1. Introduction
Based on the U.S. Centers for Disease Control and Prevention (CDC) 2009 report, 12,357 women were diagnosed with cervical cancer, and 3909 women died from cervical cancer in the United States. This huge number of patients with cervical cancer has been a critical issue and is responsible for 10%–15% of cancer-related deaths in females globally [
1]. Cervical cancer is the second most common malignant tumor in women worldwide. Among these tumors, approximately 80% are squamous cell carcinomas (SCCs), and 5%–20% are adenocarcinomas (AdCAs) [
2,
3]. Cervical SCCs are developed from a premalignant disease known as cervical intraepithelial neoplasia (CIN) Graded 1–3 with increasing atypical features. The five-year overall survival for cervical cancer is only 66%.
In addition, increasing evidence suggests that infection with high-risk subtypes of human papillomavirus (HPV) (e.g., HPV-16 and HPV-18) is the most common cause and is the primary initiator of premalignant lesions [
4]. However, only a small proportion of women infected with oncogenic HPV subtypes develop cervical cancer, which suggests that HPV infection alone is insufficient to cause cancer, and there is a possibility of other host factors linked to the development of invasive cervical cancer [
5], like genetic variation, including polymorphisms, insertions or deletions in the host genome [
6,
7]. Increasing evidence demonstrates that there was a consistent relationship between certain genetic variants (such as the tumor protein 53 (TP53) Arg72Pro polymorphism) and cervical cancer, most likely modulated by the presence of high-risk HPV during progression from squamous intraepithelial lesions (SIL) to cervical cancer.
Moreover, Mexican American women in Texas have among the highest rates of cervical cancer incidence and mortality in the country [
8] with twice the frequency as compared to their non-Mexican American counterparts. The annual death rate from cervical cancer for Mexican Americans is 24.2 out of 100,000 [
9]. However, there is a lack of studies on somatic mutation identification for cervical cancer in this population. Only four studies have reported on genetic basis of cervical cancer in Mexican American women based on a PubMed search (19 November 2015). The number of the Mexican American population was estimated at 50.5 million in the 2010 census, making Mexican American the largest minority group in the U.S., as well as a rapidly growing segment of the U.S. population.
Recent advances in genome studies have led to the discovery of one important type of variation that can be assessed with recent technology: copy number alterations (CNAs), usually for a cancer study, somatic copy number changes and/or copy number variations (CNVs), usually for a non-cancer study, or germline copy number changes, such as CNVs identified for neuropsychiatric disorders in our recent study [
10]. These CNAs or CNVs are by definition chromosomal regions with sizes of 1 kb to several Mb, which vary across individuals with regard to the number of copies of a chromosomal segment. CNVs refer to structural variations of the DNA that include insertions, deletions and duplications. Studies have found that CNVs cover as much as 14% of the human genome [
11], and there is a much higher de novo rate as many as 10–1000-fold in CNVs as compared to single nucleotide polymorphisms (SNPs) [
11,
12]. Furthermore, CNVs have been shown to account for more genomic differences between individuals than SNPs [
13,
14]. Therefore, CNVs may contribute a sizeable amount of disease phenotypic variation in each individual of a population [
15].
Analyses integrating mutation information with data on rearrangements and CNAs have revealed a higher-order organization of the seemingly random genetic events that lead to cancer [
16]. Interestingly, genes in regions subject to copy number changes appear to be organized along functional ontological terms related to cancer [
16]. Studies have also implicated a number of somatic mutations, including
TP53, phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (
PIK3CA), phosphatase and tensin homolog (
PTEN), serine/threonine kinase 11 (STK11) and V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (
KRAS) [
17,
18], and several CNAs in the pathogenesis of cervical carcinomas [
19,
20] in non-Mexican Americans.
