
Stereocalpin B, a New Cyclic Depsipeptide from the Antarctic Lichen Ramalina terebrata

 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Isolation of Compounds 1–3
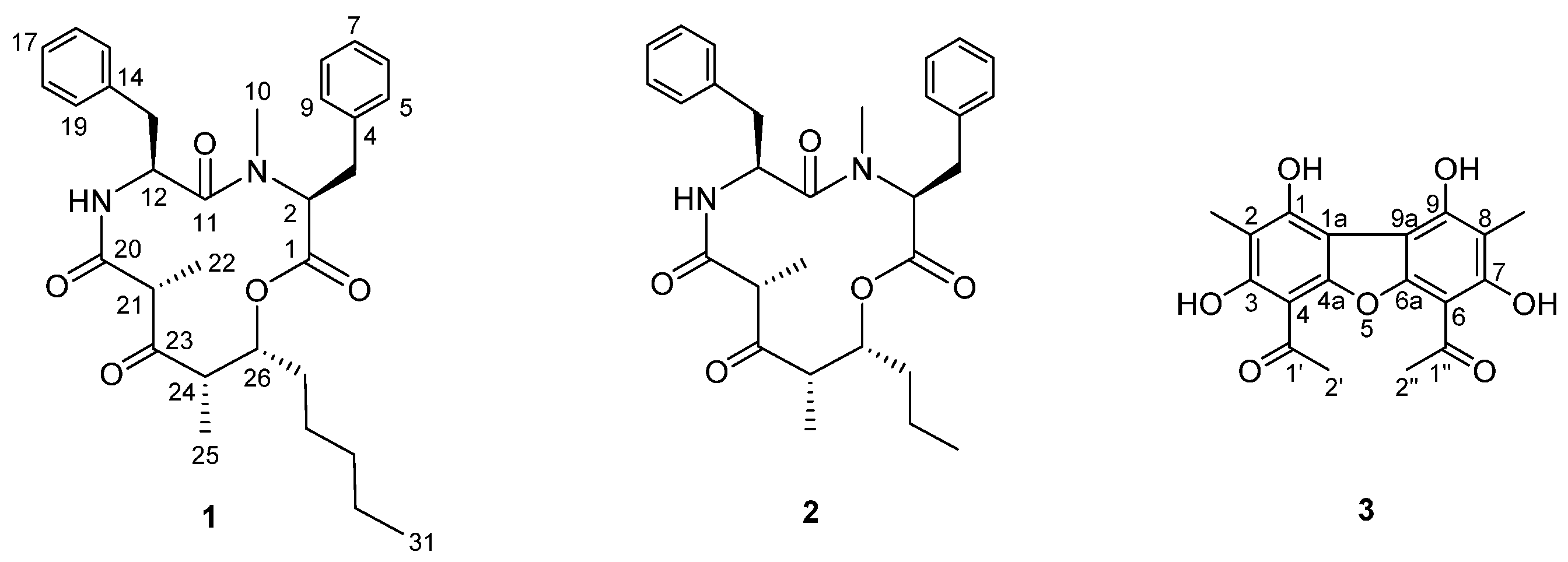
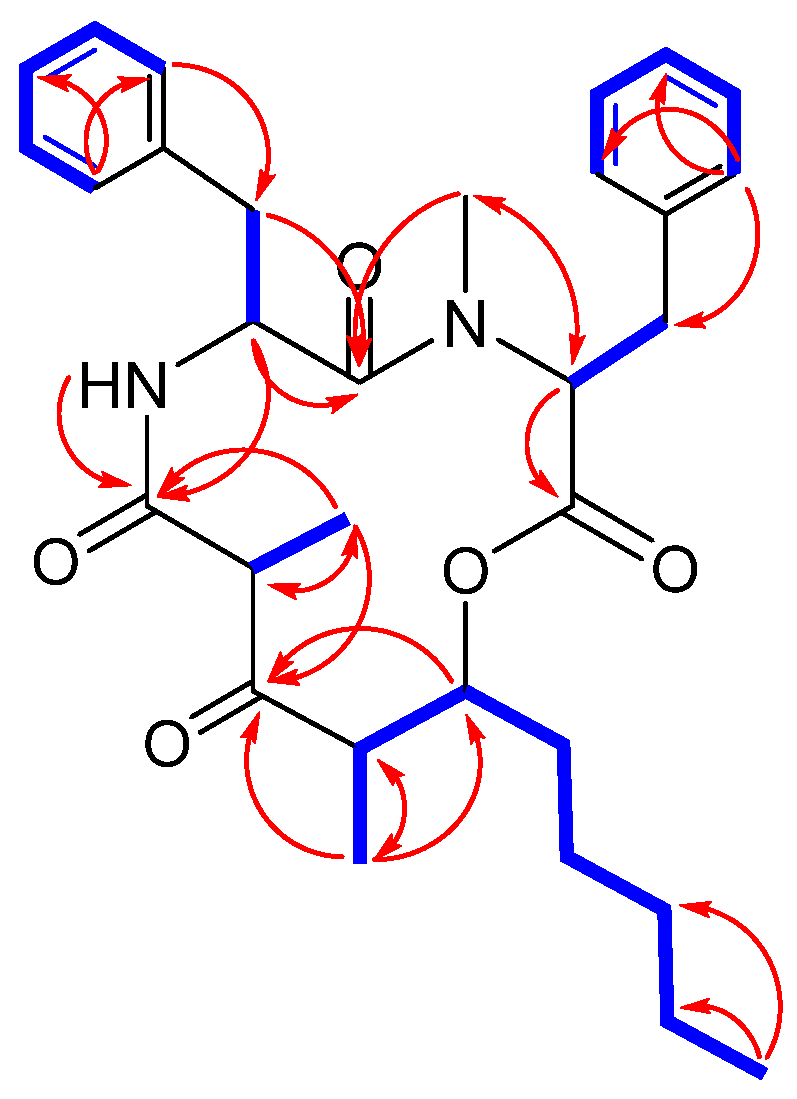
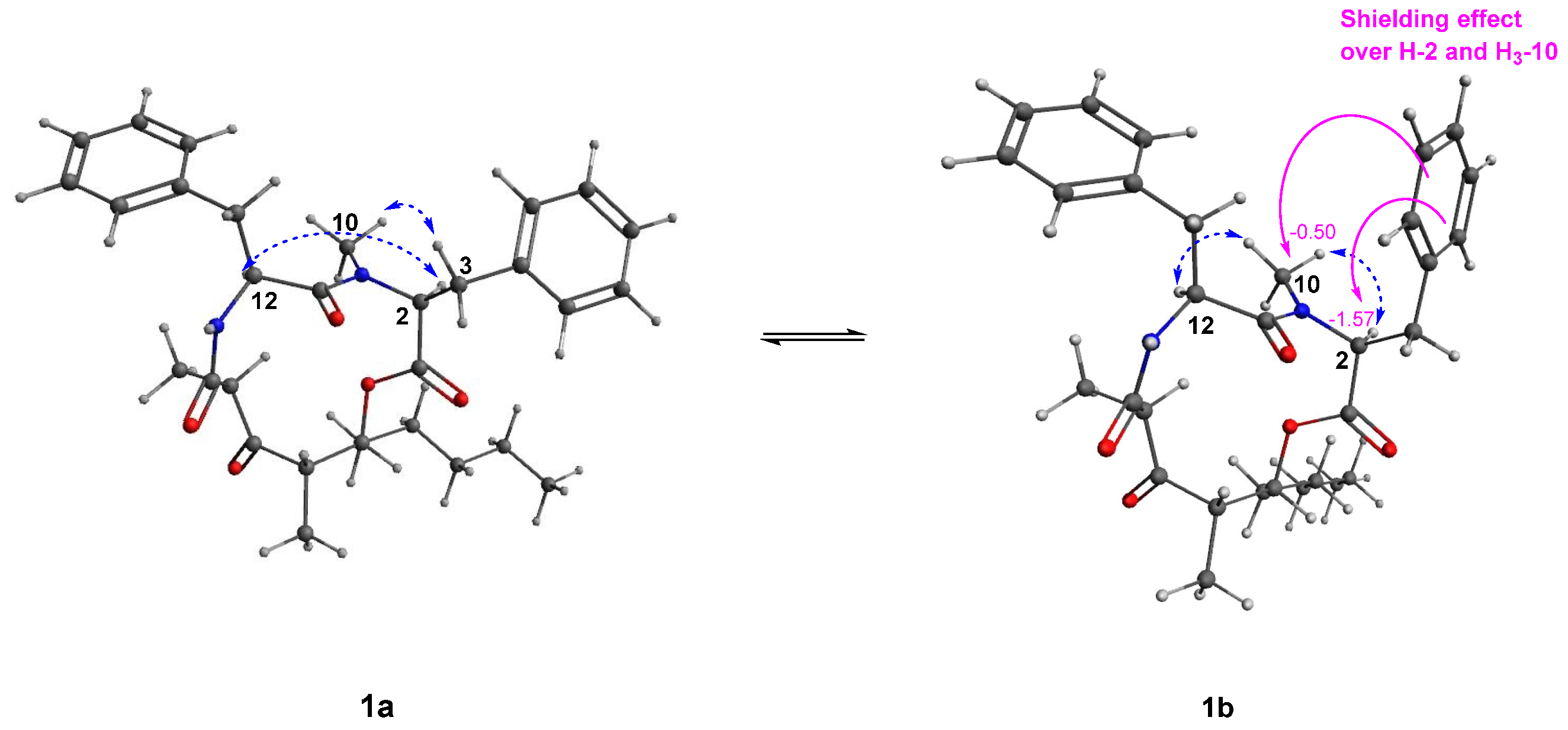
2.2. Structure Elucidation of Stereocalpin B (1)
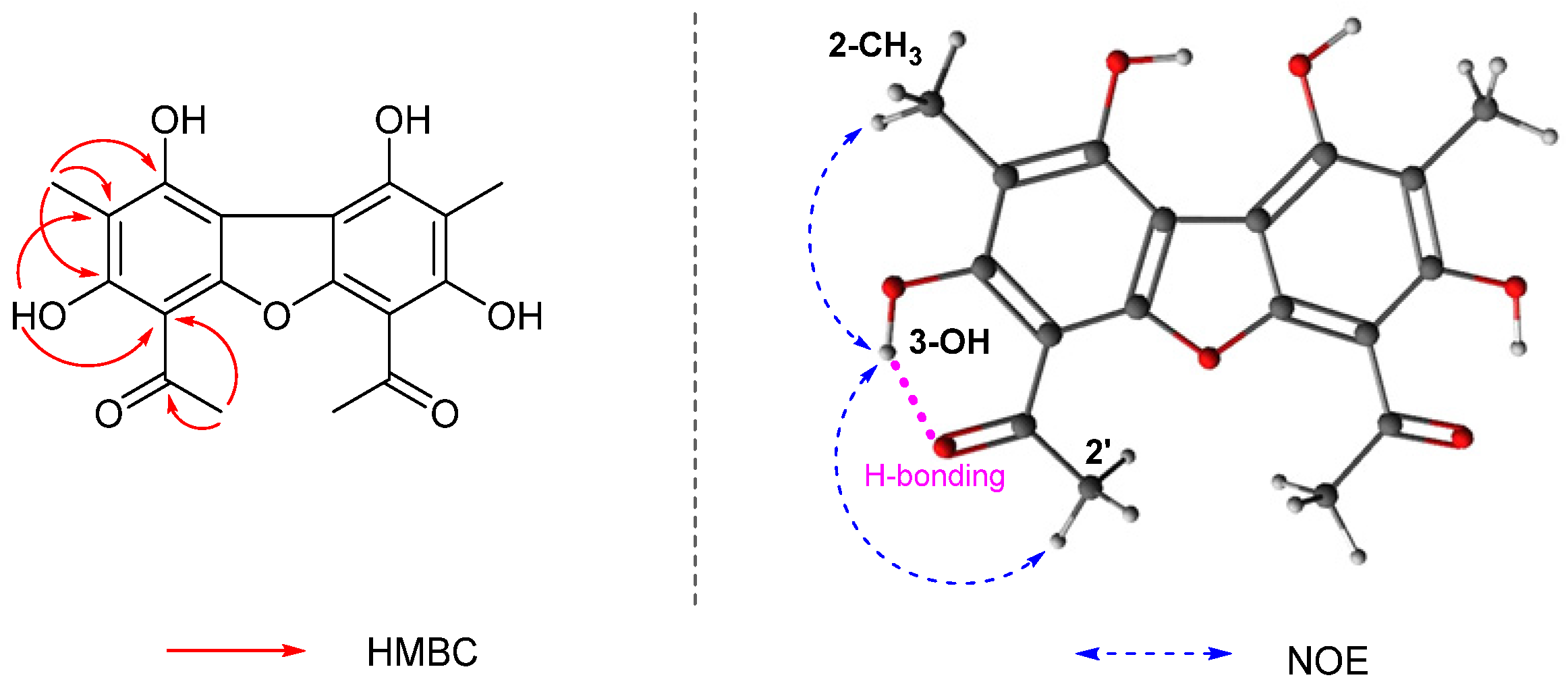
2.3. Structure Elucidation of Compound 3
2.4. Biological Activities of the Isolated Compounds (1–3)
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Lichen Material
3.3. Extraction and Isolation
3.4. Advanced Marfey’s Analyses of FDAA Derivatives Using LC-MS
3.5. Computational Details
3.6. PANIC Analysis
3.7. Antimicrobial Assay
3.8. Cell Culture
3.9. MTS Assay
3.10. Measurement of nitric oxide (NO) production
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Moreira, A.S.N.; Braz-Filho, R.; Mussi-Dias, V.; Vieira, I.J.C. Chemistry and Biological Activity of Ramalina Lichenized Fungi. Molecules 2015, 20, 8952–8987. [Google Scholar] [CrossRef] [Green Version]
- Shukla, V.; Joshi, G.P.; Rawat, M. Lichens as a Potential Natural Source of Bioactive Compounds: A Review. Phytochem. Rev. 2010, 9, 303–314. [Google Scholar] [CrossRef]
- Boustie, J.; Grube, M. Lichens—A Promising Source of Bioactive Secondary Metabolites. Plant Genet. Res. 2005, 3, 273. [Google Scholar] [CrossRef] [Green Version]
