
Analogues of Pyrimidine Nucleosides as Mycobacteria Growth Inhibitors
by
 , , and
, , and
Liudmila A. Alexandrova
*, 
Anastasia L. Khandazhinskaya
,
Elena S. Matyugina
,
Dmitriy A. Makarov
and
Sergey N. Kochetkov
Engelhardt Institute of Molecular Biology RAS, 32 Vavilov Str., 119991 Moscow, Russia
*
Author to whom correspondence should be addressed.
Microorganisms 2022, 10(7), 1299; https://doi.org/10.3390/microorganisms10071299
Submission received: 30 May 2022
/
Revised: 23 June 2022
/
Accepted: 24 June 2022
/
Published: 27 June 2022
(This article belongs to the Special Issue Mycobacterium tuberculosis Infection: Control & Treatment)
Abstract
:Tuberculosis (TB) is the oldest human infection disease. Mortality from TB significantly decreased in the 20th century, because of vaccination and the widespread use of antibiotics. However, about a third of the world’s population is currently infected with Mycobacterium tuberculosis (Mtb) and the death rate from TB is about 1.4–2 million people per year. In the second half of the 20th century, new extensively multidrug-resistant strains of Mtb were identified, which are steadily increasing among TB patients. Therefore, there is an urgent need to develop new anti-TB drugs, which remains one of the priorities of pharmacology and medicinal chemistry. The antimycobacterial activity of nucleoside derivatives and analogues was revealed not so long ago, and a lot of studies on their antibacterial properties have been published. Despite the fact that there are no clinically used drugs based on nucleoside analogues, some progress has been made in this area. This review summarizes current research in the field of the design and study of inhibitors of mycobacteria, primarily Mtb.
1. Introduction
Tuberculosis (TB) is the oldest human infectious disease, which has probably been inherited from early hominids [1]. Currently, about a third of the world’s population is infected with Mycobacterium tuberculosis (Mtb). The death rate from TB is 1.4–2 million people a year. Tuberculosis affects more than nine million people every year, and since 2000 the incidence rate has fallen by only 1.5 percent a year [2,3].
The development of drugs for the treatment of TB has remained one of the priority tasks of pharmacology and medicinal chemistry since the identification of Mtb by R. Koch in 1882 [4]. During the 20th century, many anti-tuberculosis drugs were discovered or synthesized and numerous treatment regimens were developed using first-line and second-line antibiotics [5,6,7]. As a result, the mortality from this disease was reduced tenfold [2,3] and it seemed that the final victory over TB had been achieved.
However, in the middle of the last century, drug-resistant forms of TB were identified. A rapid spread of antibiotic-resistant strains of Mtb was observed, on which standard chemotherapy has practically no effect [8,9,10,11,12,13,14,15,16,17]. In the future, worsening of the situation should be expected. According to WHO, by 2050, out of 10 million deaths that could be associated with drug resistance, about a quarter will be due to drug-resistant strains of tuberculosis [2,3]. There is an opinion that humanity is entering a post-antibiotic era, in which even common infections or minor injuries can be life-threatening [18,19].
Thus, the emergence of new multidrug-resistant strains is one of the main problems in the current century [2,8]. There is an acute need for the development of novel drugs that can act on new pathogen’s targets and are active against resistant strains of bacteria and viruses.
Nucleosides as antibacterial agents, in particular anti-tuberculosis, look very attractive. Nucleosides and nucleotides are the basic structural units of DNA and RNA which are involved in the biosynthesis of proteins. They also act as cofactors in different metabolic pathways, including lipid and polyamine biosynthesis [19,20]. It is known that in some cases even small modifications of the heterocyclic base or carbohydrate fragment of the nucleoside can have a significant effect on the recognition and inhibition of certain targets and thus block the growth of the corresponding microorganisms.
The antibacterial activity of nucleoside derivatives was revealed not so long ago; systematic studies of antimycobacterial activity have been carried out over the past two decades. Several reviews have been published on modified nucleosides that inhibit the growth of mycobacteria (Mtb, Mycobacterium bovis and Mycobacterium avium) [19,21,22,23,24,25,26,27]. Modified nucleosides inhibit enzymes involved in the metabolism of purines [26,27,28] and pyrimidines [23,24,25,26,27,29] and in the processes of iron transportation necessary for the life of mycobacteria [21,22,30] and others. Despite the fact that there is no data on clinical trials of the nucleoside agents in the literature, further study of their antimicrobial properties is forthcoming and may reveal new antibiotics of a nucleoside nature.
The present review describes the antimycobacterial activity of modified pyrimidine nucleosides and their analogues as well as possible targets of their action. It considers only derivatives and analogues of pyrimidine nucleosides, since there are detailed reviews about the anti-tuberculous activity of purine nucleoside derivatives, mainly about the synthesis and study of sulfamoyl-adenosine analogues, inhibitors of the biosynthesis of siderophores—iron carriers—which are necessary for the survival of mycobacteria [21,22,30,31].
2. Inhibitors of M. tuberculosis Growth with Unidentified Target
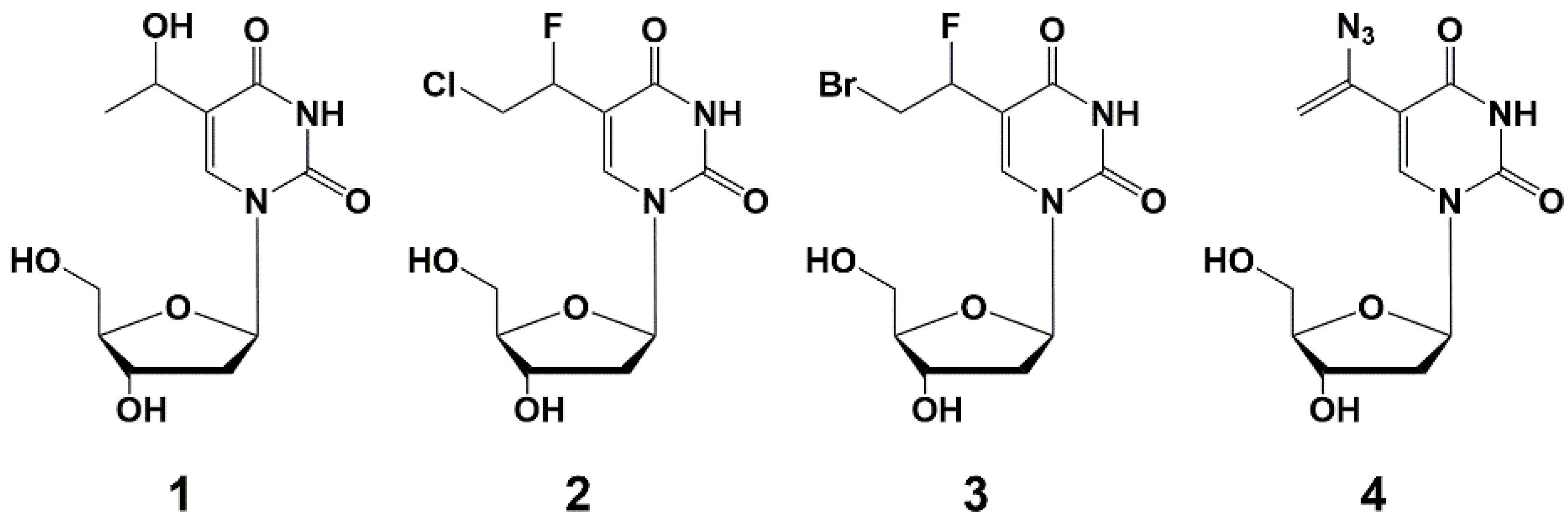
Kumar et al. firstly synthesized nucleoside analogues and accessed their ability to inhibit the growth of mycobacteria in vitro [32]. They obtained derivatives of ribo-, deoxyribo- and dideoxyribonucleosides in order to study the effect of various groups at C-5, C-2 and C-3 positions of the nucleoside on the antimycobacterial activity against M. avium. The authors suggested that such compounds are able to interact with mycobacterial enzymes involved in the process of nucleic acid synthesis as inhibitors or competitive substrates, thus affecting the synthesis of DNA or RNA [32,33], but this hypothesis was not confirmed. Among the wide range of synthesized compounds, 5-substituted derivatives 1–4 showed the highest activity (Figure 1). Compounds selectively inhibited the growth of M. avium by 90% in 1–5 µg/mL concentrations, which is the same level of values for clinically used rifampicin (MIC90 2 µg/mL) [32]. However, analogue 4 was completely inactive against M. bovis and M. tuberculosis [33].
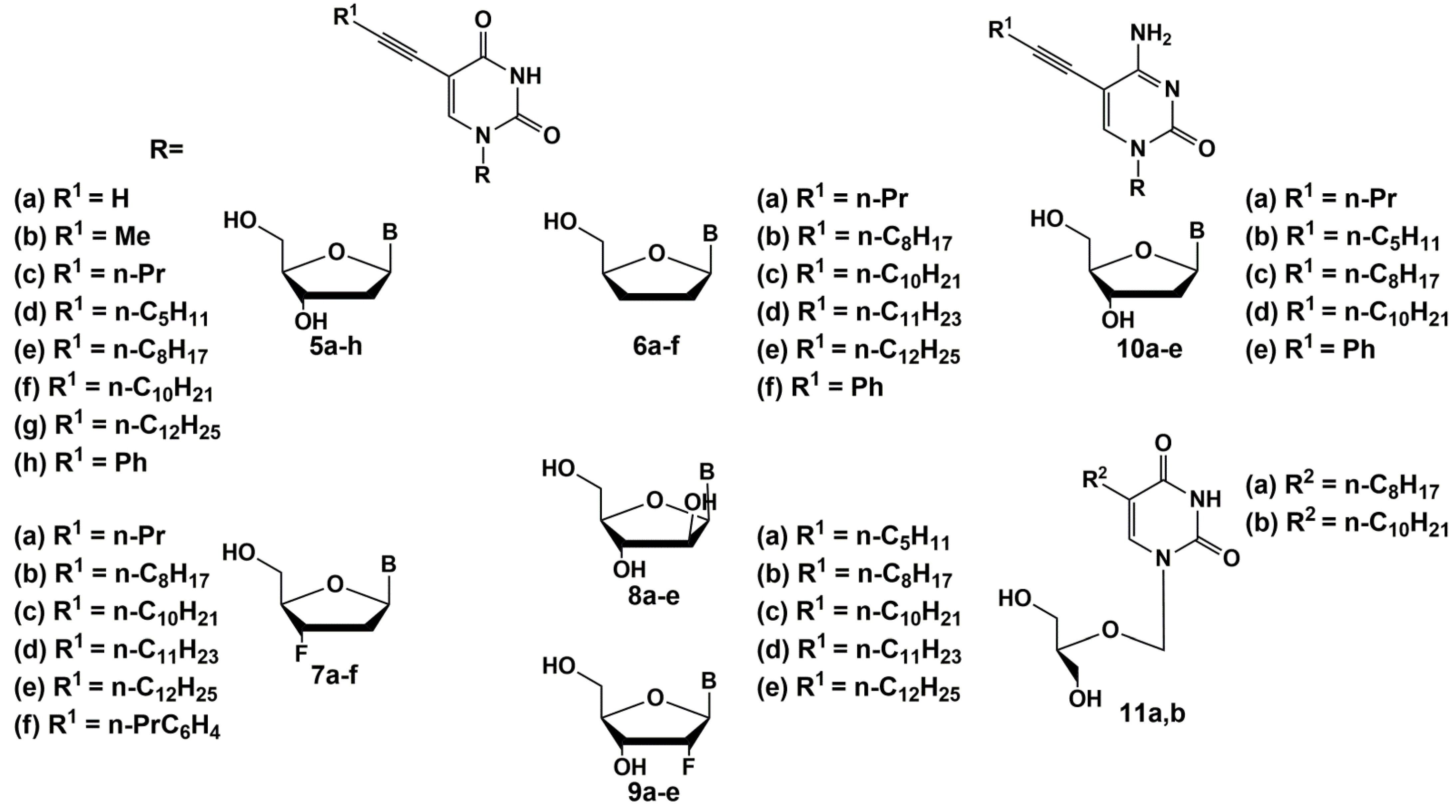
Since 2005, the same group has published a series of papers [34,35,36,37]. They have synthesized a wide range of C-5 alkynyl pyrimidine nucleoside derivatives that effectively inhibit the growth of mycobacteria, including M. tuberculosis (Figure 2) [35,36,37]. The synthesis of these compounds was carried out by the Sonogashira reaction starting from the corresponding 5-iodo derivatives of 2′-deoxyuridine, modified at the carbohydrate moiety [38,39].
The SAR studies showed that 5-(1-dodecynyl) and 5-(1-tetradecinyl) derivatives of deoxynucleosides had the best antimycobacterial activity. 5-Substituted derivatives of 2′-deoxyribouridine were more effective than their riboanalogues, while 5-modified uracil did not inhibit the growth of mycobacteria [34]. The study of SAR of modification of the carbohydrate fragment as a possible antimycobacterial pharmacophore of these nucleosides showed that almost all 2′-deoxy-5 [34], 2′,3′-dideoxy-6, 2′,3′-dideoxy-3′-fluoro-7 [36], 2′,3′-dideoxy-2′-fluoro-uridines 9, arabinouridines 8 [35] and acyclic nucleosides 11 [37] with long 1-alkynyl substituents are able to inhibit the growth of mycobacteria.
Thus, among the alkynyl derivatives of 2′-deoxynucleosides 5a–h and 10a–e, the highest activity against M. bovis and M. avium was shown by 5-dodecynyl-5f (MIC90 50 μg/mL), 5-tetradecinyl-2′-deoxyuridine 5g (MIC90 10 µg/mL) and 5-dodecynyl-2′-deoxycytidine 10d (MIC90 10 µg/mL). It should be noted that the activity increases with the length of the substituent in the 5 position of the pyrimidine base. To elucidate the role of the carbohydrate fragment, the authors also obtained 2′,3′-dideoxy derivatives 6a–f and 3′-fluoro-2′,3′-dideoxy derivatives 7a–f. The best activity against M. bovis, Mtb and M. avium was shown by compounds 6c (MIC50 1–2, 1–2, 1–50 µg/mL for each mycobacterium, respectively) and 7e (MIC50 1, 1–2, 100 μg/mL, respectively). In most cases, the studied compounds were non-toxic for different cell cultures (Huh-7, Vero or HFF cell cultures: CC50 ≥ 100 µg/mL). Thus, in general, with an increase in the length of the hydrocarbon radical, it leads to an increase in activity including one against the rifampicin-resistant Mtb strain, which is also observed for nucleosides with different modifications of the carbohydrate moiety. Among derivatives 8a–e and 9a–e, compounds 10c (MIC90 1–5, 1–5, 50–100 µg/mL) and 9d (MIC90 1, 1, 50 µg/mL) showed the highest activity against M. bovis, Mtb and M. avium (MIC90 1, 1, 50–100 μg/mL) [34,35,36,37], which confirms the importance of the long alkyl substituent.
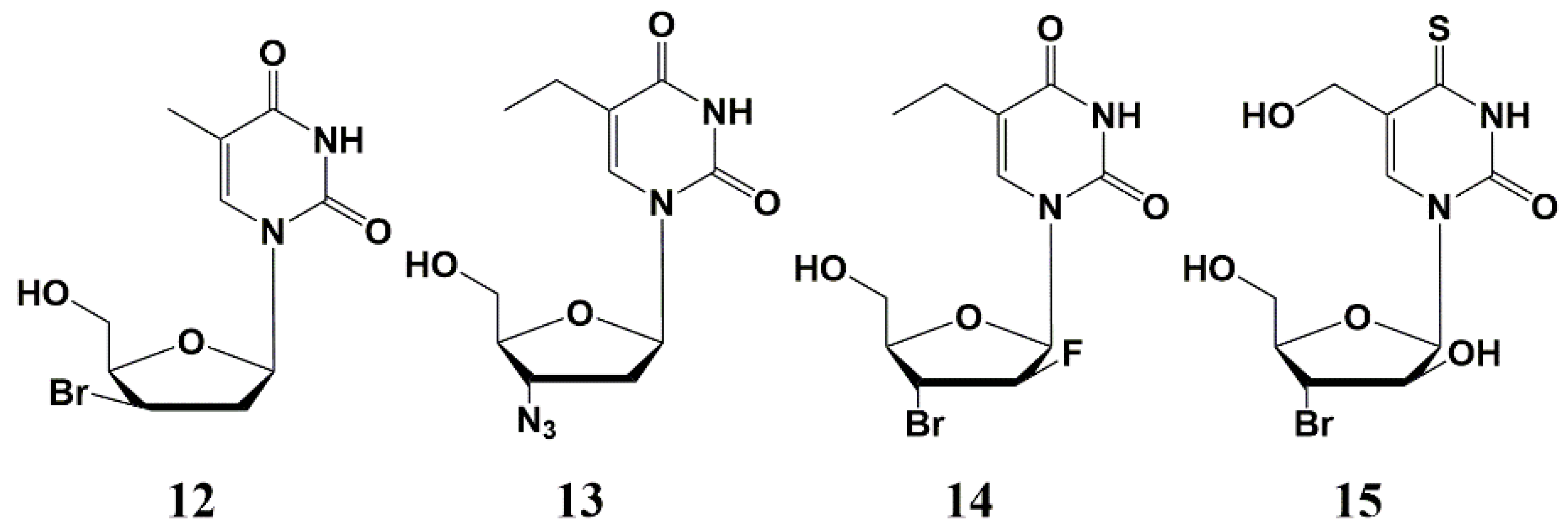
Later various 2′- or 3′-halogen derivatives of pyrimidine nucleosides containing substituents at the C-5 position of the base were synthesized by Canadian scientists [40]. Among this series, the most effective inhibitor was a 3′-bromo derivative 12 (Figure 3) with a MIC50 1–3 µg/mL against the strains of Mtb H37Rv and H37Ra. Interestingly, compound 12 also inhibits the intracellular form of Mtbon, a human monocyte cell line (THP-1) infected with H37Ra at a concentration of 25 μg/mL, showing a higher activity against the intramacrophage form of mycobacterium compared to the extra macrophage form. In addition, no cytotoxicity was found for this type of compound against human cells (CD50 > 100–200 µg/mL) [40].
Further studies led to the synthesis and study of the antimycobacterial properties of a series of 3′-azido-, 3′-amino- and 3′-halogen derivatives of pyrimidine nucleosides, among which only 3′-azido-5-ethyl-2′,3′-dideoxyuridine 13 showed significant activity against M. bovis, M. avium and Mtb (MIC50 1–5 µg/mL) (Figure 3) [41]. In the same year, Kumar et al. published paper on the synthesis and study of 5-ethyl-, 5-hydroxymethyl- and 5-methoxymethyl-substituted pyrimidines. The best inhibitory activity in this series of nucleosides was shown by 2′-fluoro derivative 14 (MIC50 5 µg/mL), as well as 4-thio derivative 15 (Figure 3), inhibiting the Mtb H37Rv strain (MIC50 0.5 µg/mL), H37Ra (MIC50 1 µg/mL), as well as M. bovis and M. avium (MIC50 1 and 10 µg/mL, respectively) [41]. No cellular cytotoxicity was found for these compounds up to the highest concentration tested (CC50 > 100 µg/mL).
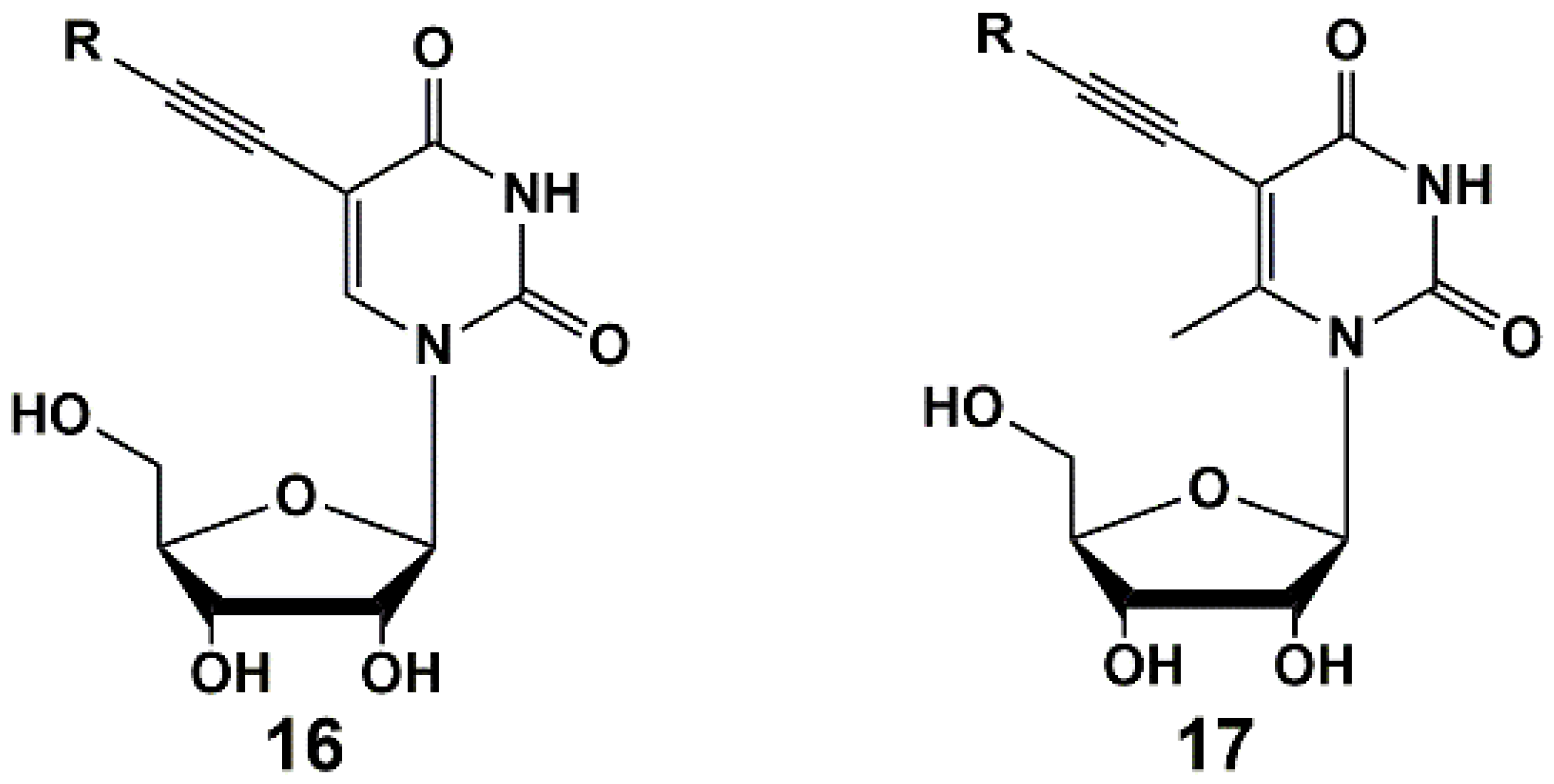
Recently, series of 5-alkynyl-substituted uridines 16 [42] and 6-methyluridines 17 [43] were synthesized (Figure 4) via Sonogashira reaction. It was found that a methyl group in C-6 position of the pyrimidine ring had no impact on the yields of target compounds as well as on the inhibitory activity. Synthesized nucleosides showed activity against M. bovis and Mtb at concentrations of 1–100 µg/mL (MABA test). The MIC50 values of some compounds were similar or close to that of the reference drug rifampicin.
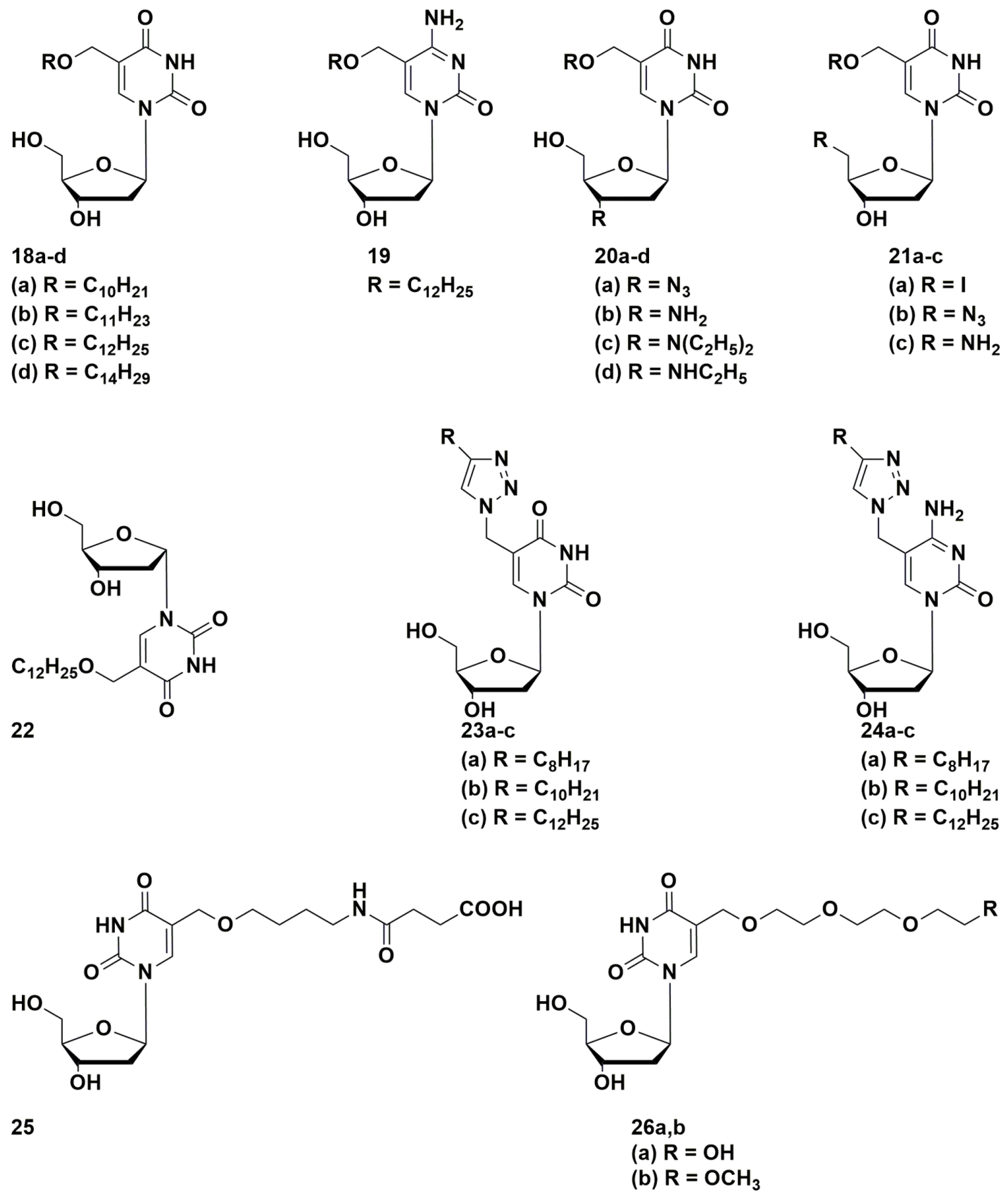
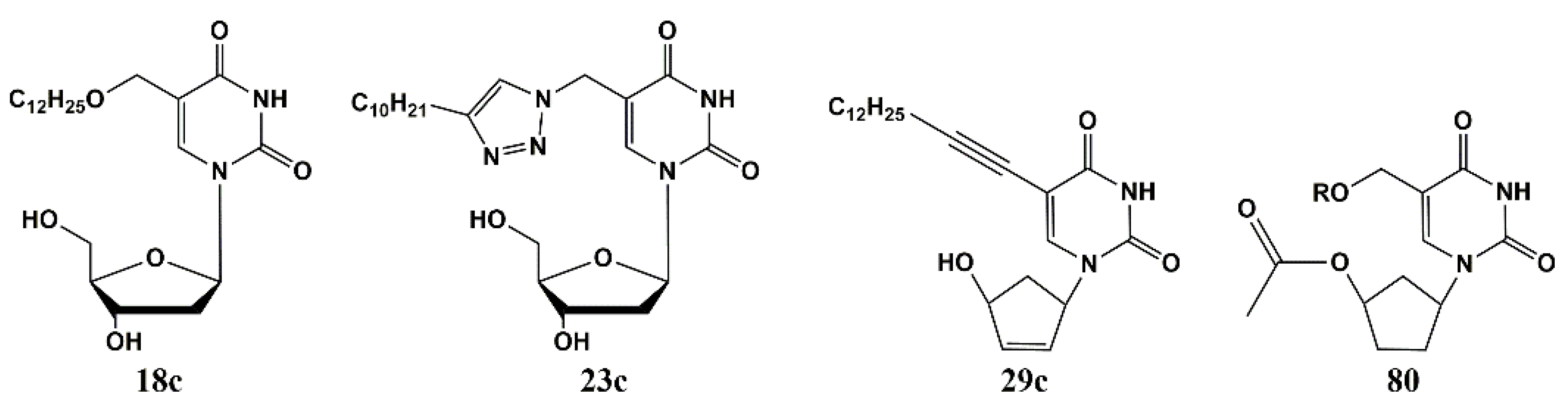