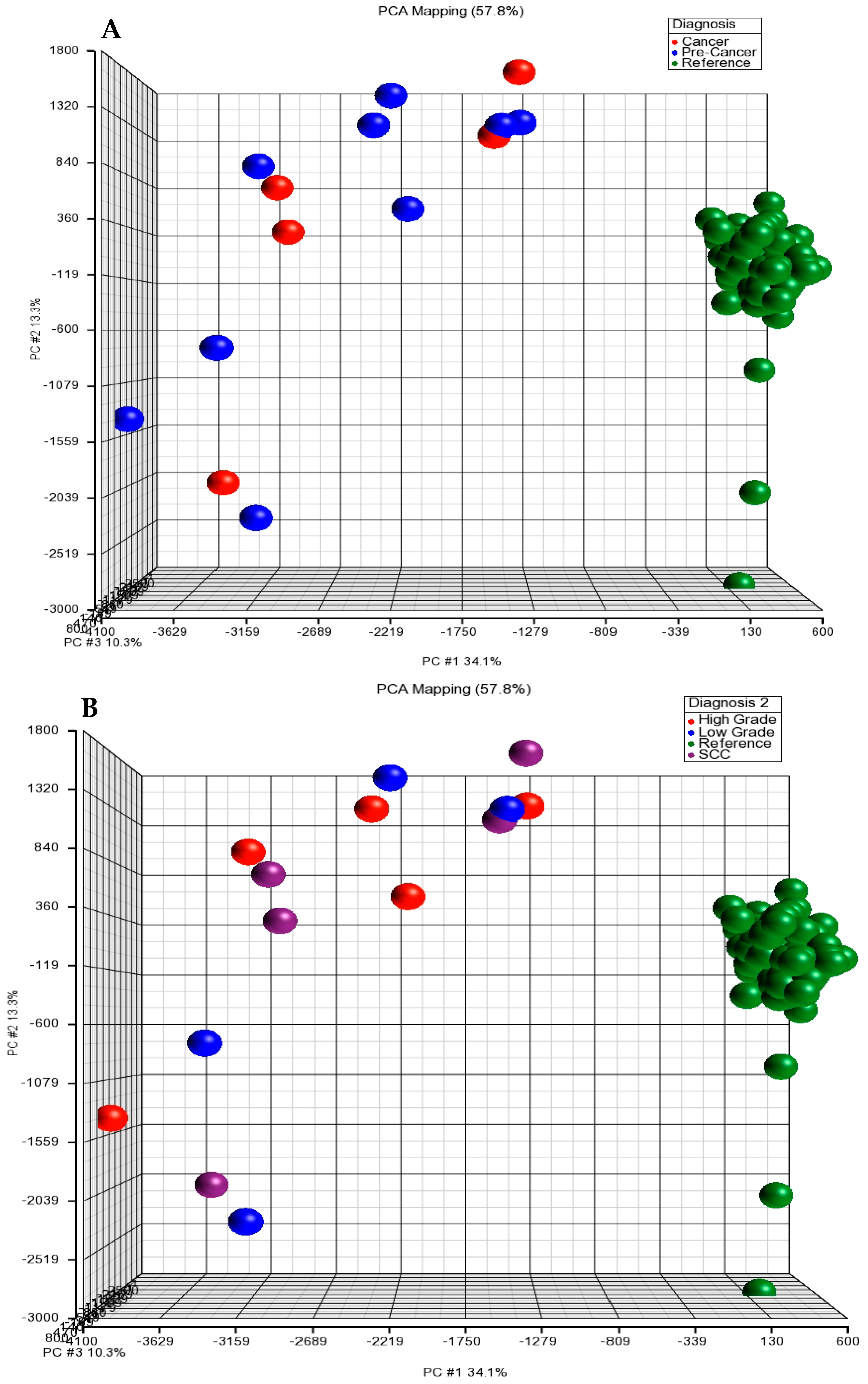
In this study, we carried out a genome-wide survey of potential somatic CNAs, including amplifications and deletions, in apparently normal tissues (n = 2), low grade dysplasia (n = 4), high grade dysplasia (n = 5), invasive carcinoma (n = 5) and blood samples (n = 125, serving as a reference group) of female subjects from the HapMap data. We are aware of the limited number of cases and the lack of a control group. Thus, in the future, a large study with a control sample and more cases as a methodological alternative is needed. We genotyped 2.5 million markers and analyzed somatic CNAs in a total of 14 tissues using the Illumina HumanOmni2.5-8 BeadChip Kit at tissue-level resolution. We mapped genomic changes: (1) between peripheral blood samples of the reference subjects and cervical tissues from the cases (cervical dysplasia and invasive carcinoma); because only two normal cervical tissue samples had insufficient statistical power, we excluded these two samples from the further analysis; (2) we also analyzed genomic changes among four diagnostic groups (normal, low, high grade and invasive carcinoma). We expect that this study: (i) will provide an estimate of the prevalence of somatic CNAs by identifying specific patterns, genes and/or biological pathways associated with different stages of cervical dysplasia in Mexican Americans; (ii) will investigate the genomic context of these somatic CNAs; and (iii) will evaluate whether the burden of somatic mutations predicts tumor progression.