- Halama, P.; Van Haluwin, C. Antifungal Activity of Lichen Extracts and Lichenic Acids. BioControl 2004, 49, 95–107. [Google Scholar] [CrossRef]
- Mayer, M.; O’Neill, M.A.; Murray, K.E.; Santos-Magalhães, N.S.; Carneiro-Leão, A.M.A.; Thompson, A.M.; Appleyard, V.C. Usnic Acid: A Non-Genotoxic Compound with Anti-Cancer Properties. Anti-Cancer Drugs 2005, 16, 805–809. [Google Scholar] [CrossRef]
- Takai, M.; Uehara, Y.; Beisler, J.A. Usnic Acid Derivatives as Potential Antineoplastic Agents. J. Med. Chem. 1979, 22, 1380–1384. [Google Scholar] [CrossRef]
- Proksa, B.; Strudikova, M.; Pronayova, N.; Liptaj, T. (-)-Usnic Acid and Its Derivatives. Their Inhibition of Fungal Growth and Enzyme Activity. Pharmazie 1996, 51, 195–196. [Google Scholar]
- Lee, S.G.; Koh, H.Y.; Oh, H.; Han, S.J.; Kim, I.-C.; Lee, H.K.; Yim, J.H. Human Dermal Fibroblast Proliferation Activity of Usimine-C from Antarctic Lichen Ramalina Terebrata. Biotechnol. Lett. 2010, 32, 471–475. [Google Scholar] [CrossRef]
- Park, H.J.; Jang, Y.J.; Yim, J.H.; Lee, H.K.; Pyo, S. Ramalin Isolated from Ramalina Terebrata Attenuates Atopic Dermatitis-Like Skin Lesions in Balb/C Mice and Cutaneous Immune Responses in Keratinocytes and Mast Cells. Phytother. Res. 2016, 30, 1978–1987. [Google Scholar] [CrossRef]
- Cornejo, A.; Areche, C. Secondary Metabolites of Lichens as Both Anti-Aggregative and Antioxidant Agents in Tauopathies. J. Clin. Cell. Immunol. 2017, 8, 2. [Google Scholar] [CrossRef] [Green Version]
