Donaldson, L.; Pittet, D. Burden of endemic health-care-associated infection in developing countries: Systematic review and meta-analysis. Lancet 2011, 377, 228–241. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Spencer, M.; Davidson, S.M.; Li, L.; Shaw, J.D.; Gulczynski, D.; Hunter, D.J.; Martha, J.F.; Miley, G.B.; Parazin, S.J.; et al. Institutional prescreening for detection and eradication of methicillin-resistant Staphylococcus aureus in patients undergoing elective orthopaedic surgery. J. Bone Jt. Surg. 2010, 92, 1820–1826. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Patkar, A.; Daskiran, M.; Levine, R.; Hinoul, P.; Nigam, S. Clinical and economic burden of surgical site infection in hysterectomy. Surg. Infect. 2014, 15, 266–273. [Google Scholar] [CrossRef]
- Owens, C.D.; Stoessel, K. Surgical site infections: Epidemiology, microbiology and prevention. J. Hosp. Infect. 2008, 70, 3–10. [Google Scholar] [CrossRef]
- Upreti, N.; Rayamajhee, B.; Sherchan, S.P.; Choudhari, M.K.; Banjara, M.R. Prevalence of methicillin resistant Staphylococcus aureus, multidrug resistant and extended spectrum β-lactamase producing gram negative bacilli causing wound infections at a tertiary care hospital of Nepal. Antimicrob. Resist. Infect. Control 2018, 7, 121. [Google Scholar] [CrossRef] [Green Version]
- Benito, D.; Gómez, P.; Aspiroz, C.; Zarazaga, M.; Lozano, C.; Torres, C. Molecular characterization of Staphylococcus aureus isolated from humans related to a livestock farm in Spain, with detection of MRSA-CC130 carrying mecC gene: A zoonotic case? Enferm. Infecc. Microbiol. Clin. 2016, 34, 280–285. [Google Scholar] [CrossRef]
- Kaya, H.; Hasman, H.; Larsen, J.; Stegger, M.; Johannesen, T.B.; Allesøe, R.L.; Lemvigh, C.K.; Aarestrup, F.M.; Lund, O.; Larsen, A.R. SCC mec Finder, a Web-Based Tool for Typing of Staphylococcal Cassette Chromosome mec in Staphylococcus aureus Using Whole-Genome Sequence Data. mSphere 2018, 3, e00612-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrade, M.M.; Luiz, W.B.; da Silva Oliveira Souza, R.; Amorim, J.H. The History of Methicillin-Resistant Staphylococcus aureus in Brazil. Can. J. Infect. Dis. Med. Microbiol. 2020, 2020, 1721936. [Google Scholar] [CrossRef] [PubMed]
- Aslam, B.; Khurshid, M.; Arshad, M.I.; Muzammil, S.; Rasool, M.; Yasmeen, N.; Shah, T.; Chaudhry, T.H.; Rasool, M.H.; Shahid, A.; et al. Antibiotic Resistance: One Health One World Outlook. Front Cell Infect Microbiol. 2021, 11, 1153. [Google Scholar] [CrossRef] [PubMed]
- Waness, A. Revisiting methicillin-resistant Staphylococcus aureus infections. J. Glob. Infect. Dis. 2010, 2, 49. [Google Scholar] [CrossRef] [PubMed]
- Bosch, T.; Pluister, G.N.; van Luit, M.; Landman, F.; van Santen-Verheuvel, M.; Schot, C.; Witteveen, S.; van der Zwaluw, K.; Heck, M.E.; Schouls, L.M. Multiple-locus variable number tandem repeat analysis is superior to spa typing and sufficient to characterize MRSA for surveillance purposes. Future Microbiol. 2015, 10, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- El-Adawy, H.; Ahmed, M.; Hotzel, H.; Monecke, S.; Schulz, J.; Hartung, J.; Ehricht, R.; Neubauer, H.; Hafez, H.M. Characterization of methicillin-resistant staphylococcus aureus isolated from healthy Turkeys and broilers using DNA microarrays. Front. Microbiol. 2016, 7, 2019. [Google Scholar] [CrossRef]
- Stefani, S.; Chung, D.R.; Lindsay, J.A.; Friedrich, A.W.; Kearns, A.M.; Westh, H.; MacKenzie, F.M. Meticillin-resistant Staphylococcus aureus (MRSA): Global epidemiology and harmonisation of typing methods. Int. J. Antimicrob. Agents 2012, 39, 273–282. [Google Scholar] [CrossRef]
