
Lifelong Aerobic Exercise Alleviates Sarcopenia by Activating Autophagy and Inhibiting Protein Degradation via the AMPK/PGC-1α Signaling Pathway

 , , and
, , and 
Abstract
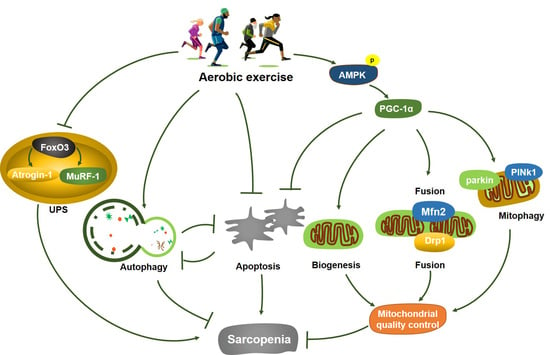
:
1. Introduction
2. Results
2.1. Lifelong Aerobic Exercise Alleviated the Reduction of Gastrocnemius Muscle Weight-Body Weight (GMW/BW) Ratio in Aged Mice
2.2. Lifelong Aerobic Exercise Ameliorated the Atrophy of Skeletal Muscle Fibers in Aged Mice
2.3. Lifelong Aerobic Exercise Suppressed Ultastructural Damage of Skeletal Muscle Fibers in Aged Mice
2.4. Lifelong Aerobic Exercise Improved Mitochondrial Function and Attenuated Oxidative Stress in Aged Skeletal Muscle
2.5. Lifelong Aerobic Exercise Suppressed E3 Ubiquitin Ligases and Decreased Akt/mTOR Signaling Pathway in Aged Skeletal Muscle
2.6. Lifelong Aerobic Exercise Activated Autophagy and Inhibited Apoptosis in Aged Skeletal Muscle
2.7. Lifelong Aerobic Exercise Improved Mitochondrial Quality Control via Activating the AMPK/PGC-1α Signaling Pathway in Aged Skeletal Muscle
3. Discussion
4. Materials and Methods
4.1. Animals, Study Design, and Ethics
4.2. Aerobic Exercise Protocol
4.3. Histological Examination of Gastrocnemius Muscle
4.4. Transmission Electron Microscopic Examination
4.5. Mitochondrial Enzyme Activity
4.6. SOD Activity and MDA Content in Serum and Gastrocnemius Muscle
4.7. Western Blotting
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cruz-Jentoft, A.J.; Sayer, A.A. Sarcopenia. Lancet 2019, 393, 2636–2646. [Google Scholar] [CrossRef]
- Anker, S.D.; Morley, J.E.; von Haehling, S. Welcome to the ICD-10 code for sarcopenia. J. Cachexia Sarcopenia Muscle 2016, 7, 512–514. [Google Scholar] [CrossRef]
- Dennison, E.M.; Sayer, A.A.; Cooper, C. Epidemiology of sarcopenia and insight into possible therapeutic targets. Nat. Rev. Rheumatol. 2017, 13, 340–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, H.N.; Kim, Y.; Erlich, A.T.; Zarrin-Khat, D.; Hood, D.A. Autophagy and mitophagy flux in young and aged skeletal muscle following chronic contractile activity. J. Physiol. 2018, 596, 3567–3584. [Google Scholar] [CrossRef]
- Liang, J.; Zeng, Z.; Zhang, Y.; Chen, N. Regulatory role of exercise-induced autophagy for sarcopenia. Exp. Gerontol. 2020, 130, 110789. [Google Scholar] [CrossRef] [PubMed]
- Del Campo, A.; Jaimovich, E.; Tevy, M.F. Mitochondria in the aging muscles of flies and mice: New perspectives for old characters. Oxidative Med. Cell. Longev. 2016, 2016, 9057593. [Google Scholar] [CrossRef] [Green Version]
- Jang, J.Y.; Blum, A.; Liu, J.; Finkel, T. The role of mitochondria in aging. J. Clin. Investig. 2018, 128, 3662–3670. [Google Scholar] [CrossRef] [Green Version]
- Romanello, V.; Sandri, M. Mitochondrial quality control and muscle mass maintenance. Front. Physiol. 2015, 6, 422. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, H.; Edgett, B.A.; Gurd, B.J. Coordination of mitochondrial biogenesis by PGC-1α in human skeletal muscle: A re-evaluation. Metabolism 2018, 79, 42–51. [Google Scholar] [CrossRef] [PubMed]