- Paudel, B.; Bhattarai, H.D.; Lee, H.K.; Oh, H.; Shin, H.W.; Yim, J.H. Antibacterial Activities of Ramalin, Usnic Acid and Its Three Derivatives Isolated from the Antarctic Lichen Ramalina Terebrata. Z. Naturforsch. C 2010, 65, 34–38. [Google Scholar] [CrossRef]
- Vo, Q.V.; Tam, N.M.; Van Bay, M.; Mechler, A. The Radical Scavenging Activity of Natural Ramalin: A Mechanistic and Kinetic Study. Chem. Phys. Lett. 2020, 739, 137004. [Google Scholar] [CrossRef]
- Cornejo, A.; Salgado, F.; Caballero, J.; Vargas, R.; Simirgiotis, M.; Areche, C. Secondary Metabolites in Ramalina Terebrata Detected by Uhplc/Esi/Ms/Ms and Identification of Parietin as Tau Protein Inhibitor. Int. J. Mol. Sci. 2016, 17, 1303. [Google Scholar] [CrossRef] [Green Version]
- Seo, C.; Yim, J.H.; Lee, H.K.; Park, S.M.; Sohn, J.-H.; Oh, H. Stereocalpin a, a Bioactive Cyclic Depsipeptide from the Antarctic Lichen Stereocaulon Alpinum. Tetrahedron Lett. 2008, 49, 29–31. [Google Scholar] [CrossRef]
- Grauso, L.; Li, Y.; Scarpato, S.; Shulha, O.; Rárová, L.; Strnad, M.; Teta, R.; Mangoni, A.; Zidorn, C. Structure and Conformation of Zosteraphenols, Tetracyclic Diarylheptanoids from the Seagrass Zostera Marina: An Nmr and Dft Study. Org. Lett. 2019, 22, 78–82. [Google Scholar] [CrossRef]
- Bhushan, R.; Brückner, H. Marfey’s Reagent for Chiral Amino Acid Analysis: A Review. Amino Acids 2004, 27, 231–247. [Google Scholar] [CrossRef]