Of considerable interest are works on the synthesis of 5-alkyloxymethyl and 5-alkyltriazolylmethyl derivatives of pyrimidine nucleosides with long alkyl substituents (Figure 5) [44,45]. The anti-tuberculosis activity of the synthesized compounds was tested on two Mtb strains: laboratory strain H37Rv and multidrug-resistant (MDR) strain MS-115 (resistant to five first-line anti-TB drugs). C-5 Alkynyl derivatives 18c, 23b, and 23c with a hydrocarbon chain length of 10 or 12 carbon atoms were the most effective; while compounds with shorter substituent lengths weakly inhibited the growth of mycobacteria. In general, 2′-deoxyuridine derivatives were more active than 2′-deoxycytidine derivatives. Analogues 18c, 23b and 23c inhibited the growth of the Mtb at concentrations comparable to currently used drugs: laboratory strain H37Rv at MIC99 20, 10, 10 µg/mL, respectively and resistant MS-115 at MIC99 50, 10, 10 µg/mL, respectively [44,45]. The value of CD50 in A549, Vero and Jurkat cell cultures was about 70–100 μg/mL.
Described compounds containing long hydrophobic groups are poorly soluble in water-organic solutions. At the same time, better soluble 5-alkyltriazolylmethyl-2′-deoxyuridines with short alkyl residues, in contrast to decyl- and dodecyl-triazolylmethyl-2′-deoxyuridines, demonstrated antibacterial activity against a number of Gram-positive bacteria [46]. On the other hand, 2′-deoxyuridine derivatives containing long hydrophilic substituents 26a,b in the 5-position (which is similar to a chain of 10–12 atoms in the C-5 position of the pyrimidine base of the nucleosides that previously showed the best anti-tuberculosis activity) were well-soluble compounds, but completely lost antimicrobial activity [47].
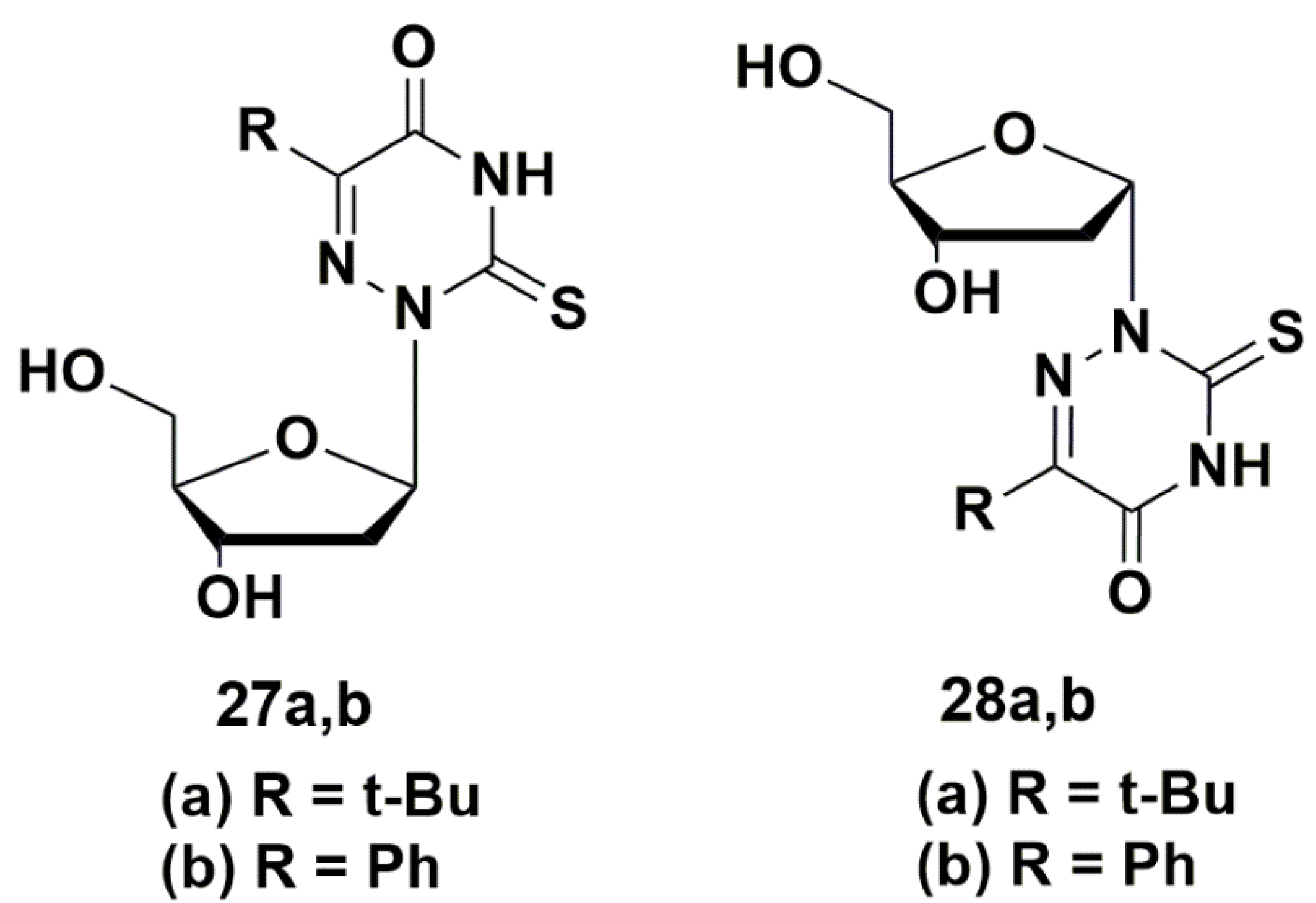
A number of 5-modified 6-aza-2′-deoxyuridines 27a,b and 28a,b (Figure 6) demonstrated inhibitory activity not only against Mycobacterium smegmatis (MIC99 0.2–0.8 mM), but also against Staphylococcus aureus (MIC99 0.03–0.9 mM). Compound 28b showed the best results, comparable with the activity of clinically used drugs [48].
5′-Norcarbocyclic Uracil Derivatives
Another interesting example of antimycobacterial nucleoside analogues are 5-alkynyl 5′-norcarbocyclic uridine derivatives [49], which were synthesized by the method described earlier [34]. Carbocyclic nucleosides are analogues of natural nucleosides in which the oxygen atom in the furanose ring is replaced with methylene group. A number of carbocyclic nucleosides, such as aristeromycine and neplanocin A, have been found in nature [50,51,52,53]. Carbocyclic analogues are more stable to phosphorylases and hydrolases but due to their structural similarity to natural nucleosides are recognized by many receptors and enzymes. Carbocyclic nucleosides showed a wide spectrum of biological activity, in particular, antiviral and anticancer properties [50,51,52,53,54]. However, in some cases being converted to triphosphate forms, which are very similar to natural nucleoside triphosphates, carbocyclic analogues exhibit significant toxicity as a result of their undesirable recognition by ATP-metabolizing enzymes [55,56,57]. In 5′-norcarbocyclic nucleoside analogues, the primary hydroxyl group is replaced by a secondary one, which prevents their recognition and phosphorylation by cellular kinases and leads to much less toxicity [53,57,58,59,60]. In this regard, 5′-norcarbocyclic nucleosides are a promising class of compounds for drug development.
The synthesized 5′-norcarbocyclic uracil derivatives (Figure 7) demonstrated anti-tuberculosis activity against both the laboratory (H37Rv) and multidrug-resistant (MS-115) Mtb strains. All compounds, except for 1-(4-hydroxy-2-cyclopenten-1-yl)-5-tetradecynyluracil (29c), showed moderate inhibitory activity on the laboratory strain H37Rv (MIC99 20–40 µg/mL). The compound 29c as racemic mixture completely suppressed the growth of mycobacteria at MIC99 10 µg/mL. Individual enantiomers of the compound 27c were isolated using enzymatic separation with Amano PS lipase [61] and tested against MDR strain (MS-115). The racemic mixture 29c as well as (+)29c enantiomer inhibited the growth of the resistant strain MS-115 at MIC99 10 μg/mL. At the same time, it was shown that the (−)29c enantiomer inhibited the growth of laboratory strain H37Rv at MIC99 5 μg/mL [49]. Compound 29c had no toxicity in Vero cell cultures up to the highest tested concentration (CC50 > 125 µg/mL).
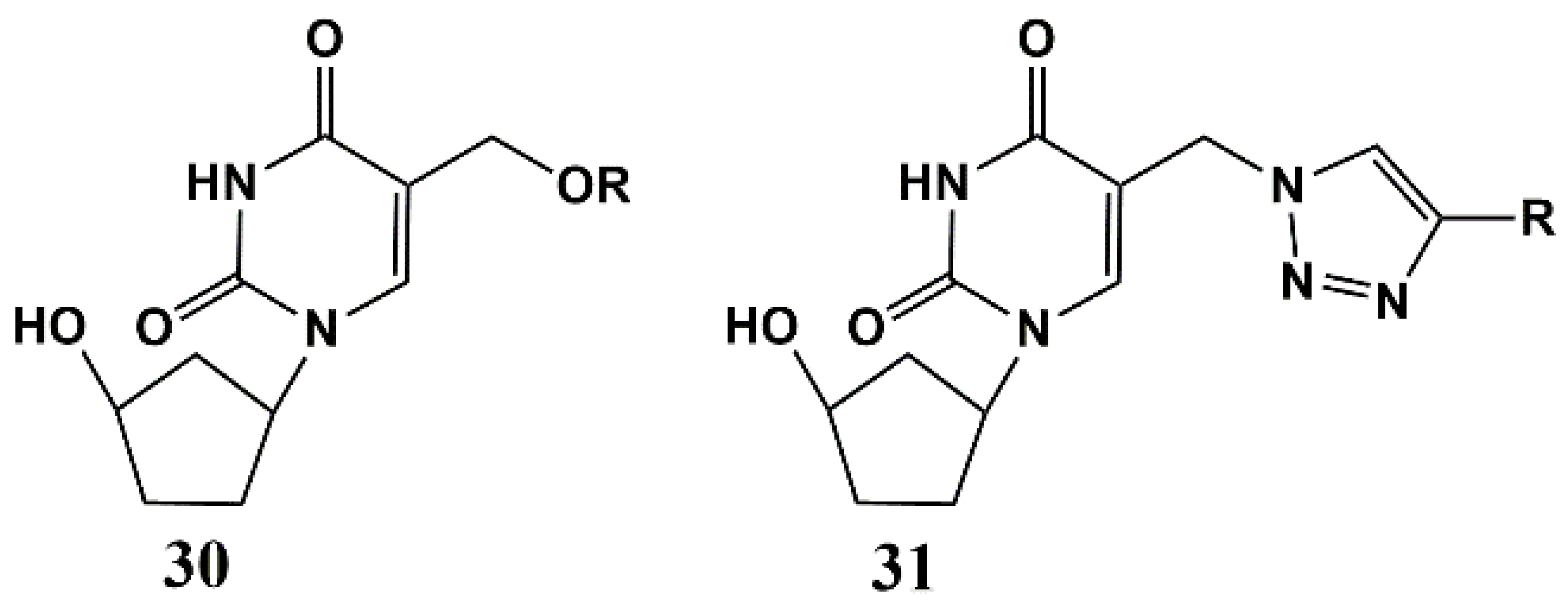
Later, 5-alkyloxymethyl and 5-alkyltriazolylmethyl 5′-norcarbocyclic uridine analogues 30 and 31 were synthesized (Figure 8) [62]. The antibacterial activity of these compounds was assessed against a number of bacteria, including both Gram-positive and Gram-negative strains. Such uracil derivatives prove to be selective inhibitors towards mycobacteria, demonstrating significant activity in vitro against two M. smegmatis strains (MIC99 67 and 6.7–67 μg/mL for mc2155 VKPM Ac 1339 strains), attenuated MtbATCC 25177 (MIC99 28–61 µg/mL) and M. bovis ATCC 35737 (MIC99 50–60 µg/mL) and two strains of Mtb, including the laboratory strain H37Rv (MIC99 20–50 µg/mL) and MDR MS-115 strain resistant to five first-line anti-TB drugs (MIC99 20–50 µg/mL). However, unlike both 5-alkyloxymethyl and 5-alkyltriazolylmethyl derivatives of 2′-deoxyuridine [44,45] and 5-alkynyl 5′-norcarbocyclic uracil derivatives [49], analogues 30 and 31 turned out to be toxic to human monocyte cells [62].
Another group of 5′-norcarbocyclic uridine analogues as Mtb inhibitors (MIC99 5–40 μg/mL) based on 5-arylaminouracil derivatives was synthesized [63,64]. The compounds showed no cytotoxicity in Vero, A549 and Huh7 cell cultures up to a concentration of 50 μg/mL.
Thus, the best activities among uridine analogues (MIC90 1–10 mg/mL) were found for 5-alkynyl (decynyl, dodecynyl, tetradecynyl, pyridylethynyl, etc.) uracil derivatives bearing various modifications in the carbohydrate moiety [23,32,33,34,35,36,37,44,45,47]. At the same time, 2′-deoxypyrimidine derivatives with long 5-alkyloxymethyl or 5-alkyltriazolylmethyl substituents (5-dodecyloxymethyl-2′-deoxyuridine, 5-decyltriazolylmethyl-2′-deoxyuridine, 5-dodecyltriazolylmethyl-2′-deoxycytidine, etc.) were able to inhibit in vitro the growth of both the laboratory strain H37Rv and the multidrug-resistant clinical isolate MtbMS-115 at MIC99 about 10–20 mg/mL [44,45,47]. Two groups of carbocyclic nucleoside analogues, namely, 5-alkynyl- and 5-alkoxymethyl-5′-norcarbocyclic uridine analogues also showed high activity against Mtb strains H37Rv and MDR MS-115 [49,62]. The observed biological activity of these new 5-substituted uracil derivatives well correlates with previously published data on the inhibition of Mtb growth by nucleoside analogues containing 5-substituted pyrimidine bases and various sugars, as well as their corresponding acyclic or carbocyclic analogues [23,32,33,34,35,36,37,47,49,62]. It can be assumed that these compounds have the same mechanism of antimicrobial action, and their effectiveness depends primarily on the length and structure of the substituent at the C-5 position of the pyrimidine base.
3. N4-Modified Cytidine Derivatives
Development of the effective inhibitors of mycobacteria among pyrimidine nucleosides have suggested that N4-modified cytidine derivatives containing extended lipophilic alkyl substituents can also exhibit antibacterial activity [65,66]. As shown earlier [23,32,33,34,35,36,37,47,49,62], derivatives with extended lipophilic alkyl substituents had the best activity against mycobacteria among the C-5-substituted pyrimidine nucleosides. To test the hypothesis, linear alkyl substituents (-CnH2n+1, n = 8–14) were introduced into the N4 position of the cytosine residue (Figure 9). A number of synthesized derivatives demonstrated inhibitory activity against M. smegmatis and some Gram-positive bacteria, including drug-resistant strains of Staphylococcus aureus MRSA [65]. It was shown that 2′-deoxy-5-methylcytidine derivatives 33 containing extended N-alkyl substituents C10H21 (33a) and C12H25 (33b) have the best activity (24–50 μM) comparable in some cases with the action of clinically used antibiotics.
N4-alkyl-2′-deoxy-5-methylcytidines (34) with additional modifications of the carbohydrate fragment also demonstrated high antibacterial activity (27–110 μM). However, these compounds, containing amino and alkylamino groups in the 3′-position of the carbohydrate fragment, showed high cytotoxicity on the HeLa cell line (6–60 μM), and couldn’t be considered as potential antibacterial drugs [66].
4. Depot Forms
4.1. Depot Forms with Increased Solubility
Low water solubility is a serious problem during the study of the antibacterial and/or antiviral properties of modified nucleosides. One of the ways to overcome this obstacle is the synthesis of depot forms (“prodrugs”) (The term depot form means a biologically inert or weakly active compound containing the parent drug, which undergoes transformation in vivo due to chemical or enzymatic cleavage to release the parent drug), which can be used to improve the pharmacokinetics, pharmacodynamics, solubility and toxicity of the drug. This approach helps in the effective delivery of the drug to the target of action and improves its pharmacokinetic characteristics due to the slower release of the active component [67,68,69,70,71,72,73].
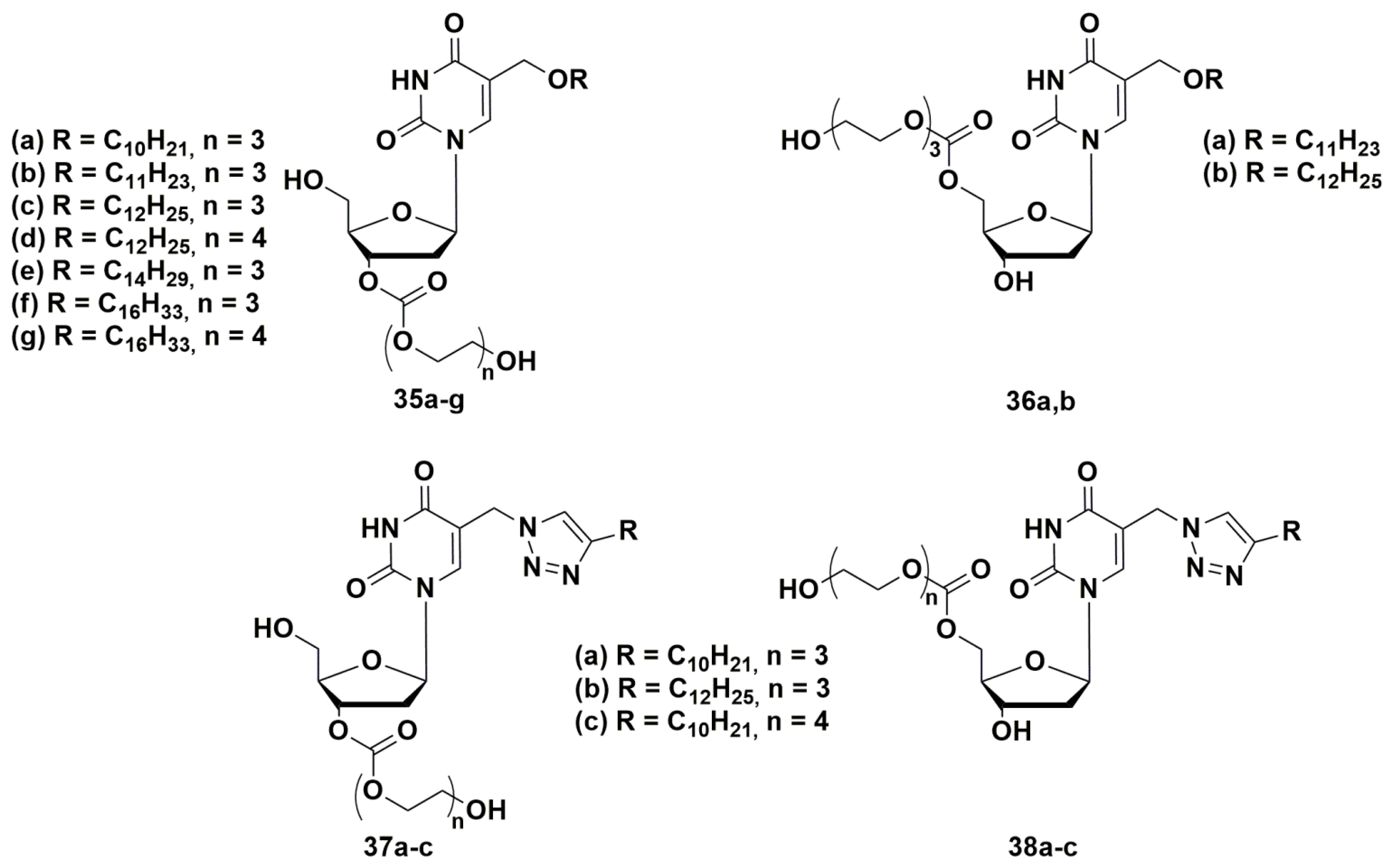
In order to develop a new set of antibacterial nucleosides, a representative library [74,75,76] of 3′- and 5′-tri- or tetraethylene glycol prodrugs of 5-alkyloxymethyl- and 5-alkyltriazolylmethyl-2′-deoxyuridines with described anti-tuberculosis activity were synthesized (Figure 10) [44,45].
The compounds were two orders of magnitude more soluble compared to the parent nucleosides, had significant inhibitory activity against a number of bacteria, including drug-resistant strains of M. smegmatis (as well as S. aureus), and exhibited low cytotoxicity [74,75,76]. The replacement of the triethylene glycol residue in C-5′ position by tetraethylene glycol led to an increase in water solubility and a significant decrease in inhibitory activity. It can be assumed that the inhibitory activity of 5-alkyloxymethyl-2′-deoxyuridines (35 and 36) depends on the substituent in C-5 position of the heterocyclic base and changes in the row: C10H21 < C11H23 < C12H25 > C14H29 > C16H33. The data obtained and the convenience of the proposed method for the synthesis of glycol prodrugs of nucleoside derivatives indicate the potential of their use as antibacterial agents.
4.2. Dual Action Nucleoside Derivatives
TB is the main opportunistic infection in HIV-infected people, which greatly raises the risk of HIV infection progression and death. HIV infection increases the chance of activation of the latent form of Mtb [77], and at the same time causes the rapid development of the disease soon after (re)infection [78]. Mtb and HIV act synergistically, which leads to a decrease the immunological status and death without treatment [79]. Therefore, it seems attractive to search for new dual-acting drugs that simultaneously inhibit the development of TB and HIV.
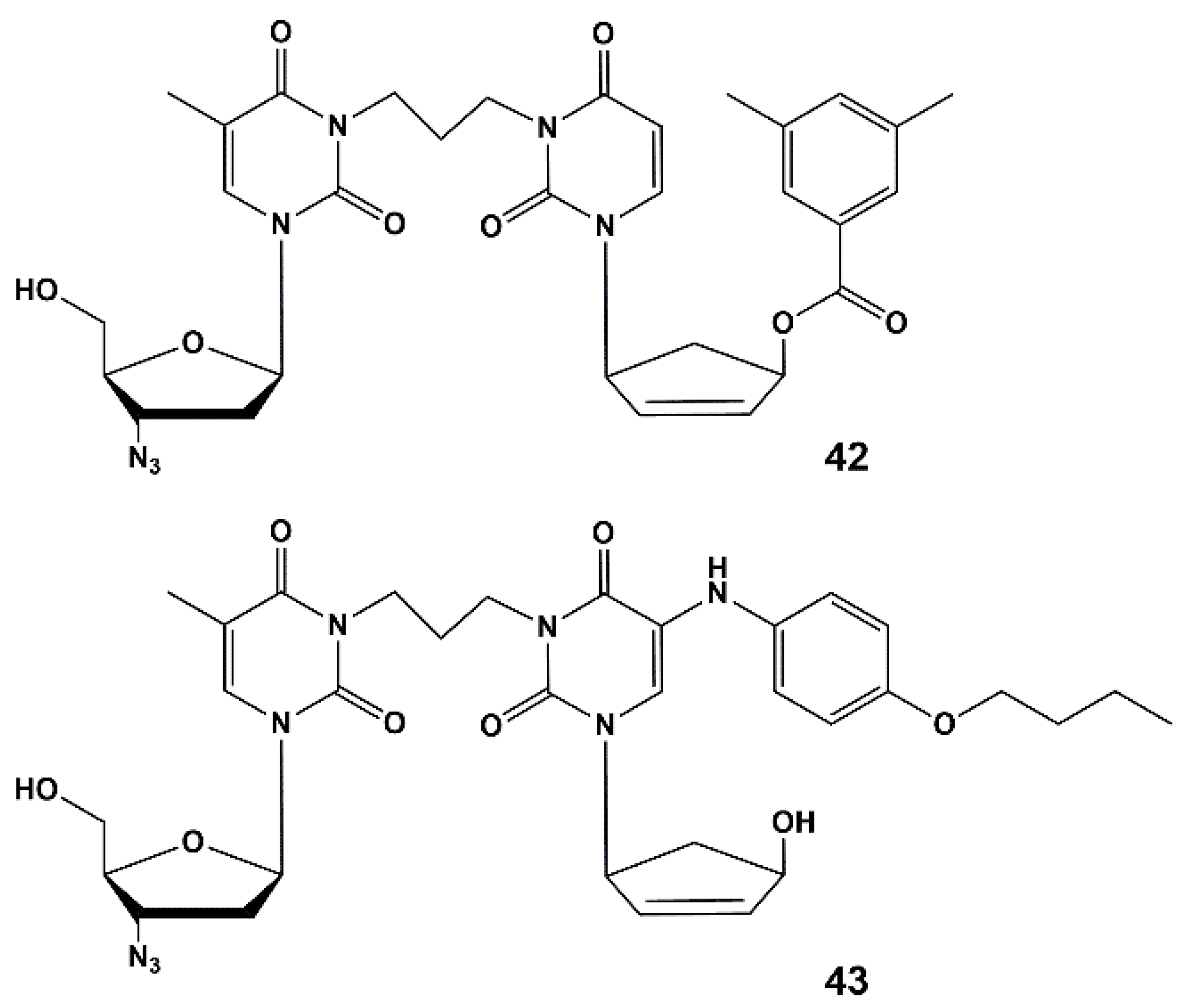
In this regard, five new compounds that can simultaneously inhibit HIV and tuberculosis in vitro and ex vivo have been designed and synthesized.
These compounds are heterodimers of 5-arylaminouracil derivative [63] or analogue of 2′-deoxyuridine 23b as anti-TB molecules [45] and of 3′-azido-3′-deoxythymidine (AZT) as anti-HIV molecules. The compounds described herein are the first examples of nucleoside derivatives having both anti-HIV and anti-TB activity.