- De Rezende, L.F.; Rey-López, J.P.; Matsudo, V.K.; do Carmo Luiz, O. Sedentary behavior and health outcomes among older adults: A systematic review. BMC Public Health 2014, 14, 333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luan, X.; Tian, X.; Zhang, H.; Huang, R.; Li, N.; Chen, P.; Wang, R. Exercise as a prescription for patients with various diseases. J. Sport Health Sci. 2019, 8, 422–441. [Google Scholar] [CrossRef] [PubMed]
- Booth, F.W.; Roberts, C.K.; Laye, M.J. Lack of exercise is a major cause of chronic diseases. Compr. Physiol. 2012, 2, 1143–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landi, F.; Marzetti, E.; Martone, A.M.; Bernabei, R.; Onder, G. Exercise as a remedy for sarcopenia. Curr. Opin. Clin. Nutr. Metab. Care 2014, 17, 25–31. [Google Scholar] [CrossRef]
- Lira, V.A.; Okutsu, M.; Zhang, M.; Greene, N.P.; Laker, R.C.; Breen, D.S.; Hoehn, K.L.; Yan, Z. Autophagy is required for exercise training-induced skeletal muscle adaptation and improvement of physical performance. FASEB J. 2013, 27, 4184–4193. [Google Scholar] [CrossRef] [Green Version]
- Dethlefsen, M.M.; Halling, J.F.; Møller, H.D.; Plomgaard, P.; Regenberg, B.; Ringholm, S.; Pilegaard, H. Regulation of apoptosis and autophagy in mouse and human skeletal muscle with aging and lifelong exercise training. Exp. Gerontol. 2018, 111, 141–153. [Google Scholar] [CrossRef]
- Starnes, J.W.; Parry, T.L.; O’Neal, S.K.; Bain, J.R.; Muehlbauer, M.J.; Honcoop, A.; Ilaiwy, A.; Christopher, P.M.; Patterson, C.; Willis, M.S. Exercise-induced alterations in skeletal muscle, heart, liver, and serum metabolome identified by non-targeted metabolomics analysis. Metabolites 2017, 7, 40. [Google Scholar] [CrossRef] [Green Version]
- Vainshtein, A.; Tryon, L.D.; Pauly, M.; Hood, D.A. Role of PGC-1α during acute exercise-induced autophagy and mitophagy in skeletal muscle. Am. J. Physiol. Cell Physiol. 2015, 308, C710–C719. [Google Scholar] [CrossRef] [Green Version]
- Liao, Z.Y.; Chen, J.L.; Xiao, M.H.; Sun, Y.; Zhao, Y.X.; Pu, D.; Lv, A.K.; Wang, M.L.; Zhou, J.; Zhu, S.Y.; et al. The effect of exercise, resveratrol or their combination on Sarcopenia in aged rats via regulation of AMPK/Sirt1 pathway. Exp. Gerontol. 2017, 98, 177–183. [Google Scholar] [CrossRef]
- Sayed, R.K.; de Leonardis, E.C.; Guerrero-Martínez, J.A.; Rahim, I.; Mokhtar, D.M.; Saleh, A.M.; Abdalla, K.E.; Pozo, M.J.; Escames, G.; López, L.C.; et al. Identification of morphological markers of sarcopenia at early stage of aging in skeletal muscle of mice. Exp. Gerontol. 2016, 83, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Rom, O.; Reznick, A.Z. The role of E3 ubiquitin-ligases MuRF-1 and MAFbx in loss of skeletal muscle mass. Free Radic. Biol. Med. 2016, 98, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, S.R.; Mizushima, N. Monitoring and measuring autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef] [PubMed]