- Takada, K.; Ninomiya, A.; Naruse, M.; Sun, Y.; Miyazaki, M.; Nogi, Y.; Okada, S.; Matsunaga, S. Surugamides a–E, Cyclic Octapeptides with Four D-Amino Acid Residues, from a Marine Streptomyces Sp.: Lc–Ms-Aided Inspection of Partial Hydrolysates for the Distinction of D-and L-Amino Acid Residues in the Sequence. J. Org. Chem. 2013, 78, 6746–6750. [Google Scholar] [CrossRef]
- Kaneda, M.; Inuki, S.; Ohno, H.; Oishi, S. Total Synthesis and Stereochemical Revision of Stereocalpin A: Mirror-Image Approach for Stereochemical Assignments of the Peptide–Polyketide Macrocycle. J. Org. Chem. 2018, 83, 3047–3060. [Google Scholar] [CrossRef]
- Mándi, A.; Swamy, M.M.; Taniguchi, T.; Anetai, M.; Monde, K. Reducing Molecular Flexibility by Cyclization for Elucidation of Absolute Configuration by Cd Calculations: Daurichromenic Acid. Chirality 2016, 28, 453–459. [Google Scholar] [CrossRef]
- Hu, H.; Krishnamurthy, K. Revisiting the Initial Rate Approximation in Kinetic Noe Measurements. J. Magn. Reson. 2006, 182, 173–177. [Google Scholar] [CrossRef]
- Butts, C.P.; Jones, C.R.; Song, Z.; Simpson, T.J. Accurate Noe-Distance Determination Enables the Stereochemical Assignment of a Flexible Molecule–Arugosin C. Chem. Comm. 2012, 48, 9023–9025. [Google Scholar] [CrossRef]
- Oh, J.; Patel, J.; Park, H.B.; Crawford, J.M. Β-Lactam Biotransformations Activate Innate Immunity. J. Org. Chem. 2018, 83, 7173–7179. [Google Scholar] [CrossRef]