- Wong, J.W.; Ip, M.; Tang, A.; Wei, V.W.; Wong, S.Y.; Riley, S.; Read, J.M.; Kwok, K.O. Prevalence and risk factors of community-associated methicillin-resistant staphylococcus aureus carriage in asia-pacific region from 2000 to 2016: A systematic review and meta-analysis. Clin. Epidemiol. 2018, 10, 1489–1501. [Google Scholar] [CrossRef] [Green Version]
- Enany, S.; Yaoita, E.; Yoshida, Y.; Enany, M.; Yamamoto, T. Molecular characterization of Panton-Valentine leukocidin-positive community-acquired methicillin-resistant Staphylococcus aureus isolates in Egypt. Microbiol. Res. 2010, 165, 152–162. [Google Scholar] [CrossRef]
- Mariem, B.J.-J.; Ito, T.; Zhang, M.; Jin, J.; Li, S.; Ilhem, B.-B.B.; Adnan, H.; Han, X.; Hiramatsu, K. Molecular characterization of methicillin-resistant Panton-valentine leukocidin positive staphylococcus aureus clones disseminating in Tunisian hospitals and in the community. BMC Microbiol. 2013, 13, 2. [Google Scholar] [CrossRef] [Green Version]
- Eiff, V. Nasal Carriage as a Sourc E of Sta Ph Yloc Occ Us Aureus Bac Ter Emia Nasal Carriage as a Source of Staphylococcus Aureus Bacteremia. N. Engl. J. 2001, 344, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Yehouenou, C.L.; Dohou, A.M.; Fiogbe, A.D.; Esse, M.; Degbey, C.; Simon, A.; Dalleur, O. Hand hygiene in surgery in Benin: Opportunities and challenges. Antimicrob. Resist. Infect. Control 2020, 9, 85. [Google Scholar] [CrossRef] [PubMed]
- Borchardt, R.A.; Tzizik, D. Update on surgical site infections: The new CDC guidelines. J. Am. Acad. Physician Assist. 2018, 31, 52–54. [Google Scholar] [CrossRef] [PubMed]
- Yehouenou, C.L.; Kpangon, A.A.; Affolabi, D.; Rodriguez-Villalobos, H.; Van Bambeke, F.; Dalleur, O.; Simon, A. Antimicrobial resistance in hospitalized surgical patients: A silently emerging public health concern in Benin. Ann. Clin. Microbiol. Antimicrob. 2020, 19, 54. [Google Scholar] [CrossRef]
- Karmakar, A.; Dua, P.; Ghosh, C. Biochemical and Molecular Analysis of Staphylococcus aureus Clinical Isolates from Hospitalized Patients. Can. J. Infect. Dis. Med. Microbiol. 2016, 2016, 9041636. [Google Scholar] [CrossRef] [Green Version]
- Boutiba-Ben Boubaker, I.; Ben Abbes, R.; Ben Abdallah, H.; Mamlouk, K.; Mahjoubi, F.; Kammoun, A.; Hammami, A.; Ben Redjeb, S. Evaluation of a cefoxitin disk diffusion test for the routine detection of methicillin-resistant Staphylococcus aureus. Clin. Microbiol. Infect. 2004, 10, 762–765. [Google Scholar] [CrossRef] [Green Version]
- Bizzini, A.; Greub, G. Matrix-assisted laser desorption ionization time-of-flight mass spectrometry, a revolution in clinical microbial identification. Clin. Microbiol. Infect. 2010, 16, 1614–1619. [Google Scholar] [CrossRef] [Green Version]
- Unal, S.; Hoskins, J.; Flokowitsch, J.E.; Wu, C.Y.; Preston, D.A.; Skatrud, P.L. Detection of Methicillin-Resistant Staphylococci by Using the Polymerase Chain Reaction. J. Clin. Microbiol. 1992, 30, 1685–1691. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes De Novo Assembler. Curr. Protoc. Bioinform. 2020, 70, e102. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Low, A.J.; Koziol, A.G.; Manninger, P.A.; Blais, B.; Carrillo, C.D. ConFindr: Rapid detection of intraspecies and cross-species contamination in bacterial whole-genome sequence data. PeerJ 2019, 2019, e6995. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Bogaerts, B.; Nouws, S.; Verhaegen, B.; Denayer, S.; Van Braekel, J.; Winand, R.; Fu, Q.; Crombé, F.; Piérard, D.; Marchal, K.; et al. Validation strategy of a bioinformatics whole genome sequencing workflow for Shiga toxin-producing Escherichia coli using a reference collection extensively characterized with conventional methods. Microb. Genom. Microbiol. Society 2021, 7, mgen000531. [Google Scholar] [CrossRef]