- Bjørkøy, G.; Lamark, T.; Pankiv, S.; Øvervatn, A.; Brech, A.; Johansen, T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009, 452, 181–197. [Google Scholar] [CrossRef]
- Hardman, S.E.; Hall, D.E.; Cabrera, A.J.; Hancock, C.R.; Thomson, D.M. The effects of age and muscle contraction on AMPK activity and heterotrimer composition. Exp. Gerontol. 2014, 55, 120–128. [Google Scholar] [CrossRef] [Green Version]
- Walburg, H.E.; Cosgrove, G.E. Ageing in irradiated and unirradiated germfree ICR mice. Exp. Gerontol. 1967, 2, 143–158. [Google Scholar] [CrossRef]
- Baumgartner, R.N.; Koehler, K.M.; Gallagher, D.; Romero, L.; Heymsfield, S.B.; Ross, R.R.; Garry, P.J.; Lindeman, R.D. Epidemiology of sarcopenia among the elderly in New Mexico. Am. J. Epidemiol. 1998, 147, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Jiao, J.; Demontis, F. Skeletal muscle autophagy and its role in sarcopenia and organismal aging. Curr. Opin. Pharmacol. 2017, 34, 1–6. [Google Scholar] [CrossRef]
- Sandri, M. Protein breakdown in muscle wasting: Role of autophagy-lysosome and ubiquitin-proteasome. Int. J. Biochem. Cell Biol. 2013, 45, 2121–2129. [Google Scholar] [CrossRef] [Green Version]
- Sandri, M. Signaling in muscle atrophy and hypertrophy. Physiology. 2008, 23, 160–170. [Google Scholar] [CrossRef] [Green Version]
- Francaux, M.; Demeulder, B.; Naslain, D.; Fortin, R.; Lutz, O.; Caty, G.; Deldicque, L. Aging reduces the activation of the mTORC1 pathway after resistance exercise and protein intake in human skeletal muscle: Potential role of REDD1 and impaired anabolic sensitivity. Nutrients 2016, 8, 47. [Google Scholar] [CrossRef] [Green Version]
- Markofski, M.M.; Dickinson, J.M.; Drummond, M.J.; Fry, C.S.; Fujita, S.; Gundermann, D.M.; Glynn, E.L.; Jennings, K.; Paddon-Jones, D.; Reidy, P.T.; et al. Effect of age on basal muscle protein synthesis and mTORC1 signaling in a large cohort of young and older men and women. Exp. Gerontol. 2015, 65, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russ, D.W.; Boyd, I.M.; McCoy, K.M.; McCorkle, K.W. Muscle-specificity of age-related changes in markers of autophagy and sphingolipid metabolism. Biogerontology 2015, 16, 747–759. [Google Scholar] [CrossRef]
- Potes, Y.; de Luxán-Delgado, B.; Rodriguez-González, S.; Guimarães, M.R.M.; Solano, J.J.; Fernández-Fernández, M.; Bermúdez, M.; Boga, J.A.; Vega-Naredo, I.; Coto-Montes, A. Overweight in elderly people induces impaired autophagy in skeletal muscle. Free Radic. Biol. Med. 2017, 110, 31–41. [Google Scholar] [CrossRef]
- Vainshtein, A.; Hood, D.A. The regulation of autophagy during exercise in skeletal muscle. J. Physiol. 2016, 120, 664–673. [Google Scholar] [CrossRef] [Green Version]