- Oh, J.; Quan, K.T.; Lee, J.S.; Park, I.; Kim, C.S.; Ferreira, D.; Thuong, P.T.; Kim, Y.H.; Na, M. Nmr-Based Investigation of Hydrogen Bonding in a Dihydroanthracen-1 (4 H) One from Rubia Philippinensis and Its Soluble Epoxide Hydrolase Inhibitory Potential. J. Nat. Prod. 2018, 81, 2429–2435. [Google Scholar] [CrossRef]
- Oh, J.; Kim, N.Y.; Chen, H.; Palm, N.W.; Crawford, J.M. An Ugi-Like Biosynthetic Pathway Encodes Bombesin Receptor Subtype-3 Agonists. J. Am. Chem. Soc. 2019, 141, 16271–16278. [Google Scholar] [CrossRef]
- Frank, É.; Körtvélyesi, T.; Czugler, M.; Mucsi, Z.; Keglevich, G. New Steroid-Fused P-Heterocycles: Part I. Synthesis and Conformational Study of Dioxaphosphorino [16, 17-D] Estrone Derivatives. Steroids 2007, 72, 437–445. [Google Scholar] [CrossRef]
- Sartillo-Piscil, F.; Cruz, S.; Sánchez, M.; Höpfl, H.; de Parrodi, C.A.; Quintero, L. Conformational Analysis of Cyclic Phosphates Derived from 5-C′ Substituted 1, 2-O-Isopropylidene-A-D-Xylofuranose Derivatives. Tetrahedron 2003, 59, 4077–4083. [Google Scholar] [CrossRef]
- Shou, Q.; Banbury, L.K.; Renshaw, D.E.; Lambley, E.H.; Mon, H.; Macfarlane, G.A.; Griesser, H.J.; Heinrich, M.M.; Wohlmuth, H. Biologically Active Dibenzofurans from Pilidiostigma Glabrum, an Endemic Australian Myrtaceae. J. Nat. Prod. 2012, 75, 1612–1617. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1a | 1b | ||
|---|---|---|---|---|