- Feldgarden, M.; Brover, V.; Gonzalez-Escalona, N.; Frye, J.G.; Haendiges, J.; Haft, D.H.; Hoffmann, M.; Pettengill, J.B.; Prasad, A.B.; Tillman, G.E.; et al. AMRFinderPlus and the Reference Gene Catalog facilitate examination of the genomic links among antimicrobial resistance, stress response, and virulence. Sci. Rep. 2021, 11, 12728. [Google Scholar] [CrossRef]
- Joensen, K.G.; Scheutz, F.; Lund, O.; Hasman, H.; Kaas, R.S.; Nielsen, E.M.; Aarestrup, F.M. Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J. Clin. Microbiol. 2014, 52, 1501–1510. [Google Scholar] [CrossRef] [Green Version]
- Ren, Y.; Chakraborty, T.; Doijad, S.; Falgenhauer, L.; Falgenhauer, J.; Goesmann, A.; Hauschild, A.-C.; Schwengers, O.; Heider, D. Prediction of antimicrobial resistance based on whole-genome sequencing and machine learning. Bioinformatics 2022, 38, 325–334. [Google Scholar] [CrossRef]
- Zhou, Z.; Alikhan, N.-F.; Sergeant, M.J.; Luhmann, N.; Vaz, C.; Francisco, A.P.; Carriço, J.A.; Achtman, M. Grapetree: Visualization of core genomic relationships among 100,000 bacterial pathogens. Genome Res. 2018, 28, 1395–1404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dallman, T.; Ashton, P.; Schafer, U.; Jironkin, A.; Painset, A.; Shaaban, S.; Hartman, H.; Myers, R.; Underwood, A.; Jenkins, C.; et al. SnapperDB: A database solution for routine sequencing analysis of bacterial isolates. Bioinformatics 2018, 34, 3028–3029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Ouedraogo, A.S.; Dunyach-Remy, C.; Kissou, A.; Sanou, S.; Poda, A.; Kyelem, C.G.; Solassol, J.; Bañuls, A.L.; Van De Perre, P.; Ouédraogo, R.; et al. High nasal carriage rate of Staphylococcus aureus containing panton-valentine leukocidin- and EDIN-encoding genes in community and hospital settings in Burkina Faso. Front. Microbiol. 2016, 7, 1406. [Google Scholar] [CrossRef] [Green Version]
- Kesah, C.; Ben Redjeb, S.; Odugbemi, T.; Boye, C.S.-B.; Dosso, M.; Achola, J.N.; Koulla-Shiro, S.; Benbachir, M.; Rahal, K.; Borg, M. Prevalence of methicillin-resistant Staphylococcus aureus in eight African hospitals and Malta. Clin. Microbiol. Infect. 2003, 9, 153–156. [Google Scholar] [CrossRef] [Green Version]
- Antri, K.; Rouzic, N.; Dauwalder, O.; Boubekri, I.; Bes, M.; Lina, G.; Vandenesch, F.; Tazir, M.; Ramdani-Bouguessa, N.; Etienne, J. High prevalence of methicillin-resistant Staphylococcus aureus clone ST80-IV in hospital and community settings in Algiers. Clin. Microbiol. Infect. 2011, 17, 526–532. [Google Scholar] [CrossRef] [Green Version]
- Alioua, M.A.; Labid, A.; Amoura, K.; Bertine, M.; Gacemi-Kirane, D.; Dekhil, M. Emergence of the European ST80 clone of community-associated methicillin-resistant Staphylococcus aureus as a cause of healthcare-associated infections in Eastern Algeria. Med. Mal. Infect. 2014, 44, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Gordon, N.C.; Price, J.R.; Cole, K.; Everitt, R.; Morgan, M.; Finney, J.; Kearns, A.M.; Pichon, B.; Young, B.; Wilson, D.J.; et al. Prediction of staphylococcus aureus antimicrobial resistance by whole-genome sequencing. J. Clin. Microbiol. 2014, 52, 1182–1191. [Google Scholar] [CrossRef] [Green Version]