- Lenhare, L.; Crisol, B.M.; Silva, V.R.R.; Katashima, C.K.; Cordeiro, A.V.; Pereira, K.D.; Luchessi, A.D.; da Silva, A.S.R.; Cintra, D.E.; Moura, L.P.; et al. Physical exercise increases Sestrin 2 protein levels and induces autophagy in the skeletal muscle of old mice. Exp. Gerontol. 2017, 97, 17–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.A.; Kim, Y.S.; Oh, S.L.; Kim, H.J.; Song, W. Autophagic response to exercise training in skeletal muscle with age. J. Physiol. Biochem. 2013, 69, 697–705. [Google Scholar] [CrossRef]
- Song, W.; Kwak, H.B.; Lawler, J.M. Exercise training attenuates age-induced changes in apoptotic signaling in rat skeletal muscle. Antioxid. Redox Signal. 2006, 8, 517–528. [Google Scholar] [CrossRef]
- Ziaaldini, M.M.; Koltai, E.; Csende, Z.; Goto, S.; Boldogh, I.; Taylor, A.W.; Radak, Z. Exercise training increases anabolic and attenuates catabolic and apoptotic processes in aged skeletal muscle of male rats. Exp. Gerontol. 2015, 67, 9–14. [Google Scholar] [CrossRef] [Green Version]
- Dupont-Versteegden, E.E. Apoptosis in skeletal muscle and its relevance to atrophy. World J. Gastroenterol. 2006, 12, 7463–7466. [Google Scholar] [CrossRef]
- Cheema, N.; Herbst, A.; McKenzie, D.; Aiken, J.M. Apoptosis and necrosis mediate skeletal muscle fiber loss in age-induced mitochondrial enzymatic abnormalities. Aging Cell 2015, 14, 1085–1093. [Google Scholar] [CrossRef]
- Marzetti, E.; Calvani, R.; Bernabei, R.; Leeuwenburgh, C. Apoptosis in skeletal myocytes: A potential target for interventions against sarcopenia and physical frailty—A mini-review. Gerontology 2012, 58, 99–106. [Google Scholar] [CrossRef]
- Chen, J.; Wong, H.S.; Leong, P.K.; Leung, H.Y.; Chan, W.M.; Ko, K.M. Ursolic acid induces mitochondrial biogenesis through the activation of AMPK and PGC-1 in C2C12 myotubes: A possible mechanism underlying its beneficial effect on exercise endurance. Food Funct. 2017, 8, 2425–2436. [Google Scholar] [CrossRef] [PubMed]
- Lantier, L.; Fentz, J.; Mounier, R.; Leclerc, J.; Treebak, J.T.; Pehmøller, C.; Sanz, N.; Sakakibara, I.; Saint-Amand, E.; Rimbaud, S.; et al. AMPK controls exercise endurance, mitochondrial oxidative capacity, and skeletal muscle integrity. FASEB J. 2014, 28, 3211–3224. [Google Scholar] [CrossRef] [PubMed]
- Hawley, J.A.; Lundby, C.; Cotter, J.D.; Burke, L.M. Maximizing cellular adaptation to endurance exercise in skeletal muscle. Cell Metab. 2018, 27, 962–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, K.; Vandecasteele, G.; Carli, C.; Romagnoli, A.; Szabadkai, G.; Rizzuto, R. Regulation of Ca2+ signalling and Ca2+-mediated cell death by the transcriptional coactivator PGC-1alpha. Cell Death Differ. 2006, 13, 586–596. [Google Scholar] [CrossRef]
- Adhihetty, P.J.; Uguccioni, G.; Leick, L.; Hidalgo, J.; Pilegaard, H.; Hood, D.A. The role of PGC-1alpha on mitochondrial function and apoptotic susceptibility in muscle. Am. J. Physiol. Cell Physiol. 2009, 297, C217–C225. [Google Scholar] [CrossRef] [Green Version]
- Alway, S.E.; Mohamed, J.S.; Myers, M.J. Mitochondria initiate and regulate sarcopenia. Exerc. Sport Sci. Rev. 2017, 45, 58–69. [Google Scholar] [CrossRef]