| δH (m, J in Hz) | δC | δH (m, J in Hz) | δC | |
| 1 | 171.2 | 169.0 | ||
| 2 | 4.90 dd (11.0, 4.6) | 59.5 | 3.33 dd (11.2, 3.2) | 67.3 |
| 3 | 3.04 b, 3.20 b | 35.3 | 3.04 b, 3.27 dd (13.8, 3.2) | 34.9 |
| 4 | 138.1 | 135.8 | ||
| 5 | 6.60 d (7.2) | 128.9 | 7.39 d (7.0) | 128.9 |
| 6 | 7.05 t (7.4) | 128.3 | 7.31 b | 129.0 |
| 7 | 7.11 t (7.4) | 126.2 | 7.24 b | 127.2 |
| 8 | 7.05 t (7.4) | 128.3 | 7.31 b | 129.0 |
| 9 | 6.60 d (7.2) | 128.9 | 7.39 d (7.0) | 128.9 |
| 10 | 3.06 s | 29.5 | 2.56 s | 39.4 |
| 11 | 170.4 | 171.7 | ||
| 12 | 5.08 ddd (9.5, 8.0, 6.6) | 50.5 | 5.18 ddd (11.0, 9.2, 4.9) | 49.8 |
| 13 | 2.89 dd (14.1, 8.0), 3.12 dd (14.1, 6.6) | 38.0 | 3.06 b, 3.18 b | 36.8 |
| 14 | 136.5 | 135.7 | ||
| 15 | 7.08 d (7.0) | 129.3 | 7.28 d (7.1) | 128.9 |
| 16 | 7.24 t (7.0) | 128.3 | 7.23 b | 128.4 |
| 17 | 7.18 t (7.0) | 126.5 | 7.39 b | 127.2 |
| 18 | 7.24 t (7.0) | 128.3 | 7.23 b | 128.4 |
| 19 | 7.08 d (7.0) | 129.3 | 7.28 d (7.1) | 128.9 |
| NH | 6.17 d (9.5) | 6.65 d (9.2) | ||
| 20 | 166.7 | 168.3 | ||
| 21 | 3.17 m b | 57.0 | 3.43 q (6.5) | 55.8 |
| 22 | 0.93 d (6.6) | 13.6 | 1.27 d (6.5) | 12.2 |
| 23 | 205.2 | 205.4 | ||
| 24 | 3.22 m b | 47.3 | 3.00 m b | 48.5 |
| 25 | 1.04 d (7.1) | 14.3 | 1.14 d (7.4) | 15.9 |
| 26 | 4.71 dt (11.8, 3.6) | 77.3 | 5.04 dt (10.4, 2.7) | 77.2 |
| 27 | 1.73 b | 29.7 | 1.83 b | 29.7 |
| 28 | 1.66 b,c | 27.3 | 1.69 b,c | 27.3 |
| 29 | 1.22 b | 31.3 | 1.22 b | 31.3 |