- Wolters, M.; Frickmann, H.; Christner, M.; Both, A.; Rohde, H.; Oppong, K.; Akenten, C.W.; May, J.; Dekker, D. Molecular characterization of staphylococcus aureus isolated from chronic infected wounds in rural Ghana. Microorganisms 2020, 8, 2052. [Google Scholar] [CrossRef]
- Akinkunmi, E.O.; Lamikanra, A. Species distribution and antibiotic resistance in coagulase-negative staphylococci colonizing the gastrointestinal tract of children in Ile-Ife, Nigeria. Trop. J. Pharm. Res. 2010, 9, 35–43. [Google Scholar] [CrossRef] [Green Version]
- Schaumburg, F.; Alabi, A.S.; Peters, G.; Becker, K. New epidemiology of Staphylococcus aureus infection in Africa. Clin. Microbiol. Infect. 2014, 20, 589–596. [Google Scholar] [CrossRef] [Green Version]
- Rasigade, J.P.; Laurent, F.; Lina, G.; Meugnier, H.; Bes, M.; Vandenesch, F.; Etienne, J.; Tristan, A. Global distribution and evolution of panton-valentine leukocidin-positive methicillin-susceptible staphylococcus aureus, 1981–2007. J. Infect. Dis. 2010, 201, 1589–1597. [Google Scholar] [CrossRef] [Green Version]
- Shittu, A.; Oyedara, O.; Abegunrin, F.; Okon, K.; Raji, A.; Taiwo, S.; Ogunsola, F.; Onyedibe, K.; Elisha, G. Characterization of methicillin-susceptible and -resistant staphylococci in the clinical setting: A multicentre study in Nigeria. BMC Infect. Dis. 2012, 12, 286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruimy, R.; Maiga, A.; Armand-Lefevre, L.; Maiga, I.; Diallo, A.; Koumaré, A.K.; Ouattara, K.; Soumaré, S.; Gaillard, K.; Lucet, J.C.; et al. The carriage population of Staphylococcus aureus from Mali is composed of a combination of pandemic clones and the divergent Panton-Valentine leukocidin-positive genotype ST152. J. Bacteriol. 2008, 190, 3962–3968. [Google Scholar] [CrossRef] [Green Version]
- Mietze, A.; Morick, D.; Köhler, H.; Harrus, S.; Dehio, C.; Nolte, I.; Goethe, R. Combined MLST and AFLP typing of Bartonella henselae isolated from cats reveals new sequence types and suggests clonal evolution. Vet. Microbiol. 2011, 148, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Jappe, U.; Heuck, D.; Strommenger, B.; Wendt, C.; Werner, G.; Altmann, D.; Witte, W. Staphylococcus aureus in dermatology outpatients with special emphasis on community-associated methicillin-resistant strains. J. Investig. Dermatol. 2008, 128, 2655–2664. [Google Scholar] [CrossRef] [Green Version]
- Sudagidan, M.; Aydin, A. Virulence properties of methicillin-susceptible Staphylococcus aureus food isolates encoding Panton-Valentine Leukocidin gene. Int. J. Food Microbiol. 2010, 138, 287–291. [Google Scholar] [CrossRef] [Green Version]
- Rosenthal, M.E.; Mediavilla, J.; Chen, L.; Sonnenfeld, J.; Pierce, L.; Shannon, A.; Boucher, H.; Pearlmutter, M.; Kreiswirth, B.; Kuo, Y.H.; et al. Molecular epidemiology of Staphylococcus aureus in post-earthquake northern Haiti. Int. J. Infect. Dis. 2014, 29, 146–151. [Google Scholar] [CrossRef] [Green Version]
- Dinges, M.M.; Orwin, P.M.; Schlievert, P.M. Exotoxins of Staphylococcus aureus. Clin. Microbiol. Rev. 2000, 13, 16–34. [Google Scholar] [CrossRef]
- Zhu, X.; Sun, X.; Zeng, Y.; Feng, W.; Li, J.; Zeng, J.; Zeng, Y. Can nasal Staphylococcus aureus screening and decolonization prior to elective total joint arthroplasty reduce surgical site and prosthesis-related infections? A systematic review and meta-analysis. J. Orthop. Surg. Res. 2020, 15, 60. [Google Scholar] [CrossRef] [PubMed]