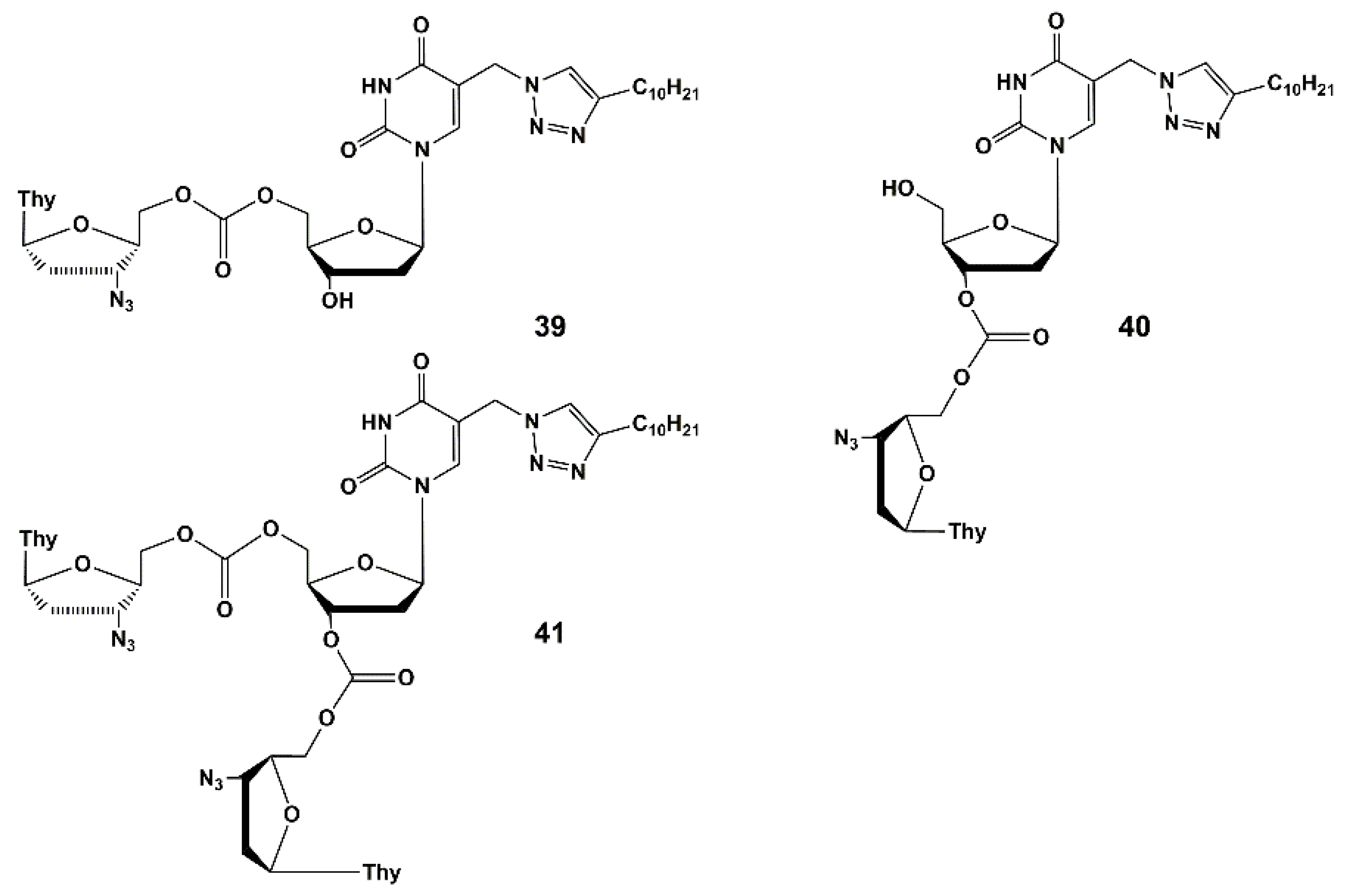
Heterodimers 39–43 (Figure 11) are depot forms of two active agents either with fast (39–41) or slow (42–43) hydrolysis rates. To optimize the structure of the active components and the linker that binds them, further studies are needed. The evaluation of the pharmacokinetic parameters of anti-HIV and anti-TB drugs released into the blood of laboratory animals should be conducted. It is worth noting that compound 39 showed the highest activity and inhibited HIV and the drug-resistant strain of tuberculosis MS-115 better than the original nucleoside 23b and a mixture of AZT and 23b (1:2 mol-equiv.). In MT-4 cell culture, moderate cytotoxicity and cytostatic effect were observed for heterodimers 39, 40, 42 and 43. However, the most active compound (39) had no toxicity and low cytostatic effect (about 4 times lower than AZT itself).
Thus, heterodimer 39 could be the candidate for future optimization and studying in vivo.
5. Target Enzymes for Nucleoside Inhibitors of M. tuberculosis
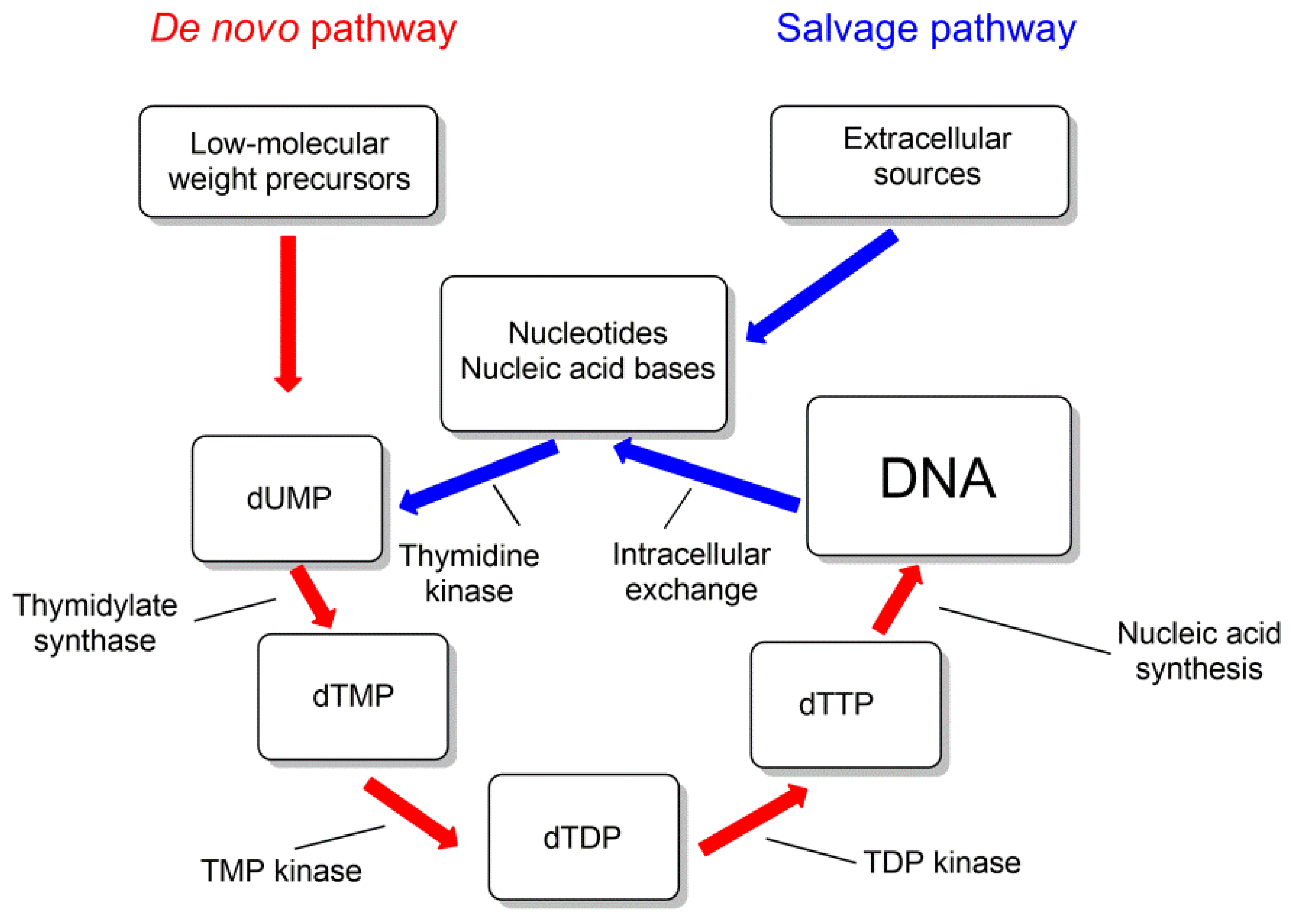
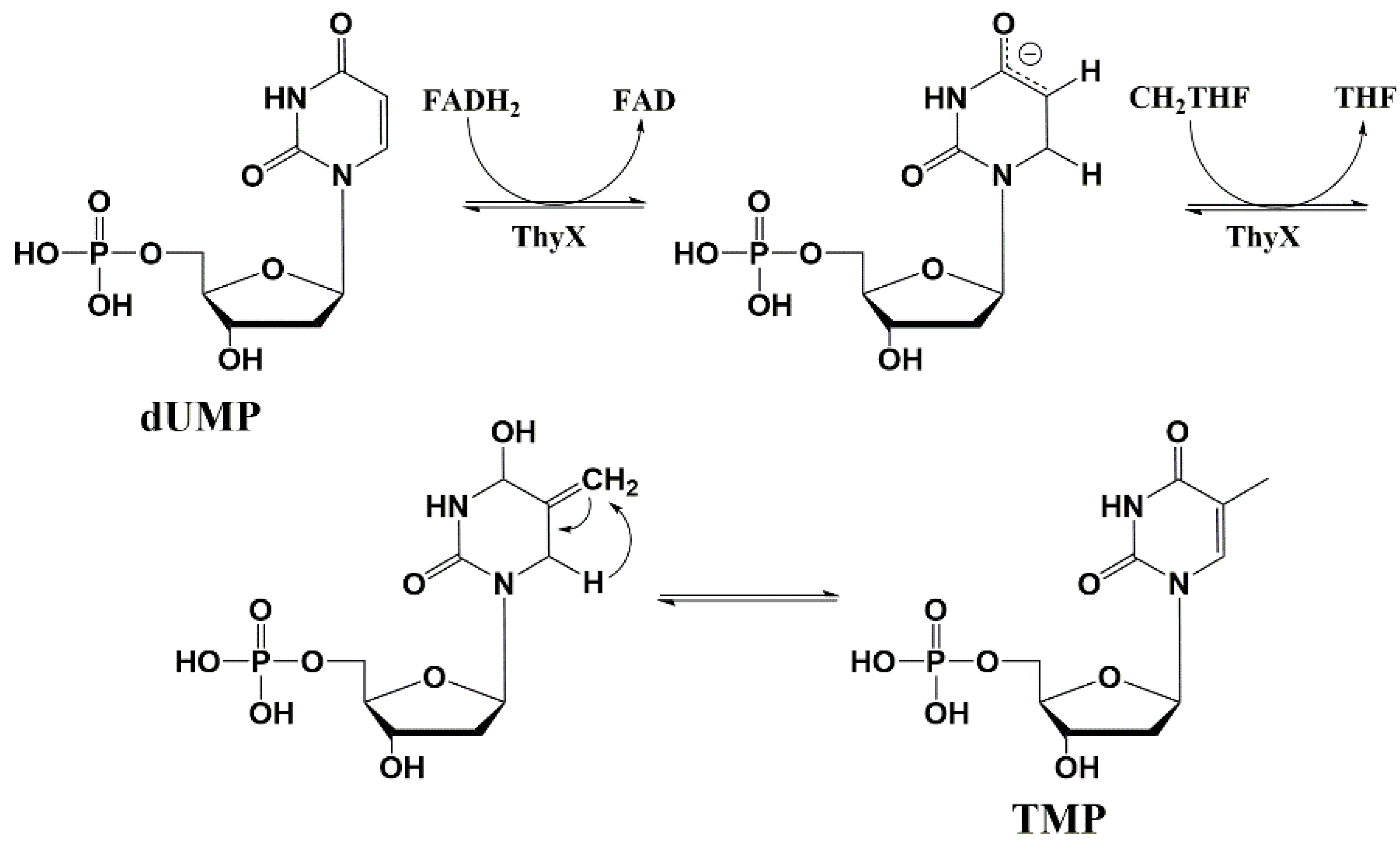
For some of the pyrimidine nucleoside derivatives that are inhibitors of mycobacteria growth, cellular targets were identified [29,80]. These are thymidylate synthase (TS) and thymidine monophosphate kinase (TMPK), catalyzing the biosynthesis of thymidine 5′-mono- and diphosphates (Figure 12).
Thymidine monophosphate kinase (TMPK) belongs to the nucleoside monophosphate kinase superfamily and catalyzes the phosphorylation of thymidine monophosphate (TMP) to thymidine diphosphate (TDP) using ATP as a donor of phosphate group. This enzyme connects the de novo and recycling pathways for the synthesis of thymidine triphosphate (Figure 12). By the de novo pathway, TMP is synthesized from deoxyuridine monophosphate (dUMP) by thymidine synthase (TS) (ThyA or ThyX); by the recycling pathway, thymidine is phosphorylated by thymidine kinase to form TMP [29,81].
To date, a significant number of thymidine derivatives have been synthesized and tested as inhibitors of Mtb growth. For some of them, the action on the enzymes mentioned above is directly shown, for others the target was not finally identified.
5.1. Thymidine Monophosphate Kinase Inhibitors
TMPK is a perspective target for the development of anti-tuberculosis drugs that are able to selectively effect the mycobacterial isoform of TMPK, which is vital for the mycobacteria growth.
TMPKmtb is a homodimer (Figure 13), each monomer of which consists of 214 amino acid residues. TMPKmtb is a thermostable enzyme (Tm 68°) (which is also characteristic of other Mtb proteins). The spatial structure of TMPKmtb is similar to other enzymes of this class, but significantly differs in amino acid sequences (26% homology with E. coli TMPK, 25% with yeast TMPK and 22% with human TMPK) [82,83].
5.2. Nucleic Base Modification
The replacement of the methyl group at the 5 position with a bromine atom resulted in 5-Br-dUMP (44), which is an active substrate of TMPKmtb (44, Km = 30 µM, Table 1) [83,84]. The introduction of bulky substituents into the 5 position led to the loss of substrate properties and resulted in inhibitory activity of these compounds (the most active were 5-hydroxymethyl-(45) and 5-furan-2-yl dUMP (46) (Ki 110 and 140 μM respectively) [85] (Table 1). Modification at the 2 position of the pyrimidine ring led to 5-methyl-iso-dCMP, which turned out to be a competitive inhibitor of the enzyme with a Ki of 130 μM [83].
5.3. Modification of the Carbohydrate Fragment
Immediately after obtaining the crystal structure of TMPKmtb [83], it was found that 3′-azido-2′,3′-dideoxythymidine 5′-monophosphate (47) is an inhibitor of this enzyme (Ki 10 µM) [83,84]. It is probable that TMPKmtb does not phosphorylate 47 due to the interaction of the terminal nitrogen atom of the azido group with the active site of the enzyme, which may lead to weaker binding to ATP [85,86,87].
As a rule, structural modifications of 47 led to a decrease in inhibitory activity, for example, 3′-aminothymidine monophosphate 48 showed only insignificant inhibitory activity, while the 3′-fluoro analogue was a substrate with Km of 30 μM [86].
Later, a number of 3′-nucleotides with branched chains were synthesized: 3′-C-azidomethyl-49, 3′-aminomethyl-50, 3′-fluoromethyl-51 and 3′-hydroxymethyl-52 derivatives of dTMP (Ki 10.5—29 μM) [88], which proves the ability of the enzyme to bind inhibitors with bulky substituents in the 3′ position. The absence of a phosphate group in these derivatives increased their selectivity, but led to loss of activity [88,89].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Interaction of thymidine derivatives with TMPKmtb.
 | |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | Ki (µM) TMPKmtb | Ref. |
| dTMP | OH | OPO32− | CH3 | 4.5 | [83,84] |
| Thymidine | OH | OH | CH3 | 27 | [83,84] |
| 44 | OH | OPO32− | Br | 30 * | [83,84] |
| 45 | OH | OPO32− | CH2OH | 110 | [85] |
| 46 | OH | OPO32− | Furan-2-yl | 140 | [85] |
| 47 | N3 | OPO32− | CH3 | 10 | [83,84,85,86] |
| 48 | NH2 | OPO32− | CH3 | 235 | [86] |
| 49 | CH2N3 | OPO32− | CH3 | 12 | [88] |
| 50 | CH2NH2 | OPO32− | CH3 | 10.5 | [88] |
| 51 | CH2F | OPO32− | CH3 | 15 | [88] |
| 52 | CH2OH | OPO32− | CH3 | 29 | [88] |
| 53 | OH | N3 | CH3 | 7 | [89] |
| 54 | OH | NH2 | CH3 | 11 | [89] |
| 55 | OH | OH | Br | 5 | [90] |
| 56 | OH | OH | Cl | 10 | [90] |
| 57 | N3 | OH | Br | 10.5 | [91] |
| 58 | N3 | OH | Cl | 16 | [91] |
* Substrate, Km (μM).
Some derivatives without a 5′-monophosphate group proved to be effective inhibitors of TMPKmtb: 5′-azido-53 and 5′-aminothymidine 54 (which showed the highest activity) [90], 5-bromo-55 and 5-chloro-2′-deoxyuridine 56 and their 3′-azido analogues 57 and 58 [91] (Table 1). Ki values for these compounds were close to the Km of the natural substrate. So, azido- and amino-modifications seem attractive for the design of potent selective enzyme inhibitors.
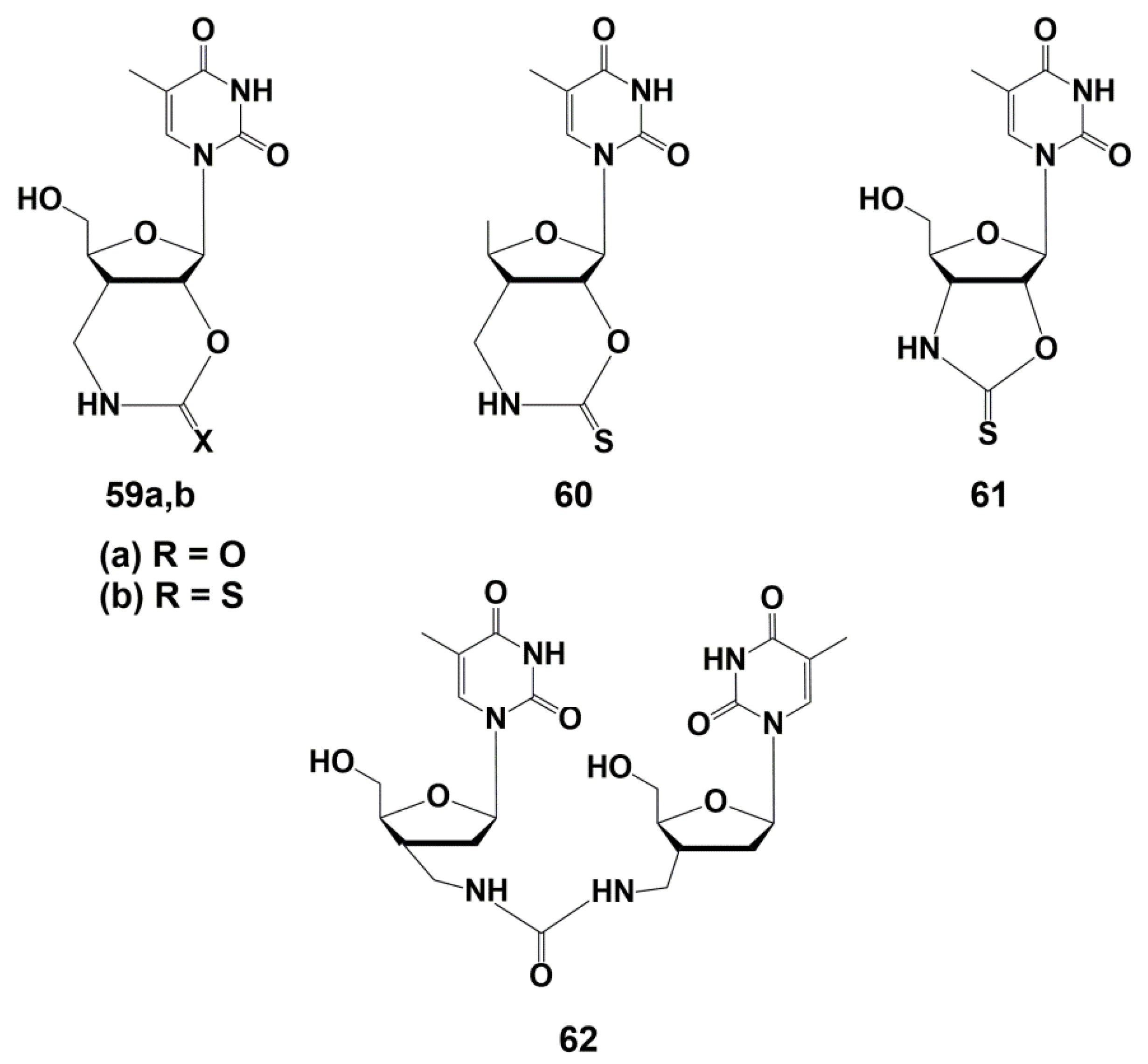
Representatives of another class of nucleoside analogues demonstrating effective and selective inhibition of TMPKmtb were bicyclic thymidine derivatives 59–61 (Figure 14). The synthesis of these compounds and their analogues, carried out by Van Calenbergh and co-workers [89,92], was discussed in detail in the earlier published reviews [24,25].
The urethane and thiourethane derivatives 59a,b showed Ki values of 13.5 and 3.5 μM on isolated TMPKmtb. However, they were inactive against M. tuberculosis, probably due to the low permeability of the bacterial cell wall for polar molecules. To solve this problem, a 5′-deoxyanalogue of the thiourethane derivative 60 was synthesized, which proved to be the most effective inhibitor of TMPKmtb (Ki 2.3 μM), showing, however, low activity against Mtb (MIC99 31 µg/mL, 100 μM). Compound 61, which is a five-membered analogue of 59b, was noticeably less active against both the enzyme (Ki 8 µM) and mycobacteria (MIC99 500 µg/mL, 1.6 mM) [89].
Revealed inhibitory activity of dinucleoside 62 was unexpected, since the binding site for the second nucleoside is absent in TMPKmtb. Further studies, however, showed that such a compound can act as a competitive inhibitor of the two substrates required for the enzyme: TMP and ATP. This promising activity of analogue 62 has pushed further synthesis of derivatives capable of increasing the inhibitory properties.
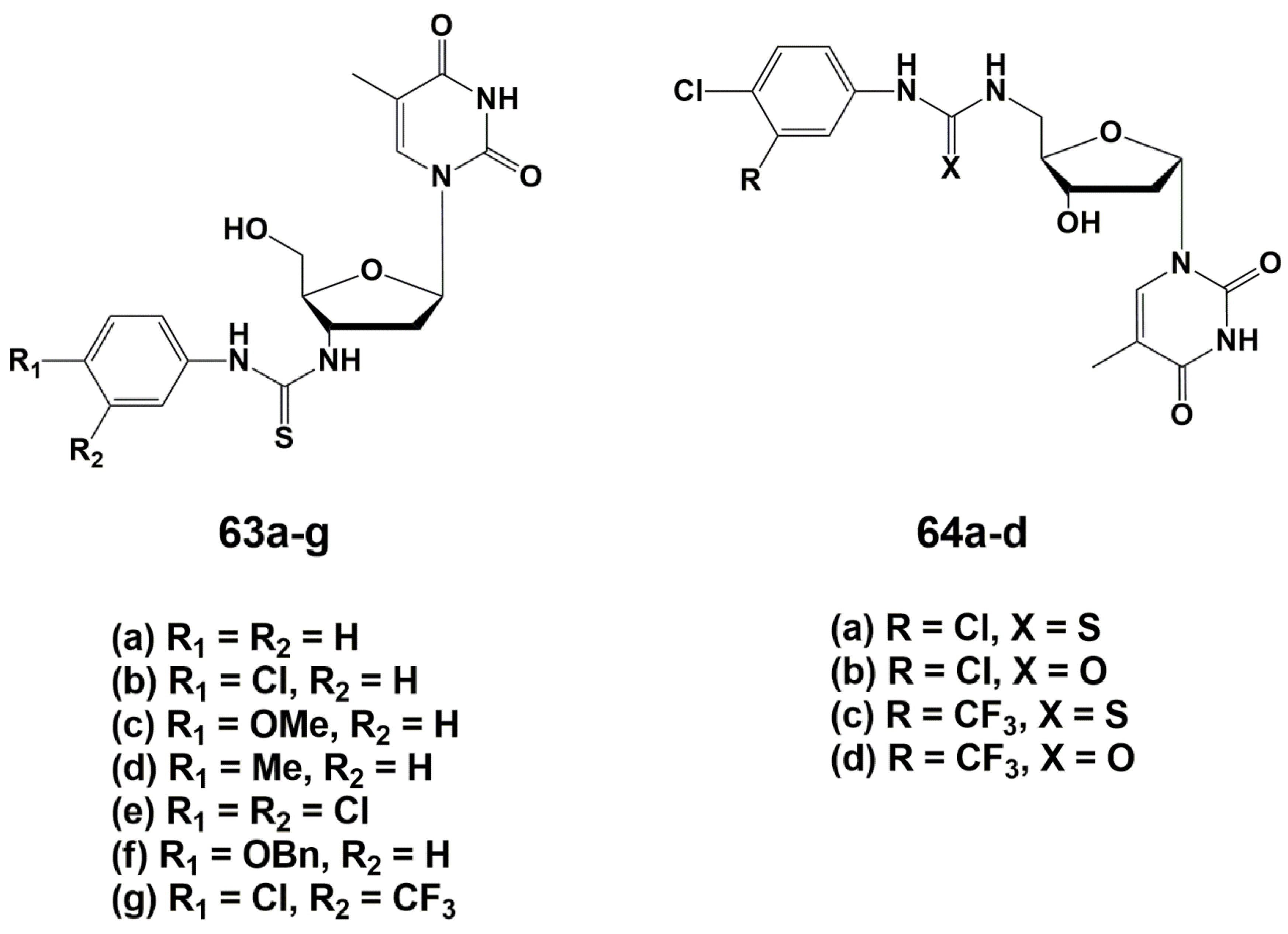
During the optimizing of the structure, one of the two monomers was replaced by different aromatic groups in order to enhance the electron-withdrawing properties of the molecule [93]. As a result, a series of 3′-arylthiourea thymidine derivatives 63a–g was synthesized (Figure 14). According to computer modeling data [89], the authors assumed that the 5′-derivatives of α-thymidine will bind to TMPKmtb similar to their β-analogues with substituents at the 3′-position, because of similarity in spatial structure. In order to confirm this hypothesis, a number of α-thymidine derivatives 64a–d were synthesized (Figure 15).
To synthesize both series of nucleosides, thymidine anomers were converted into the corresponding 3′- or 5′-amino derivatives, followed by the addition of the necessary aryl isothiocyanates. The synthesis of these compounds is described in detail in reviews [24,25].
The study of the inhibitory properties of 3′-modified β-thymidine derivatives showed that the substituents in the phenyl ring significantly affect the activity of the compounds, leading to the highest efficiency in the case of electron-withdrawing groups. The best inhibitor in this series was the 3-trifluoromethyl-4-chlorophenyl derivative 63g (Ki 5.0 μM). 5′-Modified α-thymidine derivatives were more active than their β-analogues. The same time lipophilic and electron-withdrawing substituents in the phenyl fragment significantly increased the inhibitory activity of the compounds. Compound 64c was the most active against TMPKmtb with a Ki value of 0.6 μM, a selectivity index of 600 and effective inhibition of M. bovis growth (MIC99 20 μg/mL, 50 μM). Analogue 64c was able to inhibit the Mtb growth by 39% maximum at 6.25 µg/mL concentration [93].
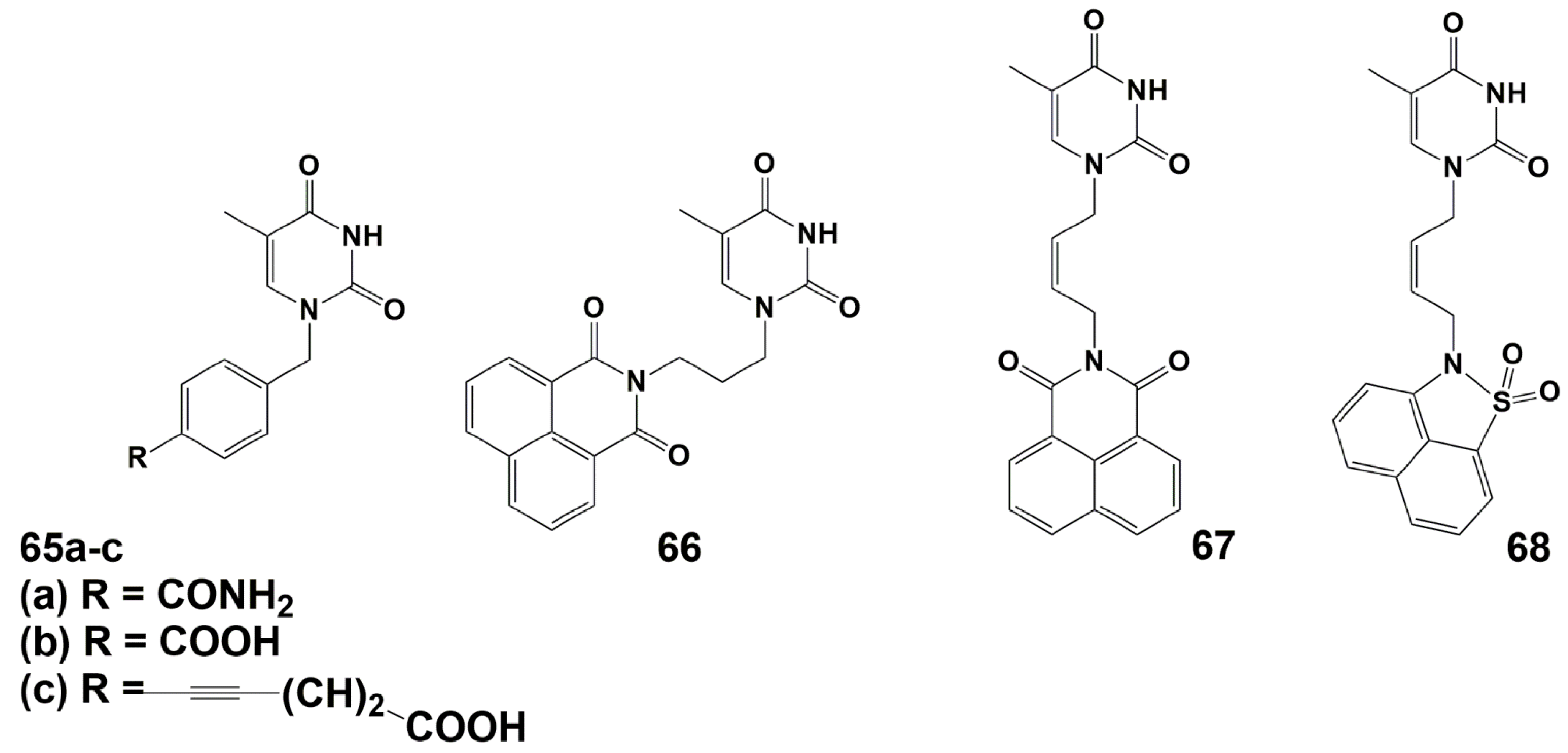
A number of arylpyrimidines have been investigated as inhibitors of TMPKmtb [94,95,96,97]. Initial screening results revealed derivative 66, which showed the best inhibition of TMPKmtb (Ki 42 μM) (Figure 16).
Further modifications of this compound, primarily the linker between the nucleic base and the naphthalimide fragment, led to a significant increase in activity (Ki 0.42 and 0.27 μM for compounds 67 and 68, respectively). (Figure 16). However, none of the synthesized compounds inhibited the growth of M. bovis BCG and Mtb cells at concentrations up to 7.5 and 32 μg/mL (19 and 80 μM), respectively.
Thus, to date, a wide range of TMPKmtb inhibitors have been obtained, some of which have demonstrated a significant inhibitory effect on Mtb. So, this enzyme can serve as a convenient model in the search for new inhibitors of mycobacteria growth.
5.4. Thymidylate Synthase Inhibitors
The thymidylate synthase enzyme (ThyA) is essential for living organisms to synthesize thymidine monophosphate de novo [98]. ThyA catalyzes the reductive methylation of 2′-deoxyuridine 5′-monophosphate (dUMP) to thymidine 5′-monophosphate (TMP).
Further studies have shown that some microorganisms do not have ThyA encoding genes, but remain viable in an environment with a lack of thymidine. Such microorganisms are capable of expressing the ThyX enzyme. This enzyme and the encoding gene ThyX are rare in eukaryotes and are completely absent in humans [99,100,101].
ThyX and ThyA catalyze the same reaction; however, these enzymes do not have structural similarity and have different mechanisms of catalysis. Despite ThyA, ThyX activity depends on the coenzyme flavin adenine dinucleotide (FADH2) as a hydride ion donor (Figure 17) [102,103,104,105].
Mtb strains are known to contain both ThyA and ThyX enzymes and the latter is absolutely necessary for mycobacteria to survive in macrophages. Since macrophages are the reservoir of Mtb in the latent phase of the disease, ThyX inhibitors can become prototypes of drugs that act on this phase. The suggestion was made based on the analysis of the crystal structure of the ThyX enzyme (Figure 18) [102,104,106].
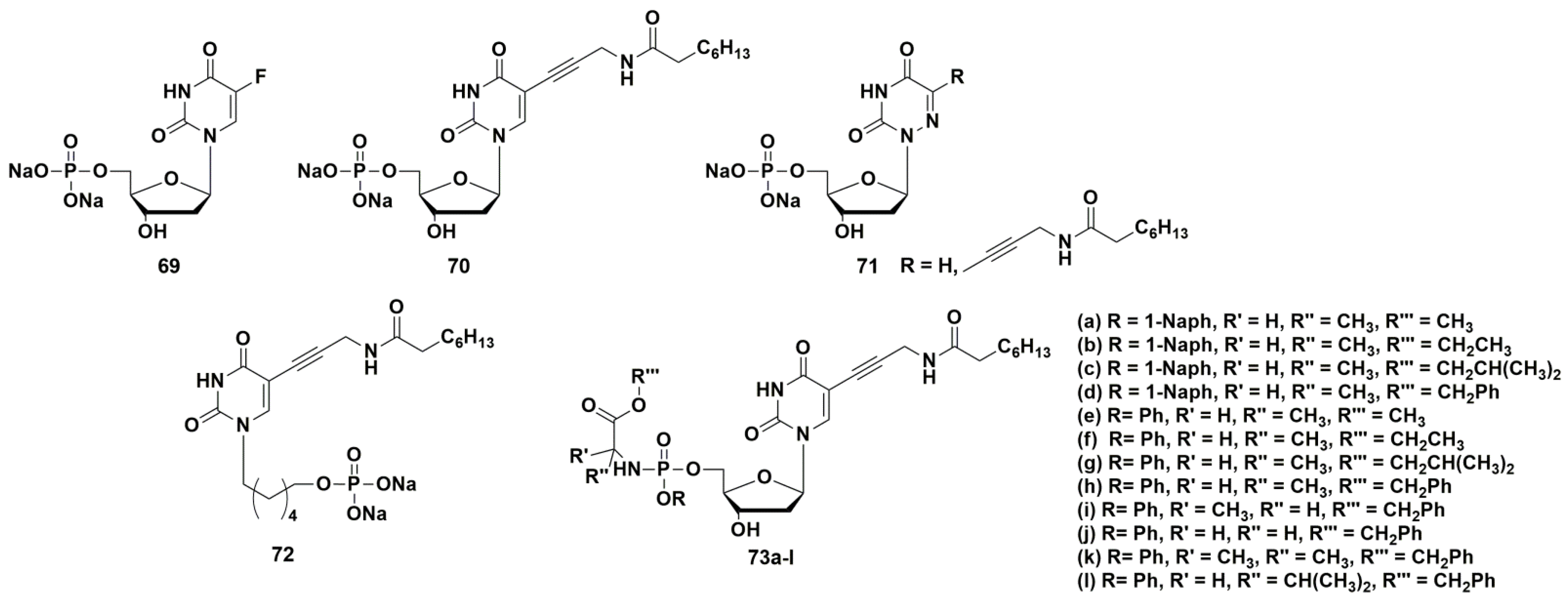