| 30 | 1.31 b | 22.2 | 1.31 b | 22.2 |
| 31 | 0.85 t (6.7) b,c | 13.9 | 0.86 t (6.7) b,c | 13.9 |
| Position | δH (m, J in Hz) | δC | Position | δH (m, J in Hz) | δC |
|---|---|---|---|---|---|
| 1a | 105.0 | 9 | 157.0 | ||
| 1 | 157.0 | 9a | 105.0 | ||
| 2 | 106.9 | 2-CH3 | 1.99 s | 7.5 | |
| 3 | 162.4 | 8-CH3 | 1.99 s | 7.5 | |
| 4 | 101.0 | 1′ | 200.8 | ||
| 4a | 155.4 | 2′ | 2.66 s | 31.0 | |
| 5 | 1′′ | 200.8 | |||
| 6a | 155.4 | 2′′ | 2.66 s | 31.0 | |
| 6 | 101.0 | 3-OH | 13.37 s | ||
| 7 | 162.4 | 7-OH | 13.37 s | ||
| 8 | 106.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.; Jeong, S.Y.; Nguyen, D.L.; So, J.E.; Kim, K.H.; Kim, J.H.; Han, S.J.; Suh, S.-S.; Lee, J.H.; Youn, U.J. Stereocalpin B, a New Cyclic Depsipeptide from the Antarctic Lichen Ramalina terebrata. Metabolites 2022, 12, 141. https://doi.org/10.3390/metabo12020141
Lee S, Jeong SY, Nguyen DL, So JE, Kim KH, Kim JH, Han SJ, Suh S-S, Lee JH, Youn UJ. Stereocalpin B, a New Cyclic Depsipeptide from the Antarctic Lichen Ramalina terebrata. Metabolites. 2022; 12(2):141. https://doi.org/10.3390/metabo12020141
Chicago/Turabian StyleLee, Seulah, Se Yun Jeong, Dieu Linh Nguyen, Jae Eun So, Ki Hyun Kim, Ji Hee Kim, Se Jong Han, Sung-Suk Suh, Jun Hyuck Lee, and Ui Joung Youn. 2022. "Stereocalpin B, a New Cyclic Depsipeptide from the Antarctic Lichen Ramalina terebrata" Metabolites 12, no. 2: 141. https://doi.org/10.3390/metabo12020141