- Haney Carr, J.; Hageman, J. Guidance on the Diagnosis and Management of PVL-Associated Staphylococcus aureus Infections (PVL-SA) in England, 2nd ed.; Health Protection Agency: London, UK, 2008.




{kind=link}
{kind=link}
{kind=link}
| ST | spa Type | Isolate | AMGs | BLAs | FOF | MLs | QA | QNs | ST | TET | TMP | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| aac(6′)-Ie aph(2″)-Ia | aph(3′)-IIIa | blaI | blaR1 | blaZ | mecA | mecI | mecR1 | fosB | glpT A100V | glpT L27F | murA E291D | murA G257D | murA T396N | erm(C) | mph(C) | msr(A) | qacC | gyrA S84L | parC S80F | parC S80Y | sat4 | tet(38) | tet(K) | dfrG | dfrS1 | |||
| 8 | t121 | 7111 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 0 | 0 |
| 8 | t121 | 961 | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 0 | 1 | 0 | 1 | 1 | 1 | 0 | 0 | 0 |
| 8 | t1476 | 1684 | 1 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 0 | 1 | 0 | 1 | 1 |
| 8 | t1476 | 2090 | 1 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 0 | 1 | 0 | 1 | 1 | 1 | 1 |
| 8 | t1476 | 32 | 0 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 0 | 1 | 1 | 1 | 1 |
| 8 | t1476 | 5232 | 1 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | 1 | 1 | 1 |
| 8 | t1476 | 998 | 1 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | 1 | 1 | 1 |
| 8 | t1476 | 1052 | 1 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 0 | 1 | 0 | 1 | 1 |
| 8 | t1476 | 1053 | 1 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 0 | 1 | 0 | 1 | 1 |
| 8 | t1476 | 1057 | 1 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | 1 | 1 | 1 |
| 8 | t1476 | 1401 | 1 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 1 | 1 | 1 |
| 8 | t1476 | 1405 | 1 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 0 | 1 | 0 | 1 | 1 |
| 8 | t1476 | 994 | 1 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | 1 | 1 | 0 |
| 8 | t1476 | 995 | 1 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 0 | 1 | 1 | 1 | 1 |
| 8 | t1476 | 996 | 1 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 0 | 1 | 0 | 1 | 1 |
| 121 | t314 | 3326 | 0 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 1 | 1 | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 |
| 121 | t314 | 2777 | 0 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 1 | 1 | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 |
| 121 | t314 | 4101 | 0 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 1 | 1 | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 |
| 121 | t314 | 562 | 0 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 1 | 1 | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 |
| 121 | t314 | 1043 | 0 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 1 | 1 | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 |
| 152 | t1096 | 905 | 0 | 0 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 |
| 152 | t1096 | 907 | 0 | 0 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 |
| 152 | t355 | 2656 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 |
| 152 | t4690 | 2007 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 |
| 152 | t4690 | 3247 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 |
| 772 | t657 | 5729 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 1 | 0 |
| 789 | t091 | 1002 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 0 |
| ST | spa Type | Isolate | Exo-Enzymes | Host Immunity | Toxins | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| aur | splA | splB | splE | ACME | sak | scn | edinB | hlgA | hlgB | hlgC | lukD | lukE | lukF-PV | lukS-PV | sea | seb | sec | seg | sei | sej | sek | sel | sem | sen | seo | sep | seq | ser | seu | tst | |||