5-Fluoro-2′-deoxyuridine 5′-monophosphate 69 (Figure 19) was the first compound, which inhibits the ThyX enzyme with an IC value of 0.57 µM. However, this compound also inhibited ThyA at the same range (IC50 0.29 µM) [101].
A series of papers on selective inhibitors of ThyX was published by Piet Herdewijn and co-workers [105,107,108]. Among the obtained nucleotides containing alkynyl or aryl substituents at the C-5 position of the heterocyclic base, 5-(3-octanoylaminoprop-1-ynyl)-2′-deoxyuridine 5′-monophosphate 70 (Figure 19) had the highest activity. This compound inhibited ThyX (IC50 0.91 μM) more effectively than ThyA (IC50 > 50 μM). Despite the noticeable activity and selectivity of the inhibitor, it was impossible to study its antibacterial properties due to the high polarity of the compound caused by the presence of the phosphate group, which limits the penetration through the cell wall of mycobacteria. In order to increase pharmacological properties, McGuigan and co-workers designed and synthesized so-called “ProTide” prodrug forms of the target compound [109]. This strategy consists in “masking” the negative charges of the monophosphate group with two lipophilic residues: an amino acid ester and an aryloxy fragment [110]. Synthesis of such prodrugs increases the possibility of penetration into the cell by passive diffusion and stability to dephosphorylation. The release of the original monophosphate occurs in two stages: enzymatic and subsequent spontaneous chemical hydrolysis. This strategy has been successfully applied in the development of anticancer drugs [111,112] and antiviral nucleoside analogues [113].
ProTide derivatives 73 (Figure 19) proved to be the most effective inhibitors of Mtb growth (strain H37Rv), showing moderate antimycobacterial activity (MIC99 from 62.5 to 250 mg/L, 83–324 μM) [109].
Herdewijn et al. synthesized a series of potential inhibitors of thymidylate synthase and thymidine monophosphate kinase, namely 5′-monophosphates of 5-modified 6-aza-2′-deoxyuridines 71 (Figure 19) [107]. However, the compounds were inactive against ThyX; only derivative 71b showed weak inhibitory activity (40.9% inhibition of ThyX and 13.8% inhibition of ThyA, respectively, at a concentration of 50 μM) [107].
Since the 5′-phosphates of nucleotides are easily cleaved by esterases, similarly to the successful development of antiviral nucleoside analogues [114,115], a series of acyclic nucleoside phosphonates (ANP) containing 5-alkynyluracil (70, Figure 19) and a number of others were synthesized [108]. An important advantage of ANPs is their catabolic stability. From all tested compounds, only (6-(5-(3-octanamidoprop-1-yn-1-yl)-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)hexyl)phosphonate 72 showed 43% inhibition of ThyX at 50 µM. So, the data indicate some possibility of ThyX inhibition by representatives of this class of acyclic nucleoside phosphonates [108].
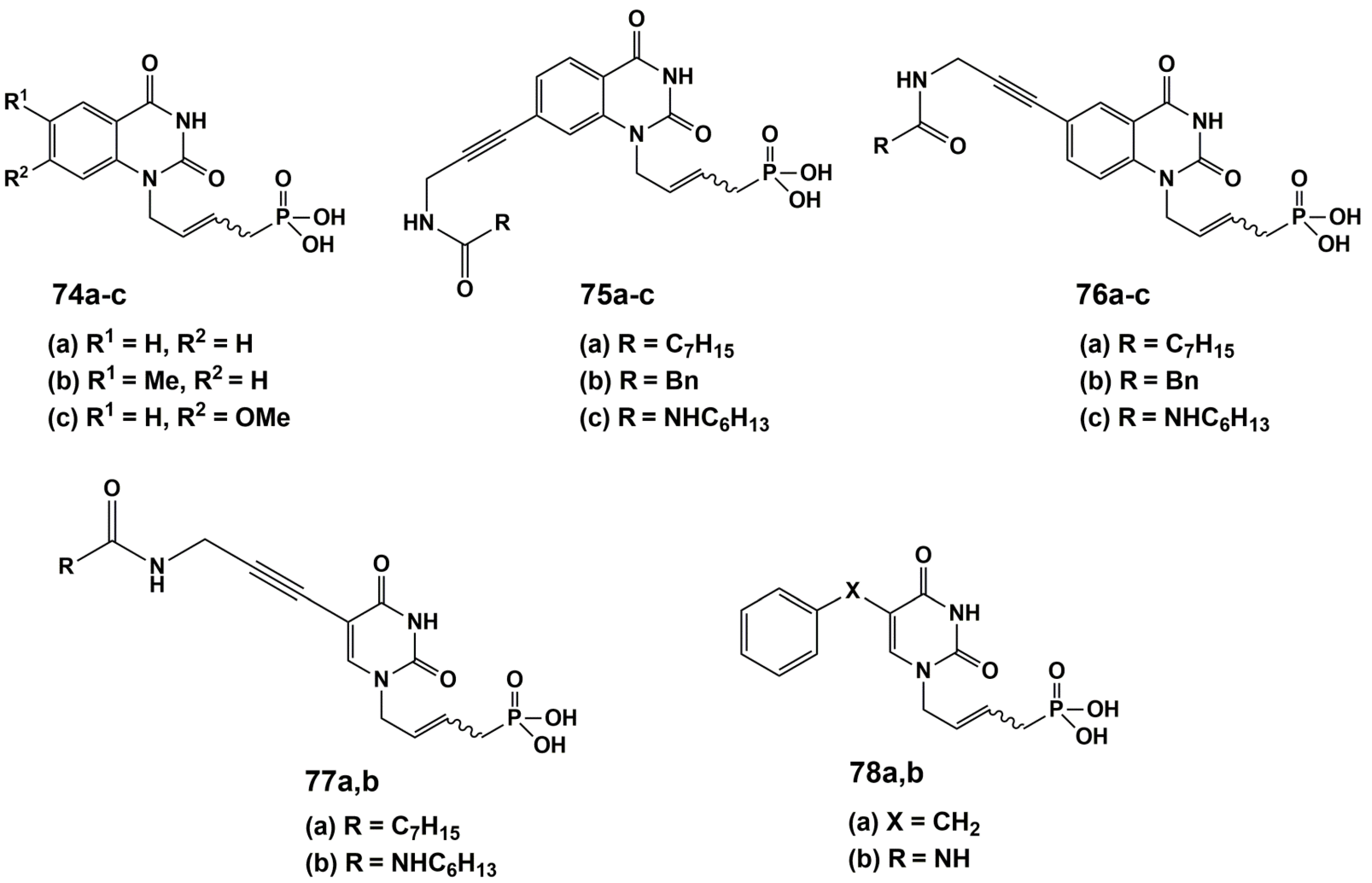
Agrofoglio and co-workers [116] have developed a new class of ANPs based on the mimic of two natural cofactors (dUMP and FAD) as inhibitors against Mtb ThyX (Figure 20).
Twelve analogues of acyclic nucleoside phosphonates were synthesized and tested for their activity against Mtb ThyX. These phosphonates were 5- and 7-substituted quinazoline and 5-substituted uracil linked at N1 to a (E)-but-2-enyl phosphonic acid moiety. Only the quinazoline analogue 75b showed a poor 31.8% inhibitory effect on ThyX at 200 μM. Nevertheless, based on 75b, future development is needed to develop an optimal acyclic chain and identify more potent compounds.
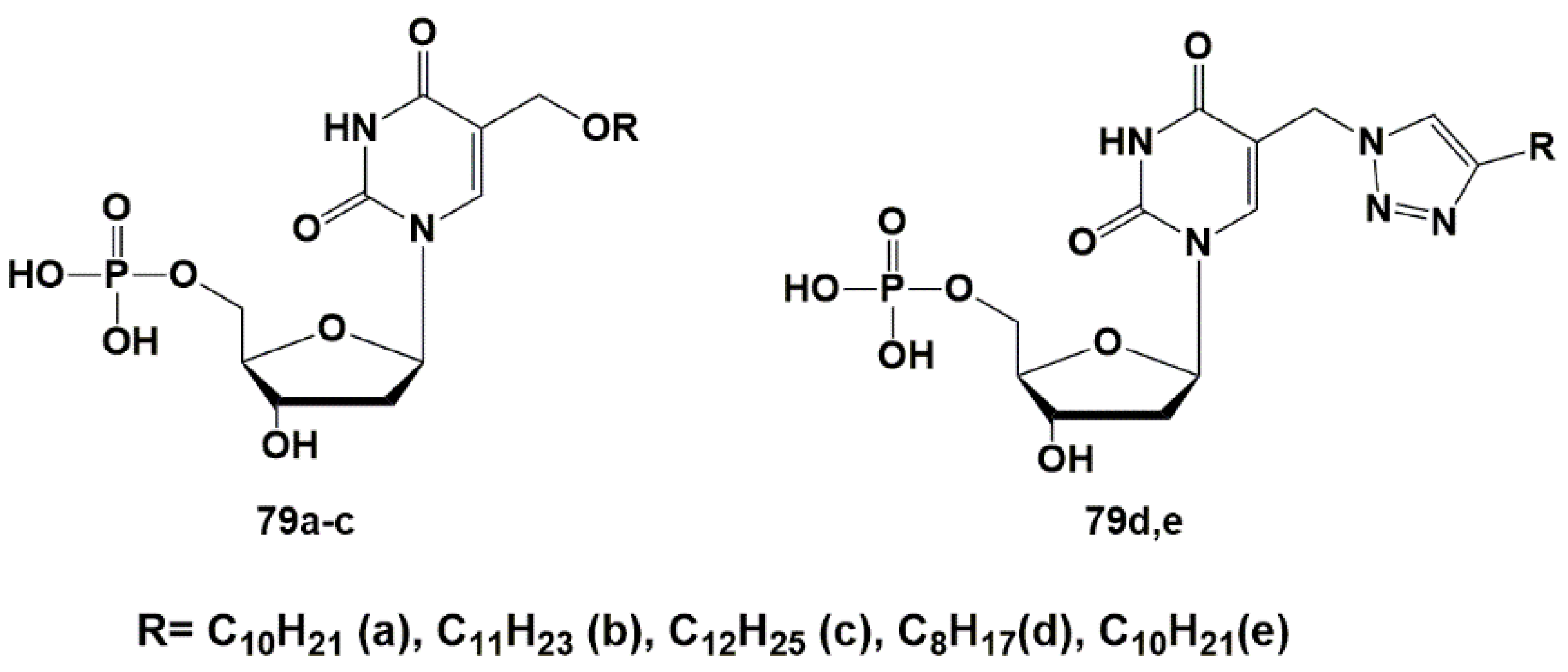
To check the hypothesis about a potential target of anti-TB action, a group of 5′-monophosphates of 2′-deoxyuridine derivatives was synthesized and their inhibitory activity against ThyX was evaluated. Extended alkyl substituents in the C-5 position were introduced through the oxymethyl (79a–c) or (1,2,3-triazol-1-yl)methyl (79d,e) linkers [117] (Figure 21). The parent nucleosides (18 and 23) previously demonstrated significant inhibition of Mtb growth [44,45].
Synthesized derivatives of 2′-deoxyuridine 5′-monophosphates 79a–c and 79d,e were tested against ThyA and ThyX according to a previously described method [105]. All compounds showed no inhibitory activity against ThyA up to concentrations of 100 μM. Inhibition of ThyX was weak and only 5-undecyloxymethyl-2′-deoxyuridine 5′-monophosphate ammonium salt 79b showed significant inhibition of ThyX (IC50 8.32 μM + 1.39 μM).
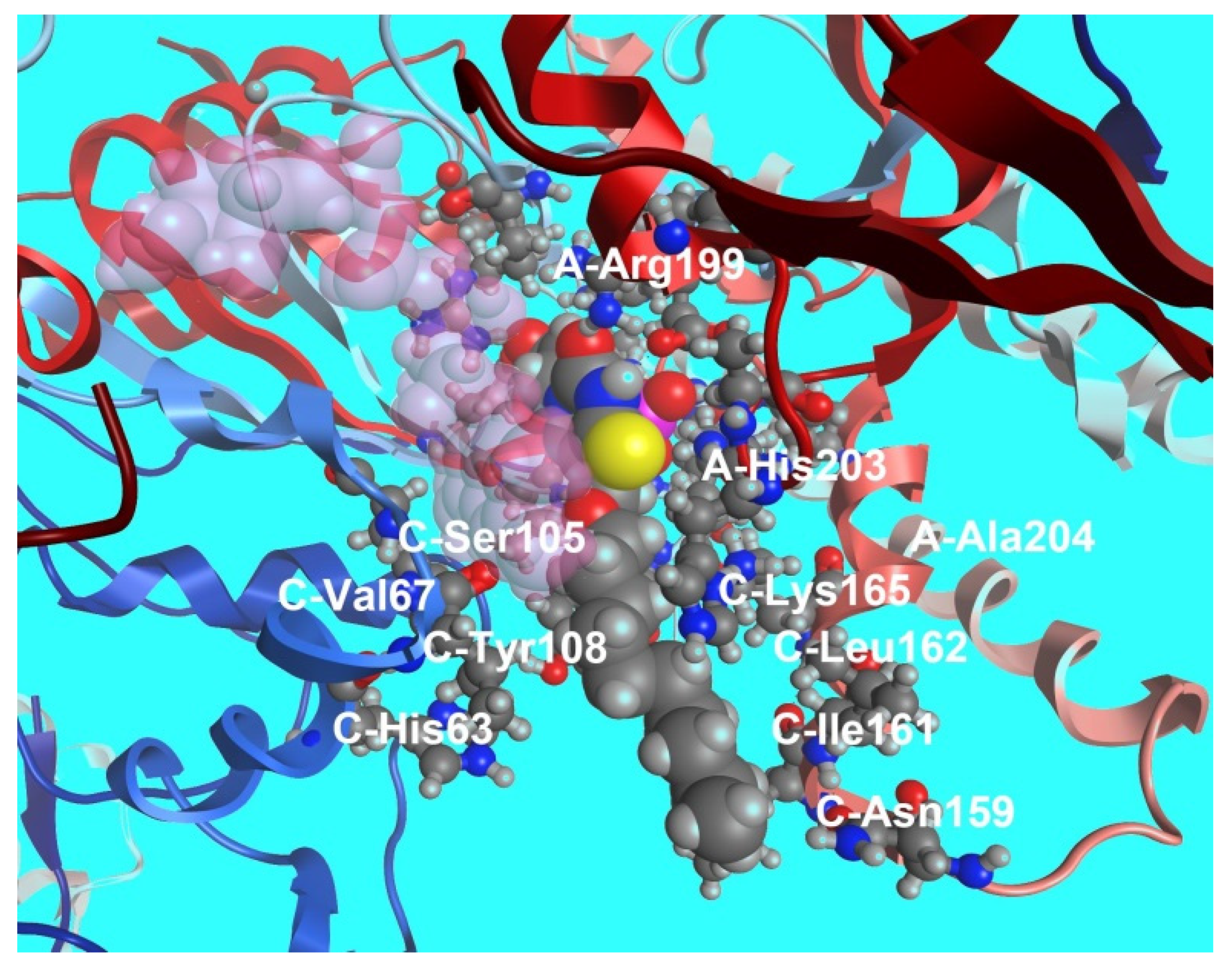
Binding of compound 79b to the ThyX active site was modeled by docking it into the structure of Mtb ThyX (PDB file 2AF6) [104], ThyA (PDB file 4FOA) [104] and human TS (PDB file 3HB8) [118] using the Molecular Operating Environment (MOE) program [119]. The results of molecular modeling provided qualitatively consistent data in the systems of parameters MMFF94x [120,121] and AMBER 99 [122]. As shown on Figure 22, the substituent at the C-5 position is located within the ThyX protein core. Similar calculations showed that the compound 72b is not able to bind to the active site of human TS, ThyA and, thus, the selectivity of the inhibitory effect of 5-alkyloxymethyl-2′-deoxyuridine. Although 79b inhibits ThyX worse than the previously described inhibitor N-(3-(5-(2′-deoxyuridine-5′-monophosphate))prop-2-ynyl) octanamide 70 (IC50 0.91 μM) [103], high yield synthesis of 5-undecyloxymethyl-dUMP (79b) makes it promising for further studies. Thus, to date, some selective effective inhibitors of ThyX have been synthesized.
6. Potential Targets of the Antimicrobial Action for 5-Modified 2′-Deoxynucleoside Derivatives and Their 5′-Norcarbocyclic Analogues
Despite the intensive research, the biological targets and mechanism of action of 5-modified 2′-deoxynucleoside derivatives and their 5′-norcarbocyclic analogues have not been finally identified. As mentioned above, Herdewijn et al. have shown that a number of 5′-monophosphates of 5-modified 2′-deoxyuridines effectively inhibit the flavin-dependent ThyX thymidylate synthase of Mtb (a unique enzyme for mycobacteria [99,100,104], practically not interacting with the main bacterial enzyme ThyA (also close to eukaryotic thymidylate synthases) [105,107,108,117]. So, it can be assumed that ThyX could be at least one of the possible targets of action for 5-modified 2′-deoxyuridines [105,107,108,117]. On the other hand, as mentioned above, compounds that couldn’t be phosphorylated in vitro, namely 5′-iodo-, azido- and amino-derivatives of 5-dodecyloxymethyl-2′-deoxyuridine [45] and 5-substituted carbocyclic 5′-norcarbocyclic uridine analogues with extended 1-alkynyl [49], alkyloxymethyl and alkyltriazolylmethyl [60] substituents have significant inhibitory activity against Mtb.
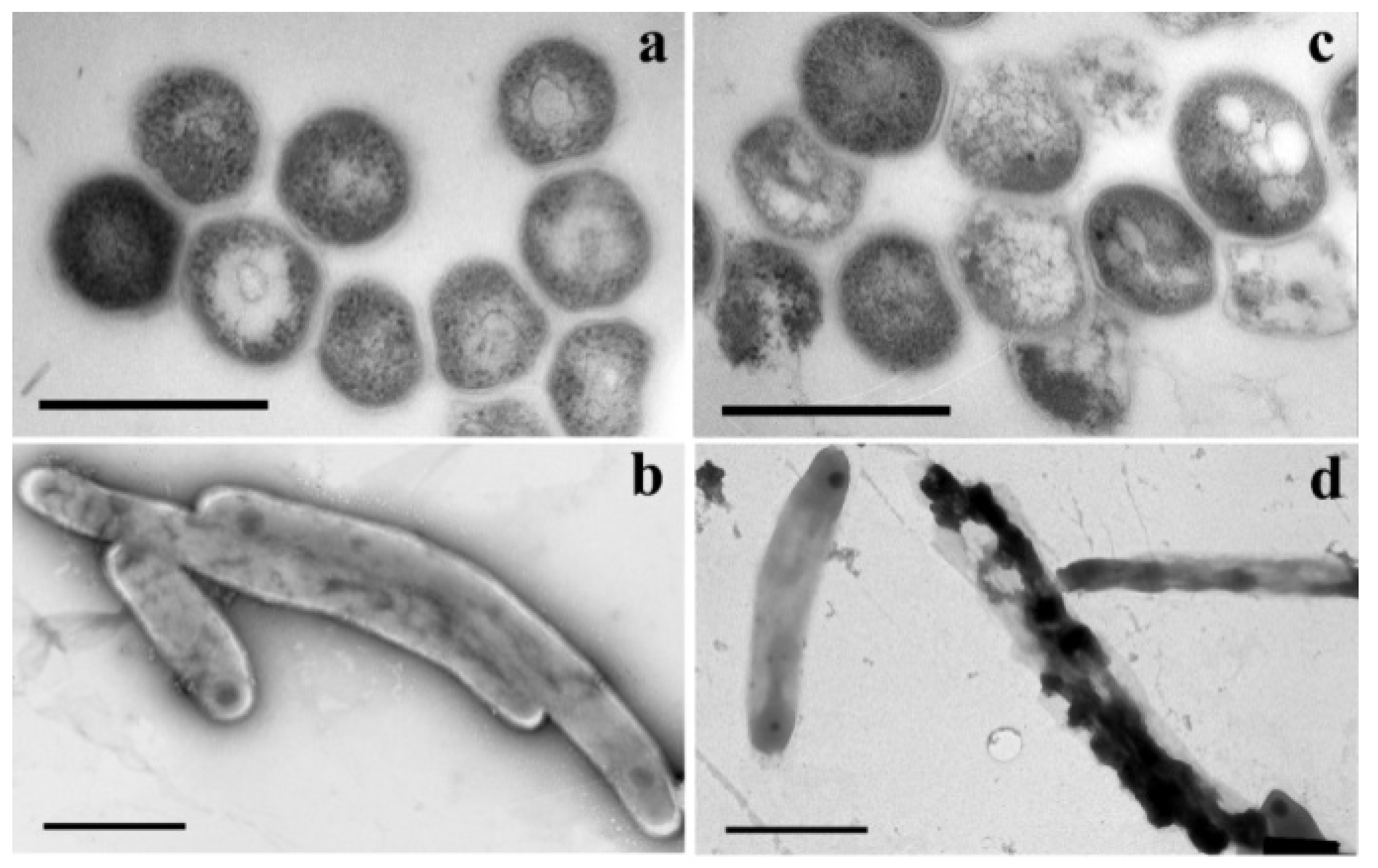
In an attempt to better understand the mode of action of 5-modified pyrimidine nucleosides, an electron microscopic study (TEM transmission electron microscopy) of mycobacteria Mtb (strain H37Rv), incubated with active representatives of 5-alkyloxymethyl and alkyltriazolylmethyl derivatives of 2′-deoxyuridine and its 5′-norcarbocyclic analogues, was carried out [62,123] (Figure 23).
Authors studied changes in the morphology of Mtb cells on (i) ultrathin sections and (ii) fixed whole bacteria stained with uranyl acetate [123]. In the control experiment, the absolute majority (~97%) of cells had a standard shape (length 2.3–4.5 μm and diameter 270–550 nm) and were surrounded by a three-layer bacterial wall ~14 nm thick. The addition of 5-substituted pyrimidine nucleosides to the incubation medium led to dramatic changes in the morphology of bacterial cells. All cells observed in randomly selected areas of ultrathin sections were divided into several groups according to their morphology, with cells with intact morphology being the minority (less than 8.5–22.3%). The number of vacuolated cells increased sharply to 31.1–37.9% in the presence of compounds 18c, 23c, 29c and 80.
Compounds 18c, 23c, 29c and 80 effectively inhibited the growth of Mtb H37Rv cells and caused a number of morphological changes (Figure 24). Firstly, lipid accumulation: intracellular vacuole-like inclusions were observed in cells. Several electron microscopy studies have shown previously, that the accumulation of such lipid inclusions in mycobacteria allows them to survive under unfavorable conditions and during the dormant period [124]. In particular, Mtb cells have been shown to accumulate lipid inclusions under oxidative stress, iron deficiency, isoniazid exposure and a number of other factors, including poor treatment outcomes [125,126,127]. These data correlate well with the observations that in the control experiment, when mycobacteria cells were grown in a medium containing only DMSO/Tween-80 solvent, the proportion of vacuolated cells increased to ~19%, but there was no inhibition of cell growth. On the contrary, after treatment with studied compounds, the proportion of vacuolated cells with accumulation of lipid inclusions increased to 30–35%, and the cell wall in 41–61% of cells was destroyed or damaged.
The thick three-layered cell wall is a unique structural feature of mycobacteria. It was observed microscopically in several laboratory strains of mycobacteria and clinical isolates of Mtb [128,129]. The cell wall is essential for the survival of Mtb, as it acts as a permeability barrier to drug entry and modulates the host’s immune response [130]. Treatment of H37Rv cells with compounds 18c, 23c, 29c and 80 resulted not only in protrusions and/or indentations on the surface, but also in the destruction of bacterial cells. It should be noted that the total number of lesions in Mtb cells after their treatment with compounds 18c, 23c, 29c and 80 correlates with their anti-tuberculosis activity (MIC) values. Thus, the total number of damages caused by the action of studied compounds is 41.5; 44.7; 61.8 and 45.2 while MIC99 is 20; 20; 10 and 20, respectively.
As reported earlier [131,132], the anti-TB drug isoniazid causes the destruction of the H37Rv cell wall by inhibiting the synthesis of mycolic acids. However, the morphology of H37Rv cells treated with isoniazid is significantly different from H37Rv cells treated with 5-substituted pyrimidine nucleosides. It suggests different mechanisms of action of isoniazid and 5-substituted pyrimidine nucleosides.
Thus, several mechanisms of action can be suggested for 5-modified pyrimidine nucleosides against Mtb, including (1) inhibition of Mtb flavin-dependent ThyX and (2) destruction of the cell wall or inhibition of enzymes associated with its biosynthesis.
7. Conclusions
As mentioned above, the antimycobacterial activity of nucleoside derivatives and analogues has been revealed recently, but a number of studies on their antibacterial properties have been published. Despite the fact that there are no drugs based on the described compounds, certain successes have been achieved in this area:
- Two specific enzymes of Mtb were identified as targets for compounds with antimycobacterial activity: thymidine monophosphate kinase and thymidylate synthase Thy-X. A number of effective inhibitors were synthesized.
- Several series of nucleoside derivatives and analogues have been synthesized with high antimycobacterial activity, comparable to the activity of some anti-TB drugs currently used in clinic and effective against drug-resistant strains of Mtb. Some compounds can be considered as drug prototypes.
- Rational methods have been developed to increase the bioavailability of nucleoside derivatives and analogues with high antimycobacterial activity by design of their depot forms.