- Chen, C.C.W.; Erlich, A.T.; Crilly, M.J.; Hood, D.A. Parkin is required for exercise-induced mitophagy in muscle: Impact of aging. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E404–E415. [Google Scholar] [CrossRef]
- Coen, P.M.; Musci, R.V.; Hinkley, J.M.; Miller, B.F. Mitochondria as a target for mitigating sarcopenia. Front. Physiol. 2018, 9, 1883. [Google Scholar] [CrossRef] [Green Version]
- Sebastián, D.; Sorianello, E.; Segalés, J.; Irazoki, A.; Ruiz-Bonilla, V.; Sala, D.; Planet, E.; Berenguer-Llergo, A.; Muñoz, J.P.; Sánchez-Feutrie, M.; et al. Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J. 2016, 35, 1677–1693. [Google Scholar] [CrossRef]
- Otera, H.; Mihara, K. Molecular mechanisms and physiologic functions of mitochondrial dynamics. J. Biochem. 2011, 149, 241–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedford, T.G.; Tipton, C.M.; Wilson, N.C.; Oppliger, R.A.; Gisolfi, C.V. Maximum oxygen consumption of rats and its changes with various experimental procedures. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1979, 47, 1278–1283. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Zhang, Y.; Zhang, M.; Chang, J.; Zeng, Z.; Kou, X.; Chen, N. Exercise attenuates brain aging by rescuing down-regulated Wnt/β-Catenin signaling in aged rats. Front. Aging Neurosci. 2020, 12, 105. [Google Scholar] [CrossRef] [Green Version]
- Kou, X.; Li, J.; Liu, X.; Yang, X.; Fan, J.; Chen, N. Ampelopsin attenuates the atrophy of skeletal muscle from d-gal-induced aging rats through activating AMPK/SIRT1/PGC-1α signaling cascade. Biomed. Pharmacother. 2017, 90, 311–320. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Body Weight (g) | Gastrocnemius Muscle (g) | GMW/BW Ratio (×100) |
|---|---|---|---|
| YC | 46.733 ± 2.375 | 0.433 ± 0.0296 | 0.9261 ± 0.064 |
| OC | 57.851 ± 4.324 | 0.451 ± 0.0384 | 0.7801 ± 0.050 *** |
| OE | 51.709 ± 3.284 | 0.434 ± 0.0377 | 0.8395 ± 0.051 ## |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, J.; Zhang, H.; Zeng, Z.; Wu, L.; Zhang, Y.; Guo, Y.; Lv, J.; Wang, C.; Fan, J.; Chen, N. Lifelong Aerobic Exercise Alleviates Sarcopenia by Activating Autophagy and Inhibiting Protein Degradation via the AMPK/PGC-1α Signaling Pathway. Metabolites 2021, 11, 323. https://doi.org/10.3390/metabo11050323
Liang J, Zhang H, Zeng Z, Wu L, Zhang Y, Guo Y, Lv J, Wang C, Fan J, Chen N. Lifelong Aerobic Exercise Alleviates Sarcopenia by Activating Autophagy and Inhibiting Protein Degradation via the AMPK/PGC-1α Signaling Pathway. Metabolites. 2021; 11(5):323. https://doi.org/10.3390/metabo11050323
Chicago/Turabian StyleLiang, Jiling, Hu Zhang, Zhengzhong Zeng, Liangwen Wu, Ying Zhang, Yanju Guo, Jun Lv, Cenyi Wang, Jingjing Fan, and Ning Chen. 2021. "Lifelong Aerobic Exercise Alleviates Sarcopenia by Activating Autophagy and Inhibiting Protein Degradation via the AMPK/PGC-1α Signaling Pathway" Metabolites 11, no. 5: 323. https://doi.org/10.3390/metabo11050323