| 8 | t121 | 7111 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| 8 | t121 | 961 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 8 | t1476 | 1684 | 1 | 1 | 0 | 1 | 0 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 |
| 8 | t1476 | 2090 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| 8 | t1476 | 32 | 1 | 1 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 |
| 8 | t1476 | 5232 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| 8 | t1476 | 998 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| 8 | t1476 | 1052 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| 8 | t1476 | 1053 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 |
| 8 | t1476 | 1057 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| 8 | t1476 | 1401 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 |
| 8 | t1476 | 1405 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 8 | t1476 | 994 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| 8 | t1476 | 995 | 1 | 1 | 0 | 1 | 0 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| 8 | t1476 | 996 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 (2) | 0 | 0 |
| 121 | t314 | 3326 | 1 | 0 | 1 | 0 | 0 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 1 | 0 |
| 121 | t314 | 2777 | 1 | 0 | 1 | 0 | 0 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 | 0 |
| 121 | t314 | 4101 | 1 | 0 | 1 | 0 | 0 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 | 0 |
| 121 | t314 | 562 | 1 | 0 | 1 | 0 | 0 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 | 0 |
| 121 | t314 | 1043 | 1 | 0 | 1 | 0 | 0 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 | 0 |
| 152 | t1096 | 905 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 152 | t1096 | 907 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 152 | t355 | 2656 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 1 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 152 | t4690 | 2007 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 152 | t4690 | 3247 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 772 | t657 | 5729 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 0 | 0 | 1 | 1 | 1 | 0 | 1 | 0 (1) | 1 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 1 | 0 |
| 789 | t091 | 1002 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laurence Yehouenou, C.; Bogaerts, B.; Vanneste, K.; De Keersmaecker, S.C.J.; Roosens, N.H.C.; Kpangon, A.A.; Affolabi, D.; Simon, A.; Dossou, F.M.; Dalleur, O. Whole-Genome Sequencing-Based Screening of MRSA in Patients and Healthcare Workers in Public Hospitals in Benin. Microorganisms 2023, 11, 1954. https://doi.org/10.3390/microorganisms11081954
Laurence Yehouenou C, Bogaerts B, Vanneste K, De Keersmaecker SCJ, Roosens NHC, Kpangon AA, Affolabi D, Simon A, Dossou FM, Dalleur O. Whole-Genome Sequencing-Based Screening of MRSA in Patients and Healthcare Workers in Public Hospitals in Benin. Microorganisms. 2023; 11(8):1954. https://doi.org/10.3390/microorganisms11081954
Chicago/Turabian StyleLaurence Yehouenou, Carine, Bert Bogaerts, Kevin Vanneste, Sigrid C. J. De Keersmaecker, Nancy H. C. Roosens, Arsène A. Kpangon, Dissou Affolabi, Anne Simon, Francis Moise Dossou, and Olivia Dalleur. 2023. "Whole-Genome Sequencing-Based Screening of MRSA in Patients and Healthcare Workers in Public Hospitals in Benin" Microorganisms 11, no. 8: 1954. https://doi.org/10.3390/microorganisms11081954