- An original approach was proposed for the synthesis of depot forms with dual-action, which was used for obtaining compounds simultaneously effective against HIV and Mtb.
Thus, the use of nucleoside derivatives and analogues as inhibitors of mycobacteria is of considerable interest for microbiology, biochemistry, medicinal chemistry and pharmacology.
Author Contributions
L.A.A., A.L.K., E.S.M., D.A.M. and S.N.K. contributed to the original writing and editing of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Russian Foundation for Basic Research (RFBR) (Project No. 20-04-00094) and Russian Science Foundation (Project No. 19-74-10048).
Data Availability Statement
Data discussed in the review are all from, and available through, cited published literatures.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gutierrez, M.C.; Brisse, S.; Brosch, R.; Fabre, M.; Omaïs, B.; Marmiesse, M.; Supply, P.; Vincent, V. Ancient Origin and Gene Mosaicism of the Progenitor of Mycobacterium Tuberculosis. PLoS Pathog. 2005, 1, e5. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Tuberculosis Report. Available online: www.who.int/health-topics/tuberculosis (accessed on 19 February 2022).
- WHO. Tuberculosis Factsheet, Online. Available online: https://www.who.int/publications/m/item/factsheet-global-tb-report-2021 (accessed on 21 February 2022).
- Martini, M.; Besozzi, G.; Barberis, I. The Never-Ending Story of the Fight against Tuberculosis: From Koch’s Bacillus to Global Control Programs. J. Prev. Med. Hyg. 2018, 59, E241–E247. [Google Scholar] [CrossRef] [PubMed]
- Bansal, R.; Sharma, D.; Singh, R. Tuberculosis and Its Treatment: An Overview. Mini-Rev. Med. Chem. 2017, 18, 58–71. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Dwivedi, S.P.; Gaharwar, U.S.; Meena, R.; Rajamani, P.; Prasad, T. Recent Updates on Drug Resistance in Mycobacterium Tuberculosis. J. Appl. Microbiol. 2020, 128, 1547–1567. [Google Scholar] [CrossRef] [Green Version]
- Makarov, V.; Salina, E.; Reynolds, R.C.; Kyaw Zin, P.P.; Ekins, S. Molecule Property Analyses of Active Compounds for Mycobacterium Tuberculosis. J. Med. Chem. 2020, 63, 8917–8955. [Google Scholar] [CrossRef]
- Hoffman, P.S. Antibacterial Discovery: 21st Century Challenges. Antibiotics 2020, 9, 213. [Google Scholar] [CrossRef]
- Ventola, C.L. The Antibiotic Resistance Crisis: Part 1: Causes and Threats. Pharm. Ther. 2015, 40, 277–283. [Google Scholar] [PubMed]
- Brown, E.D.; Wright, G.D. Antibacterial Drug Discovery in the Resistance Era. Nature 2016, 529, 336–343. [Google Scholar] [CrossRef]
- Cohen, A.; Mathiasen, V.D.; Schön, T.; Wejse, C. The Global Prevalence of Latent Tuberculosis: A Systematic Review and Meta-Analysis. Eur. Respir. J. 2019, 54, 1900655. [Google Scholar] [CrossRef]
- Datta, S.; Evans, C.A. Healthy Survival after Tuberculosis. Lancet Infect. Dis. 2019, 19, 1045–1047. [Google Scholar] [CrossRef]
- Martinez, L.; Verma, R.; Croda, J.; Horsburgh, C.R.; Walter, K.S.; Degner, N.; Middelkoop, K.; Koch, A.; Hermans, S.; Warner, D.F.; et al. Detection, Survival and Infectious Potential of Mycobacterium Tuberculosis in the Environment: A Review of the Evidence and Epidemiological Implications. Eur. Respir. J. 2019, 53, 1802302. [Google Scholar] [CrossRef] [PubMed]
- Joean, O.; Thiele, T.; Schütz, K.; Schwerk, N.; Sedlacek, L.; Kalsdorf, B.; Baumann, U.; Stoll, M. Multidrug-Resistant Mycobacterium Tuberculosis: A Report of Cosmopolitan Microbial Migration and an Analysis of Best Management Practices. BMC Infect. Dis. 2020, 20, 678. [Google Scholar] [CrossRef] [PubMed]
- Heyckendorf, J.; Lange, C.; Martensen, J. Multidrug-Resistant Tuberculosis. In Emerging Infectious Diseases; Elsevier: Amsterdam, The Netherlands, 2014; pp. 239–253. [Google Scholar] [CrossRef]
- Khan, M.T.; Junaid, M.; Mao, X.; Wang, Y.; Hussain, A.; Malik, S.I.; Wei, D. Pyrazinamide Resistance and Mutations L19R, R140H, and E144K in Pyrazinamidase of Mycobacterium Tuberculosis. J. Cell. Biochem. 2019, 120, 7154–7166. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.; Khan, A.; Ali, S.S.; Ali, S.; Wei, D.-Q. A Computational Perspective on the Dynamic Behaviour of Recurrent Drug Resistance Mutations in the PncA Gene from Mycobacterium Tuberculosis. RSC Adv. 2021, 11, 2476–2486. [Google Scholar] [CrossRef]
- Alanis, A.J. Resistance to Antibiotics: Are We in the Post-Antibiotic Era? Arch. Med. Res. 2005, 36, 697–705. [Google Scholar] [CrossRef]
- Yssel, A.E.J.; Vanderleyden, J.; Steenackers, H.P. Repurposing of Nucleoside- and Nucleobase-Derivative Drugs as Antibiotics and Biofilm Inhibitors. J. Antimicrob. Chemother. 2017, 72, 2156–2170. [Google Scholar] [CrossRef]
- Winn, M.; Goss, R.J.M.; Kimura, K.; Bugg, T.D.H. Antimicrobial Nucleoside Antibiotics Targeting Cell Wall Assembly: Recent Advances in Structure–Function Studies and Nucleoside Biosynthesis. Nat. Prod. Rep. 2010, 27, 279–304. [Google Scholar] [CrossRef]
- Duckworth, B.P.; Nelson, K.M.; Aldrich, C.C. Adenylating Enzymes in Mycobacterium Tuberculosis as Drug Targets. Curr. Top. Med. Chem. 2012, 12, 766–796. [Google Scholar] [CrossRef]
- Duckworth, B.P.; Wilson, D.J.; Nelson, K.M.; Boshoff, H.I.; Barry, C.E.; Aldrich, C.C. Development of a Selective Activity-Based Probe for Adenylating Enzymes: Profiling MbtA Involved in Siderophore Biosynthesis from Mycobacterium Tuberculosis. ACS Chem. Biol. 2012, 7, 1653–1658. [Google Scholar] [CrossRef] [Green Version]
- Shakya, N.; Garg, G.; Agrawal, B.; Kumar, R. Chemotherapeutic Interventions Against Tuberculosis. Pharmaceuticals 2012, 5, 690–718. [Google Scholar] [CrossRef] [Green Version]
- Shmalenyuk, E.R.; Kochetkov, S.N.; Alexandrova, L.A. Novel Inhibitors of Mycobacterium tuberculosis Growth Based on Modified Pyrimidine Nucleosides and Their Analogues. Russ. Chem. Rev. 2013, 82, 896–915. [Google Scholar] [CrossRef]
- Van Calenbergh, S.; Pochet, S.; Munier-Lehmann, H. Drug Design and Identification of Potent Leads Against Mycobacterium tuberculosis Thymidine Monophosphate Kinase. Curr. Top. Med. Chem. 2012, 12, 694–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serpi, M.; Ferrari, V.; Pertusati, F. Nucleoside Derived Antibiotics to Fight Microbial Drug Resistance: New Utilities for an Established Class of Drugs? J. Med. Chem. 2016, 59, 10343–10382. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, V.; Serpi, M. Nucleoside Analogs and Tuberculosis: New Weapons against an Old Enemy. Future Med. Chem. 2015, 7, 291–314. [Google Scholar] [CrossRef]
- Ducati, R.G.; Breda, A.; Basso, L.A.; Santos, D.S. Purine Salvage Pathway in Mycobacterium tuberculosis. Curr. Med. Chem. 2011, 18, 1258–1275. [Google Scholar] [CrossRef]
- Villela, A.D.; Sanchez-Quitian, Z.A.; Ducati, R.G.; Santos, D.S.; Basso, L.A. Pyrimidine Salvage Pathway in Mycobacterium tuberculosis. Curr. Med. Chem. 2011, 18, 1286–1298. [Google Scholar] [CrossRef]
- Dawadi, S.; Kawamura, S.; Rubenstein, A.; Remmel, R.; Aldrich, C.C. Synthesis and Pharmacological Evaluation of Nucleoside Prodrugs Designed to Target Siderophore Biosynthesis in Mycobacterium tuberculosis. Bioorg. Med. Chem. 2016, 24, 1314–1321. [Google Scholar] [CrossRef] [Green Version]
- Dawadi, S.; Boshoff, H.I.M.; Park, S.W.; Schnappinger, D.; Aldrich, C.C. Conformationally Constrained Cinnolinone Nucleoside Analogues as Siderophore Biosynthesis Inhibitors for Tuberculosis. ACS Med. Chem. Lett. 2018, 9, 386–391. [Google Scholar] [CrossRef]
- Johar, M.; Manning, T.; Kunimoto, D.Y.; Kumar, R. Synthesis and in Vitro Anti-Mycobacterial Activity of 5-Substituted Pyrimidine Nucleosides. Bioorg. Med. Chem. 2005, 13, 6663–6671. [Google Scholar] [CrossRef]
- Kumar, R.; Kunimoto, D.; Manning, T.; Srivastav, N. In Vitro Anti-Mycobacterial Activities of Various 2′-Deoxyuridine, 2′-Arabinouridine and 2′-Arabinofluoro-2′-Deoxyuridine Analogues: Synthesis and Biological Studies. Med. Chem. 2006, 2, 287–293. [Google Scholar] [CrossRef]
- Rai, D.; Johar, M.; Manning, T.; Agrawal, B.; Kunimoto, D.Y.; Kumar, R. Design and Studies of Novel 5-Substituted Alkynylpyrimidine Nucleosides as Potent Inhibitors of Mycobacteria. J. Med. Chem. 2005, 48, 7012–7017. [Google Scholar] [CrossRef] [PubMed]
- Johar, M.; Manning, T.; Tse, C.; Desroches, N.; Agrawal, B.; Kunimoto, D.Y.; Kumar, R. Growth Inhibition of Mycobacterium bovis, Mycobacterium tuberculosis and Mycobacterium avium In Vitro: Effect of 1-β-d-2′-Arabinofuranosyl and 1-(2′-Deoxy-2′-Fluoro-β-d-2′-Ribofuranosyl) Pyrimidine Nucleoside Analogs. J. Med. Chem. 2007, 50, 3696–3705. [Google Scholar] [CrossRef]
- Rai, D.; Johar, M.; Srivastav, N.C.; Manning, T.; Agrawal, B.; Kunimoto, D.Y.; Kumar, R. Inhibition of Mycobacterium tuberculosis, Mycobacterium bovis, and Mycobacterium avium by Novel Dideoxy Nucleosides. J. Med. Chem. 2007, 50, 4766–4774. [Google Scholar] [CrossRef] [PubMed]
- Srivastav, N.C.; Manning, T.; Kunimoto, D.Y.; Kumar, R. Studies on Acyclic Pyrimidines as Inhibitors of Mycobacteria. Bioorg. Med. Chem. 2007, 15, 2045–2053. [Google Scholar] [CrossRef]
- Sonogashira, K.; Tohda, Y.; Hagihara, N. A Convenient Synthesis of Acetylenes: Catalytic Substitutions of Acetylenic Hydrogen with Bromoalkenes, Iodoarenes and Bromopyridines. Tetrahedron Lett. 1975, 16, 4467–4470. [Google Scholar] [CrossRef]
- Sonogashira, K. Development of Pd–Cu Catalyzed Cross-Coupling of Terminal Acetylenes with Sp2-Carbon Halides. J. Organomet. Chem. 2002, 653, 46–49. [Google Scholar] [CrossRef]
- Shakya, N.; Srivastav, N.C.; Desroches, N.; Agrawal, B.; Kunimoto, D.Y.; Kumar, R. 3′-Bromo Analogues of Pyrimidine Nucleosides as a New Class of Potent Inhibitors of Mycobacterium tuberculosis. J. Med. Chem. 2010, 53, 4130–4140. [Google Scholar] [CrossRef] [PubMed]
- Shakya, N.; Srivastav, N.C.; Bhavanam, S.; Tse, C.; Desroches, N.; Agrawal, B.; Kunimoto, D.Y.; Kumar, R. Discovery of Novel 5-(Ethyl or Hydroxymethyl) Analogs of 2′-‘up’ Fluoro (or Hydroxyl) Pyrimidine Nucleosides as a New Class of Mycobacterium tuberculosis, Mycobacterium bovis and Mycobacterium avium Inhibitors. Bioorg. Med. Chem. 2012, 20, 4088–4097. [Google Scholar] [CrossRef]
- Platonova, Y.B.; Volov, A.N.; Tomilova, L.G. The Synthesis and Antituberculosis Activity of 5-Alkynyl Uracil Derivatives. Bioorg. Med. Chem. Lett. 2020, 30, 127351. [Google Scholar] [CrossRef]
- Volov, A.N.; Volov, N.A.; Platonova, Y.B. Design and Synthesis of Novel 5-Alkynyl Pyrimidine Nucleosides Derivatives: Influence of C-6-Substituent on Antituberculosis Activity. Bioorg. Med. Chem. Lett. 2021, 48, 128261. [Google Scholar] [CrossRef]
- Alexandrova, L.A.; Shmalenyuk, E.R.; Kochetkov, S.N.; Erokhin, V.V.; Smirnova, T.G.; Andreevskaia, S.N.; Chernousova, L.N. New 5-Modified Pyrimidine Nucleoside Inhibitors of Mycobacterial Growth. Acta Naturae 2010, 2, 108–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shmalenyuk, E.R.; Chernousova, L.N.; Karpenko, I.L.; Kochetkov, S.N.; Smirnova, T.G.; Andreevskaya, S.N.; Chizhov, A.O.; Efremenkova, O.V.; Alexandrova, L.A. Inhibition of Mycobacterium tuberculosis Strains H37Rv and MDR MS-115 by a New Set of C5 Modified Pyrimidine Nucleosides. Bioorg. Med. Chem. 2013, 21, 4874–4884. [Google Scholar] [CrossRef]
- Alexandrova, L.A.; Efremenkova, O.V.; Andronova, V.L.; Galegov, G.A.; Solyev, P.N.; Karpenko, I.L.; Kochetkov, S.N. 5-(4-Alkyl-1,2,3-Triazol-1-Yl)Methyl Derivatives of 2′-Deoxyuridine as Inhibitors of Viral and Bacterial Growth. Russ. J. Bioorg. Chem. 2016, 42, 677–684. [Google Scholar] [CrossRef]
- Shmalenyuk, E.R.; Karpenko, I.L.; Chernousova, L.N.; Chizhov, A.O.; Smirnova, T.G.; Andreevskaya, S.N.; Alexandrova, L.A. New 5-Modified 2′-Deoxyuridine Derivatives: Synthesis and Antituberculosis Activity. Russ. Chem. Bull. 2014, 63, 1197–1200. [Google Scholar] [CrossRef]
- Negrya, S.D.; Efremenkova, O.V.; Solyev, P.N.; Chekhov, V.O.; Ivanov, M.A.; Sumarukova, I.G.; Karpenko, I.L.; Kochetkov, S.N.; Alexandrova, L.A. Novel 5-Substituted Derivatives of 2′-Deoxy-6-Azauridine with Antibacterial Activity. J. Antibiot. 2019, 72, 535–544. [Google Scholar] [CrossRef]
- Matyugina, E.; Khandazhinskaya, A.; Chernousova, L.; Andreevskaya, S.; Smirnova, T.; Chizhov, A.; Karpenko, I.; Kochetkov, S.; Alexandrova, L. The Synthesis and Antituberculosis Activity of 5′-nor Carbocyclic Uracil Derivatives. Bioorg. Med. Chem. 2012, 20, 6680–6686. [Google Scholar] [CrossRef]
- Kusaka, T.; Yamamoto, H.; Shibata, M.; Muroi, M.; Kishi, T.; Mizuno, K. Streptomyces citricolor nov. Sp. and a new antibiotic, aristeromycin. J. Antibiot. 1968, 21, 255–263. [Google Scholar] [CrossRef]
- Hayashi, M.; Yaginuma, S.; Yoshioka, H.; Nakatsu, K. Studies on Neplanocin A, New Antitumor Antibiotic. II. Structure Determination. J. Antibiot. 1981, 34, 675–680. [Google Scholar] [CrossRef] [Green Version]
- Nieto, M.I.; Caamaño, O.; Fernández, F.; Gómez, M.; Balzarini, J.; De Clercq, E. Synthesis, antiviral and cytostatic activities, of carbocyclic nucleosides incorporating a modified cyclopentane ring. IV. Adenosine and uridine analogues. Nucleosides Nucleotides Nucleic Acids 2002, 21, 243–255. [Google Scholar] [CrossRef]
- Matyugina, E.S.; Khandazhinskaya, A.L.; Kochetkov, S.N. Carbocyclic Nucleoside Analogues: Classification, Target Enzymes, Mechanisms of Action and Synthesis. Russ. Chem. Rev. 2012, 81, 729–746. [Google Scholar] [CrossRef]
- Novikov, M.S.; Valuev-Elliston, V.T.; Babkov, D.A.; Paramonova, M.P.; Ivanov, A.V.; Gavryushov, S.A.; Khandazhinskaya, A.L.; Kochetkov, S.N.; Pannecouque, C.; Andrei, G.; et al. N1,N3-Disubstituted Uracils as Nonnucleoside Inhibitors of HIV-1 Reverse Transcriptase. Bioorg. Med. Chem. 2013, 21, 1150–1158. [Google Scholar] [CrossRef] [PubMed]
- Borchardt, R.T. S-Adenosyl-L-Methionine-Dependent Macromolecule Methyltransferases: Potential Targets for the Design of Chemotherapeutic Agents. J. Med. Chem. 1980, 23, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Borchardt, R.T.; Creveling, C.R.; Ueland, P.M. (Eds.) Biological Methylation and Drug Design; Humana Press: Totowa, NJ, USA, 1986; ISBN 978-1-4612-9398-9. [Google Scholar]
- Yuan, C.-S.; Liu, S.; Wnuk, S.F.; Robins, M.J.; Borchardt, R.T. Design and Synthesis of S-Adenosylhomocysteine Hydrolase Inhibitors as Broad-Spectrum Antiviral Agents. In Advances in Antiviral Drug Design; De Clercq, E., Ed.; Elsevier: Paris, France, 1996; pp. 41–88. ISBN 1-55938-693-2. [Google Scholar]
- Schneller, S.W.; Seley, K.L.; Hegde, V.R.; Rajappan, V.P. 5′-Norcarbanucleosides in L-Like Configurations. In Recent Advances in Nucleosides: Chemistry and Chemotherapy; Chu, C.K., Ed.; Elsevier: Amsterdam, The Netherlands, 2002; pp. 291–297. ISBN 0-444-50951-8. [Google Scholar]
- Hegde, V.R.; Seley, K.L.; Schneller, S.W. Carbocyclic 5′-Noruridine. Nucleosides Nucleotides Nucleic Acids 2000, 19, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Siddiqi, S.M.; Chen, X.; Schneller, S.W.; Ikeda, S.; Snoeck, R.; Andrei, G.; Balzarini, J.; De Clercq, E. Antiviral Enantiomeric Preference for 5′-Noraristeromycin. J. Med. Chem. 1994, 37, 551–554. [Google Scholar] [CrossRef]
- Jeong, L.S.; Yoo, S.J.; Lee, K.M.; Koo, M.J.; Choi, W.J.; Kim, H.O.; Moon, H.R.; Lee, M.Y.; Park, J.G.; Lee, S.K.; et al. Design, Synthesis, and Biological Evaluation of Fluoroneplanocin A as the Novel Mechanism-Based Inhibitor of S-Adenosylhomocysteine Hydrolase. J. Med. Chem. 2003, 46, 201–203. [Google Scholar] [CrossRef]
- Khandazhinskaya, A.L.; Alexandrova, L.A.; Matyugina, E.S.; Solyev, P.N.; Efremenkova, O.V.; Buckheit, K.W.; Wilkinson, M.; Buckheit, R.W.; Chernousova, L.N.; Smirnova, T.G.; et al. Novel 5′-Norcarbocyclic Pyrimidine Derivatives as Antibacterial Agents. Molecules 2018, 23, 3069. [Google Scholar] [CrossRef] [Green Version]
- Matyugina, E.; Novikov, M.; Babkov, D.; Ozerov, A.; Chernousova, L.; Andreevskaya, S.; Smirnova, T.; Karpenko, I.; Chizhov, A.; Murthu, P.; et al. 5-Arylaminouracil Derivatives: New Inhibitors of Mycobacterium Tuberculosis. Chem. Biol. Drug Des. 2015, 86, 1387–1396. [Google Scholar] [CrossRef]
- Matyugina, E.S.; Novikov, M.S.; Babkov, D.A.; Valuev-Elliston, V.T.; Vanpouille, C.; Zicari, S.; Corona, A.; Tramontano, E.; Margolis, L.B.; Khandazhinskaya, A.L.; et al. 5-Arylaminouracil Derivatives as Potential Dual-Action Agents. Acta Nat. 2015, 7, 113–115. [Google Scholar] [CrossRef] [Green Version]
- Alexandrova, L.A.; Jasko, M.V.; Negrya, S.D.; Solyev, P.N.; Shevchenko, O.V.; Solodinin, A.P.; Kolonitskaya, D.P.; Karpenko, I.L.; Efremenkova, O.V.; Glukhova, A.A.; et al. Discovery of Novel N4-Alkylcytidines as Promising Antimicrobial Agents. Eur. J. Med. Chem. 2021, 215, 113212. [Google Scholar] [CrossRef]
- Alexandrova, L.A.; Shevchenko, O.V.; Jasko, M.V.; Solyev, P.N.; Karpenko, I.L.; Negrya, S.D.; Efremenkova, O.V.; Vasilieva, B.F.; Efimenko, T.A.; Avdanina, D.A.; et al. 3′-Amino Modifications Enhance the Antifungal Properties of N 4-Alkyl-5-Methylcytidines for Potential Biocides. New J. Chem. 2022, 46, 5614–5626. [Google Scholar] [CrossRef]
- Rautio, J.; Kumpulainen, H.; Heimbach, T.; Oliyai, R.; Oh, D.; Järvinen, T.; Savolainen, J. Prodrugs: Design and Clinical Applications. Nat. Rev. Drug Discov. 2008, 7, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Huttunen, K.M.; Raunio, H.; Rautio, J. Prodrugs—From Serendipity to Rational Design. Pharmacol. Rev. 2011, 63, 750–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stella, V.J.; Nti-Addae, K.W. Prodrug Strategies to Overcome Poor Water Solubility. Adv. Drug Deliv. Rev. 2007, 59, 677–694. [Google Scholar] [CrossRef] [PubMed]
- Khandazhinskaya, A.L.; Matyugina, E.S.; Shirokova, E.A. Anti-HIV therapy with AZT prodrugs: AZT phosphonate derivatives, current state and prospects. Expert Opin. Drug Metab. Toxicol. 2010, 6, 701–714. [Google Scholar] [CrossRef]
- Khandazhinskaya, A.L.; Yasko, M.V.; Shirokova, E.A. The synthesis and antiviral properties of acyclic nucleoside analogues with a phosphonomethoxy fragment in the side chain. Curr. Med. Chem. 2006, 13, 2953–2980. [Google Scholar] [CrossRef]
- Jornada, D.; dos Santos Fernandes, G.; Chiba, D.; de Melo, T.; dos Santos, J.; Chung, M. The Prodrug Approach: A Successful Tool for Improving Drug Solubility. Molecules 2015, 21, 42. [Google Scholar] [CrossRef] [Green Version]
- Mori, G.; Chiarelli, L.R.; Riccardi, G.; Pasca, M.R. New Prodrugs against Tuberculosis. Drug Discov. Today 2017, 22, 519–525. [Google Scholar] [CrossRef]
- Negrya, S.D.; Jasko, M.V.; Solyev, P.N.; Karpenko, I.L.; Efremenkova, O.V.; Vasilyeva, B.F.; Sumarukova, I.G.; Kochetkov, S.N.; Alexandrova, L.A. Synthesis of Water-Soluble Prodrugs of 5-Modified 2′-Deoxyuridines and Their Antibacterial Activity. J. Antibiot. 2020, 73, 236–246. [Google Scholar] [CrossRef]
- Negrya, S.D.; Jasko, M.V.; Makarov, D.A.; Solyev, P.N.; Karpenko, I.L.; Shevchenko, O.V.; Chekhov, O.V.; Glukhova, A.A.; Vasilyeva, B.F.; Efimenko, T.A.; et al. Glycol and Phosphate Depot Forms of 4- and/or 5-Modified Nucleosides Exhibiting Antibacterial Activity. Mol. Biol. 2021, 55, 143–153. [Google Scholar] [CrossRef]
- Negrya, S.D.; Jasko, M.V.; Makarov, D.A.; Karpenko, I.L.; Solyev, P.N.; Chekhov, V.O.; Efremenkova, O.V.; Vasilieva, B.F.; Efimenko, T.A.; Kochetkov, S.N.; et al. Oligoglycol carbonate prodrugs of 5-modified 2′-deoxyuridines: Synthesis and antibacterial activity. Mendeleev Commun. 2022. In press. [Google Scholar]
- Bucher, H.C.; Griffith, L.E.; Guyatt, G.H.; Sudre, P.; Naef, M.; Sendi, P.; Battegay, M. Isoniazid Prophylaxis for Tuberculosis in HIV Infection: A Meta-Analysis of Randomized Controlled Trials. Aids 1999, 13, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Daley, C.L.; Small, P.M.; Schecter, G.F.; Schoolnik, G.K.; McAdam, R.A.; Jacobs, W.R.; Hopewell, P.C. An Outbreak of Tuberculosis with Accelerated Progression among Persons Infected with the Human Immunodeficiency Virus. N. Engl. J. Med. 1992, 326, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Pawlowski, A.; Jansson, M.; Sköld, M.; Rottenberg, M.E.; Källenius, G. Tuberculosis and HIV Co-Infection. PLoS Pathog. 2012, 8, e1002464. [Google Scholar] [CrossRef]
- Sassetti, C.M.; Boyd, D.H.; Rubin, E.J. Genes Required for Mycobacterial Growth Defined by High Density Mutagenesis. Mol. Microbiol. 2003, 48, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Sclafani, R.A.; Fangman, W.L. Yeast Gene CDC8 Encodes Thymidylate Kinase and Is Complemented by Herpes Thymidine Kinase Gene TK. Proc. Natl. Acad. Sci. USA 1984, 81, 5821–5825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crystal Structure of Inactive Mycobacterium Tuberculosis Thymidylate Kinase Complexed with Thymidine Monophosphate (TMP) at pH 4.6 (Resolution 1.85 Å). Available online: https://www.rcsb.org/structure/1N5I (accessed on 17 January 2022).
- Li de la Sierra, I.; Munier-Lehmann, H.; Gilles, A.; Bârzu, O.; Delarue, M. X-ray Structure of TMP Kinase from Mycobacterium tuberculosis Complexed with TMP at 1.95 Å Resolution. J. Mol. Biol. 2001, 311, 87–100. [Google Scholar] [CrossRef]
- Munier-Lehmann, H.; Chaffotte, A.; Pochet, S.; Labesse, G. Thymidylate Kinase of Mycobacterium tuberculosis: A Chimera Sharing Properties Common to Eukaryotic and Bacterial Enzymes. Protein Sci. 2001, 10, 1195–1205. [Google Scholar] [CrossRef] [Green Version]
- Haouz, A.; Vanheusden, V.; Munier-Lehmann, H.; Froeyen, M.; Herdewijn, P.; Van Calenbergh, S.; Delarue, M. Enzymatic and Structural Analysis of Inhibitors Designed against Mycobacterium tuberculosis Thymidylate Kinase. J. Biol. Chem. 2003, 278, 4963–4971. [Google Scholar] [CrossRef] [Green Version]
- Vanheusden, V.; Munier-Lehmann, H.; Pochet, S.; Herdewijn, P.; Van Calenbergh, S. Synthesis and Evaluation of Thymidine-5′-O-Monophosphate Analogues as Inhibitors of Mycobacterium tuberculosis Thymidylate Kinase. Bioorg. Med. Chem. Lett. 2002, 12, 2695–2698. [Google Scholar] [CrossRef]
- Fioravanti, E.; Adam, V.; Munier-Lehmann, H.; Bourgeois, D. The Crystal Structure of Mycobacterium Tuberculosis Thymidylate Kinase in Complex with 3’-Azidodeoxythymidine Monophosphate Suggests a Mechanism for Competitive Inhibition. Biochemistry 2005, 44, 130–137. [Google Scholar] [CrossRef]
- Vanheusden, V.; Munier-Lehmann, H.; Froeyen, M.; Dugué, L.; Heyerick, A.; De Keukeleire, D.; Pochet, S.; Busson, R.; Herdewijn, P.; Van Calenbergh, S. 3′-C-Branched-Chain-Substituted Nucleosides and Nucleotides as Potent Inhibitors of Mycobacterium tuberculosis Thymidine Monophosphate Kinase. J. Med. Chem. 2003, 46, 3811–3821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanheusden, V.; Munier-Lehmann, H.; Froeyen, M.; Busson, R.; Rozenski, J.; Herdewijn, P.; Van Calenbergh, S. Discovery of Bicyclic Thymidine Analogues as Selective and High-Affinity Inhibitors of Mycobacterium tuberculosis Thymidine Monophosphate Kinase. J. Med. Chem. 2004, 47, 6187–6194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pochet, S.; Dugue, L.; Douguet, D.; Labesse, G.; Munier-Lehmann, H. Nucleoside Analogues as Inhibitors of Thymidylate Kinases: Possible Therapeutic Applications. ChemBioChem 2002, 3, 108–110. [Google Scholar] [CrossRef]
- Pochet, S.; Dugué, L.; Labesse, G.; Delepierre, M.; Munier-Lehmann, H. Comparative Study of Purine and Pyrimidine Nucleoside Analogues Acting on the Thymidylate Kinases of Mycobacterium tuberculosis and of Humans. ChemBioChem 2003, 4, 742–747. [Google Scholar] [CrossRef] [Green Version]
- Van Daele, I.; Munier-Lehmann, H.; Hendrickx, P.M.S.; Marchal, G.; Chavarot, P.; Froeyen, M.; Qing, L.; Martins, J.C.; Van Calenbergh, S. Synthesis and Biological Evaluation of Bicyclic Nucleosides as Inhibitors OfM. Tuberculosis Thymidylate Kinase. ChemMedChem 2006, 1, 1081–1090. [Google Scholar] [CrossRef] [PubMed]
- Van Daele, I.; Munier-Lehmann, H.; Froeyen, M.; Balzarini, J.; Van Calenbergh, S. Rational Design of 5′-Thiourea-Substituted α-Thymidine Analogues as Thymidine Monophosphate Kinase Inhibitors Capable of Inhibiting Mycobacterial Growth. J. Med. Chem. 2007, 50, 5281–5292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasse, C.; Huteau, V.; Douguet, D.; Munier-Lehmann, H.; Pochet, S. A New Family of Inhibitors of Mycobacterium tuberculosis Thymidine Monophosphate Kinase. Nucleosides Nucleotides Nucleic Acids 2007, 26, 1057–1061. [Google Scholar] [CrossRef]
- Gasse, C.; Douguet, D.; Huteau, V.; Marchal, G.; Munier-Lehmann, H.; Pochet, S. Substituted Benzyl-Pyrimidines Targeting Thymidine Monophosphate Kinase of Mycobacterium tuberculosis: Synthesis and in Vitro Anti-Mycobacterial Activity. Bioorg. Med. Chem. 2008, 16, 6075–6085. [Google Scholar] [CrossRef]
- Familiar, O.; Munier-Lehmann, H.; Negri, A.; Gago, F.; Douguet, D.; Rigouts, L.; Hernández, A.; Camarasa, M.; Pérez-Pérez, M. Exploring Acyclic Nucleoside Analogues as Inhibitors of Mycobacterium tuberculosis Thymidylate Kinase. ChemMedChem 2008, 3, 1083–1093. [Google Scholar] [CrossRef] [Green Version]
- Familiar, O.; Munier-Lehmann, H.; Aínsa, J.A.; Camarasa, M.-J.; Pérez-Pérez, M.-J. Design, Synthesis and Inhibitory Activity against Mycobacterium tuberculosis Thymidine Monophosphate Kinase of Acyclic Nucleoside Analogues with a Distal Imidazoquinolinone. Eur. J. Med. Chem. 2010, 45, 5910–5918. [Google Scholar] [CrossRef]
- Carreras, C.W.; Santi, D.V. The Catalytic Mechanism and Structure of Thymidylate Synthase. Annu. Rev. Biochem. 1995, 64, 721–762. [Google Scholar] [CrossRef] [PubMed]
- Myllykallio, H.; Lipowski, G.; Leduc, D.; Filee, J.; Forterre, P.; Liebl, U. An Alternative Flavin-Dependent Mechanism for Thymidylate Synthesis. Science 2002, 297, 105–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koehn, E.M.; Fleischmann, T.; Conrad, J.A.; Palfey, B.A.; Lesley, S.A.; Mathews, I.I.; Kohen, A. An Unusual Mechanism of Thymidylate Biosynthesis in Organisms Containing the ThyX Gene. Nature 2009, 458, 919–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graziani, S.; Xia, Y.; Gurnon, J.R.; Van Etten, J.L.; Leduc, D.; Skouloubris, S.; Myllykallio, H.; Liebl, U. Functional Analysis of FAD-Dependent Thymidylate Synthase ThyX from Paramecium Bursaria Chlorella Virus-1. J. Biol. Chem. 2004, 279, 54340–54347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathews, I.I.; Deacon, A.M.; Canaves, J.M.; McMullan, D.; Lesley, S.A.; Agarwalla, S.; Kuhn, P. Functional Analysis of Substrate and Cofactor Complex Structures of a Thymidylate Synthase-Complementing Protein. Structure 2003, 11, 677–690. [Google Scholar] [CrossRef] [Green Version]
- Choi, M.; Karunaratne, K.; Kohen, A. Flavin-Dependent Thymidylate Synthase as a New Antibiotic Target. Molecules 2016, 21, 654. [Google Scholar] [CrossRef] [Green Version]
- Sampathkumar, P.; Turley, S.; Ulmer, J.E.; Rhie, H.G.; Sibley, C.H.; Hol, W.G.J. Structure of the Mycobacterium tuberculosis Flavin Dependent Thymidylate Synthase (MtbThyX) at 2.0Å Resolution. J. Mol. Biol. 2005, 352, 1091–1104. [Google Scholar] [CrossRef]
- Kögler, M.; Vanderhoydonck, B.; De Jonghe, S.; Rozenski, J.; Van Belle, K.; Herman, J.; Louat, T.; Parchina, A.; Sibley, C.; Lescrinier, E.; et al. Synthesis and Evaluation of 5-Substituted 2′-Deoxyuridine Monophosphate Analogues As Inhibitors of Flavin-Dependent Thymidylate Synthase in Mycobacterium tuberculosis. J. Med. Chem. 2011, 54, 4847–4862. [Google Scholar] [CrossRef] [Green Version]
- Crystal Structure of the Mtb ThyA in Complex with 5-Fluoro-DUMP. Available online: https://www.rcsb.org/structure/4FOA (accessed on 21 January 2022).
- Kögler, M.; Busson, R.; De Jonghe, S.; Rozenski, J.; Van Belle, K.; Louat, T.; Munier-Lehmann, H.; Herdewijn, P. Synthesis and Evaluation of 6-Aza-2′-Deoxyuridine Monophosphate Analogs as Inhibitors of Thymidylate Synthases, and as Substrates or Inhibitors of Thymidine Monophosphate Kinase in Mycobacterium tuberculosis. Chem. Biodivers. 2012, 9, 536–556. [Google Scholar] [CrossRef]
- Parchina, A.; Froeyen, M.; Margamuljana, L.; Rozenski, J.; De Jonghe, S.; Briers, Y.; Lavigne, R.; Herdewijn, P.; Lescrinier, E. Discovery of an Acyclic Nucleoside Phosphonate That Inhibits Mycobacterium tuberculosis ThyX Based on the Binding Mode of a 5-Alkynyl Substrate Analogue. ChemMedChem 2013, 8, 1373–1383. [Google Scholar] [CrossRef]
- McGuigan, C.; Derudas, M.; Gonczy, B.; Hinsinger, K.; Kandil, S.; Pertusati, F.; Serpi, M.; Snoeck, R.; Andrei, G.; Balzarini, J.; et al. ProTides of N-(3-(5-(2′-Deoxyuridine))Prop-2-Ynyl)Octanamide as Potential Anti-Tubercular and Anti-Viral Agents. Bioorg. Med. Chem. 2014, 22, 2816–2824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehellou, Y.; Balzarini, J.; McGuigan, C. Aryloxy Phosphoramidate Triesters: A Technology for Delivering Monophosphorylated Nucleosides and Sugars into Cells. ChemMedChem 2009, 4, 1779–1791. [Google Scholar] [CrossRef] [PubMed]
- McGuigan, C.; Murziani, P.; Slusarczyk, M.; Gonczy, B.; Vande Voorde, J.; Liekens, S.; Balzarini, J. Phosphoramidate ProTides of the Anticancer Agent FUDR Successfully Deliver the Preformed Bioactive Monophosphate in Cells and Confer Advantage over the Parent Nucleoside. J. Med. Chem. 2011, 54, 7247–7258. [Google Scholar] [CrossRef] [PubMed]
- Slusarczyk, M.; Lopez, M.H.; Balzarini, J.; Mason, M.; Jiang, W.G.; Blagden, S.; Thompson, E.; Ghazaly, E.; McGuigan, C. Application of ProTide Technology to Gemcitabine: A Successful Approach to Overcome the Key Cancer Resistance Mechanisms Leads to a New Agent (NUC-1031) in Clinical Development. J. Med. Chem. 2014, 57, 1531–1542. [Google Scholar] [CrossRef]
- McGuigan, C.; Madela, K.; Aljarah, M.; Gilles, A.; Brancale, A.; Zonta, N.; Chamberlain, S.; Vernachio, J.; Hutchins, J.; Hall, A.; et al. Design, Synthesis and Evaluation of a Novel Double pro-Drug: INX-08189. A New Clinical Candidate for Hepatitis C Virus. Bioorg. Med. Chem. Lett. 2010, 20, 4850–4854. [Google Scholar] [CrossRef]
- Holy, A. Phosphonomethoxyalkyl Analogs of Nucleotides. Curr. Pharm. Des. 2003, 9, 2567–2592. [Google Scholar] [CrossRef]
- De Clercq, E.; Holý, A. Acyclic Nucleoside Phosphonates: A Key Class of Antiviral Drugs. Nat. Rev. Drug Discov. 2005, 4, 928–940. [Google Scholar] [CrossRef]
- Biteau, N.G.; Roy, V.; Lambry, J.-C.; Becker, H.F.; Myllykallio, H.; Agrofoglio, L.A. Synthesis of Acyclic Nucleoside Phosphonates Targeting Flavin-Dependent Thymidylate Synthase in Mycobacterium tuberculosis. Bioorg. Med. Chem. 2021, 46, 116351. [Google Scholar] [CrossRef]
- Alexandrova, L.A.; Chekhov, V.O.; Shmalenyuk, E.R.; Kochetkov, S.N.; El-Asrar, R.A.; Herdewijn, P. Synthesis and Evaluation of C-5 Modified 2′-Deoxyuridine Monophosphates as Inhibitors of M. tuberculosis Thymidylate Synthase. Bioorg. Med. Chem. 2015, 23, 7131–7137. [Google Scholar] [CrossRef]
- Gibson, L.M.; Celeste, L.R.; Lovelace, L.L.; Lebioda, L. Structures of Human Thymidylate Synthase R163K with DUMP, FdUMP and Glutathione Show Asymmetric Ligand Binding. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 60–66. [Google Scholar] [CrossRef]
- Chemical Computing Group Inc. Molecular Operating Environment (MOE). 2016. Available online: https://www.chemcomp.com/Products.htm (accessed on 21 January 2015).
- Halgren, T.A. Merck Molecular Force Field. V. Extension of MMFF94 Using Experimental Data, Additional Computational Data, and Empirical Rules. J. Comput. Chem. 1996, 17, 616–641. [Google Scholar] [CrossRef]
- Halgren, T.A. MMFF VII. Characterization of MMFF94, MMFF94s, and Other Widely Available Force Fields for Conformational Energies and for Intermolecular-Interaction Energies and Geometries. J. Comput. Chem. 1999, 20, 730–748. [Google Scholar] [CrossRef]
- Wang, J.; Cieplak, P.; Kollman, P.A. How Well Does a Restrained Electrostatic Potential (RESP) Model Perform in Calculating Conformational Energies of Organic and Biological Molecules? J. Comput. Chem. 2000, 21, 1049–1074. [Google Scholar] [CrossRef]
- Khandazhinskaya, A.L.; Matyugina, E.S.; Alexandrova, L.A.; Kezin, V.A.; Chernousova, L.N.; Smirnova, T.G.; Andreevskaya, S.N.; Popenko, V.I.; Leonova, O.G.; Kochetkov, S.N. Interaction of 5-Substituted Pyrimidine Nucleoside Analogues and M.tuberculosis: A View through an Electron Microscope. Biochimie 2020, 171–172, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Barisch, C.; Soldati, T. Mycobacterium Marinum Degrades Both Triacylglycerols and Phospholipids from Its Dictyostelium Host to Synthesise Its Own Triacylglycerols and Generate Lipid Inclusions. PLoS Pathog. 2017, 13, e1006095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijay, S.; Hai, H.T.; Thu, D.D.A.; Johnson, E.; Pielach, A.; Phu, N.H.; Thwaites, G.E.; Thuong, N.T.T. Ultrastructural Analysis of Cell Envelope and Accumulation of Lipid Inclusions in Clinical Mycobacterium tuberculosis Isolates from Sputum, Oxidative Stress, and Iron Deficiency. Front. Microbiol. 2018, 8, 2681. [Google Scholar] [CrossRef]
- Peyron, P.; Vaubourgeix, J.; Poquet, Y.; Levillain, F.; Botanch, C.; Bardou, F.; Daffé, M.; Emile, J.-F.; Marchou, B.; Cardona, P.-J.; et al. Foamy Macrophages from Tuberculous Patients’ Granulomas Constitute a Nutrient-Rich Reservoir for M. tuberculosis Persistence. PLoS Pathog. 2008, 4, e1000204. [Google Scholar] [CrossRef]
- Kayigire, X.A.; Friedrich, S.O.; van der Merwe, L.; Donald, P.R.; Diacon, A.H. Simultaneous Staining of Sputum Smears for Acid-Fast and Lipid-Containing Myobacterium tuberculosis Can Enhance the Clinical Evaluation of Antituberculosis Treatments. Tuberculosis 2015, 95, 770–779. [Google Scholar] [CrossRef]
- Velayati, A.A.; Farnia, P.; Ibrahim, T.A.; Haroun, R.Z.; Kuan, H.O.; Ghanavi, J.; Farnia, P.; Kabarei, A.N.; Tabarsi, P.; Omar, A.R.; et al. Differences in Cell Wall Thickness between Resistant and Nonresistant Strains of Mycobacterium tuberculosis: Using Transmission Electron Microscopy. Chemotherapy 2009, 55, 303–307. [Google Scholar] [CrossRef]
- Vijay, S.; Anand, D.; Ajitkumar, P. Unveiling Unusual Features of Formation of Septal Partition and Constriction in Mycobacteria—an Ultrastructural Study. J. Bacteriol. 2012, 194, 702–707. [Google Scholar] [CrossRef] [Green Version]
- Torrelles, J.B.; Schlesinger, L.S. Diversity in Mycobacterium tuberculosis Mannosylated Cell Wall Determinants Impacts Adaptation to the Host. Tuberculosis 2010, 90, 84–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Seo, H.; Mahmud, H.A.; Islam, M.I.; Kim, Y.-S.; Lyu, J.; Nam, K.-W.; Lee, B.-E.; Lee, K.-I.; Song, H.-Y. In Vitro Effect of DFC-2 on Mycolic Acid Biosynthesis in Mycobacterium tuberculosis. J. Microbiol. Biotechnol. 2017, 27, 1932–1941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parumasivam, T.; Kumar, H.S.N.; Mohamad, S.; Ibrahim, P.; Sadikun, A. Effects of a Lipophilic Isoniazid Derivative on the Growth and Cellular Morphogenesis of Mycobacterium tuberculosis H37Rv. Int. J. Pharm. Pharm. Sci. 2013, 5, 43–50. [Google Scholar]
Figure 1.
The first selective inhibitors of M. avium 1–4 among nucleoside derivatives.

Figure 2.
C-5 alkynyl derivatives of pyrimidine nucleosides.

Figure 3.
Pyrimidine nucleosides modified at the carbohydrate moiety 12–15—inhibitors of mycobacteria growth.
Figure 3.
Pyrimidine nucleosides modified at the carbohydrate moiety 12–15—inhibitors of mycobacteria growth.

Figure 4.
C-5 alkynyl derivatives of uridine and 6-methyluridine.

Figure 5.
Pyrimidine derivatives of 2′-deoxy-, 3′-azido and 3′-amino-2′,3′-dideoxynucleosides [42,43].

Figure 6.
Structures of 5-modified 6-aza-2′-deoxyuridines.

Figure 7.
5′-Norcarbocyclic uracil derivatives.

Figure 8.
5-Alkyloxymethyl and 5-alkyltriazolylmethyl substituted 5′-norcarbocyclic uridine analogues. R: C10H21 (30a, 31a), C11H23 (30b, 31b), C12H25 (30c, 31c).
Figure 8.
5-Alkyloxymethyl and 5-alkyltriazolylmethyl substituted 5′-norcarbocyclic uridine analogues. R: C10H21 (30a, 31a), C11H23 (30b, 31b), C12H25 (30c, 31c).

Figure 9.
N4-Alkyl derivatives of 2′-deoxycytidine and 2′,3′-dideoxy-3′-modified cytidine.

Figure 10.
Depot forms of 5-alkyloxymethyl—35, 36 и 5-alkyltriazolylmethyl; 37, 38 2′-deoxyuridines.
Figure 10.
Depot forms of 5-alkyloxymethyl—35, 36 и 5-alkyltriazolylmethyl; 37, 38 2′-deoxyuridines.

Figure 11.
Heterodimers of AZT and 5-(4-decyl-1,2,3-triazol-1-yl-methyl)- 2′-deoxyuridine (39–41) and the 5′-norcarbocyclic analogues of 2′,3′-dideoxy-2′,3′-didehydrouridine (42–43).
Figure 11.
Heterodimers of AZT and 5-(4-decyl-1,2,3-triazol-1-yl-methyl)- 2′-deoxyuridine (39–41) and the 5′-norcarbocyclic analogues of 2′,3′-dideoxy-2′,3′-didehydrouridine (42–43).


Figure 12.
Metabolism of thymidine nucleotides.

Figure 13.
Structure of TMPKmtb–TMP complex (protein is presented in the form of a ribbon [82]. https://www.rcsb.org/structure/1N5I, (accessed on 17 January 2022).
Figure 13.
Structure of TMPKmtb–TMP complex (protein is presented in the form of a ribbon [82]. https://www.rcsb.org/structure/1N5I, (accessed on 17 January 2022).

Figure 14.
Bicyclic inhibitors of TMPKmtb 59–62 and dinucleoside inhibitor of TMPKmtb 62.

Figure 15.
Arylurea- и arylthiourea- α- and β-thymidine derivatives 63a–g and 64a–d.

Figure 16.
Arylpyrimidine inhibitors of TMPKmtb 65–68.

Figure 17.
Synthesis of thymidine monophosphate from 2′-deoxyuridine monophosphate by ThyX catalysis. dUMP—deoxyuridine monophosphate, FAD—flavin adenine dinucleotide, FADH2—reduced form of flavin adenine dinucleotide, ThyX—flavin-dependent thymidylate synthase, CH2THF—5,10-methylenetetrahydrofolate, THF—tetrahydrofolate, TMP—thymidine monophosphate.
Figure 17.
Synthesis of thymidine monophosphate from 2′-deoxyuridine monophosphate by ThyX catalysis. dUMP—deoxyuridine monophosphate, FAD—flavin adenine dinucleotide, FADH2—reduced form of flavin adenine dinucleotide, ThyX—flavin-dependent thymidylate synthase, CH2THF—5,10-methylenetetrahydrofolate, THF—tetrahydrofolate, TMP—thymidine monophosphate.

Figure 18.
Structure of ThyX with 5-F-dUMP complex [106]. https://www.rcsb.org/structure/4FOA, (accessed on 21 January 2022).
Figure 18.
Structure of ThyX with 5-F-dUMP complex [106]. https://www.rcsb.org/structure/4FOA, (accessed on 21 January 2022).

Figure 19.
Structures of ThyX inhibitors.

Figure 20.
Structures of acyclic nucleoside analogues’ phosphonates—inhibitors of ThyX.

Figure 21.
5′-Monophosphates of 5-oxymethyl and 5-triazolylmethyl-2′-deoxyuridine.

Figure 22.
The proposed structure of the complex of 5-undecyloxymethyl-dUMP (79b) with ThyX (based on docking).
Figure 22.
The proposed structure of the complex of 5-undecyloxymethyl-dUMP (79b) with ThyX (based on docking).

Figure 23.
Structures of 5-substituted uracil derivatives, effective inhibitors of Mtb growth.

Figure 24.
Transmission electron microscopy (TEM) of Mtb H37Rv cells. (a,b)—intact control cells; (c,d)—cells cultured in a medium containing the studied nucleoside derivatives; (a,c)—ultrathin sections; (b,d)—cells adsorbed on supporting film and stained with uranyl acetate. Scale (a,c)—0,5 μm; (b,d)—1 μm.
Figure 24.
Transmission electron microscopy (TEM) of Mtb H37Rv cells. (a,b)—intact control cells; (c,d)—cells cultured in a medium containing the studied nucleoside derivatives; (a,c)—ultrathin sections; (b,d)—cells adsorbed on supporting film and stained with uranyl acetate. Scale (a,c)—0,5 μm; (b,d)—1 μm.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Alexandrova, L.A.; Khandazhinskaya, A.L.; Matyugina, E.S.; Makarov, D.A.; Kochetkov, S.N. Analogues of Pyrimidine Nucleosides as Mycobacteria Growth Inhibitors. Microorganisms 2022, 10, 1299. https://doi.org/10.3390/microorganisms10071299
AMA Style
Alexandrova LA, Khandazhinskaya AL, Matyugina ES, Makarov DA, Kochetkov SN. Analogues of Pyrimidine Nucleosides as Mycobacteria Growth Inhibitors. Microorganisms. 2022; 10(7):1299. https://doi.org/10.3390/microorganisms10071299
Chicago/Turabian StyleAlexandrova, Liudmila A., Anastasia L. Khandazhinskaya, Elena S. Matyugina, Dmitriy A. Makarov, and Sergey N. Kochetkov. 2022. "Analogues of Pyrimidine Nucleosides as Mycobacteria Growth Inhibitors" Microorganisms 10, no. 7: 1299. https://doi.org/10.3390/microorganisms10071299
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.