4. Discussion
It has been demonstrated that somatic structural alterations (e.g., amplifications or deletions) of human chromosomes represent a common class of mutations, which may cause gene disruption (e.g., deletion or rearrangement), gene activation (e.g., CNAs, gain or amplification) or the formation of novel oncogenic gene products (gene fusions). Many of these events actively drive carcinogenesis [
35,
36]. In our initial cervical cancer cohort, some CNA patterns identified were previously found to be correlated with cervical cancer.
We screened whole genomes with 2.5 million markers of an array to discover any recurrent copy number alterations in cases (pre-cancer and SCC). We identified a total of 98 CNA regions >100 kb in the case group, including low grade dysplasia, high grade dysplasia and SCC. These CNAs occurred in two or more cases, including two large deletions (1591 kb and 1123 kb) (
Table 3). Of the top 20 CNA regions >100 kb deletions in cases only, six CNAs occurred on 16p13 that have been reported in cervical cancer [
37], and mutation analysis of the
AXIN1 gene located at 16p13 was reported to be involved in the Wnt pathway in cervical carcinomas [
38]. The CNA containing the SET and MYND domain-containing protein 4 (
SMYD4) gene on 17p13.3 was also reported previously in this cancer [
37]. The
SMYD4 gene is demonstrated to be a potential tumor suppressor and plays a critical role in breast carcinogenesis, at least partly through inhibiting the expression of platelet-derived growth factor receptor-alpha (
PDGFRA), and this gene could be a novel target for improving the treatment of breast cancer [
39].
A number of previous studies demonstrate that numerical chromosomal aberrations were found to progress to invasive cancer from precursor lesions [
25,
37]; however, we did not observe this feature in our current study sample. This might be due to the small sample size or any other unknown or not yet identified factors, such as ethnicity, since most previous study populations are non-Hispanic populations.
Among two amplifications identified in CIN-III and SCC, one recurrent CNA region with a 220-kb amplification on 21q11.2 was observed in CIN-III and co-occurred with previous findings, where the amplification had been identified in breast cancer subjects with tamoxifen resistance [
40].
The recurrent CNA regions we identified contain a number of tumor suppressor genes, oncogenes and cancer-related genes. A CNA that contains the interferon-induced transmembrane protein 1 (
IFITM1) gene at 11p15.5 was identified in eight cases. This gene has been reported to be involved in cervical carcinogenesis [
41]. A total of nine cases with the CNA containing the fucosyltransferase 3 (
FUT3) gene at 19p13.3 were observed in the current study, and this gene is associated with breast cancer [
42]. A CNA region that includes the naked cuticle 2 (
NKD2) gene at 5p15.35 was detected in six cases. This gene was found to suppress breast cancer proliferation by inhibiting Wnt signaling [
43]. The ovarian cancer-associated gene 2 protein (OVCA2) at 17p13.3 was repeatedly reported to be associated with ovarian cancer, and we observed five cases carrying the CNA at this gene region. A cluster of three tumor suppressor genes, cadherin 1 (
CDH1), death-associated protein kinase (
DAPK) and hypermethylated in cancer 1 (
HIC1), displayed a significantly increased frequency of promoter methylation with progressively more severe cervical neoplasia. In addition, the Hes family BHLH Transcription Factor 5 (
HES5) gene was reported to be associated with cervical carcinoma cells using immunocytochemistry, Western blot and methyl thiazolyl tetrazolium assays [
44], and the CNAs on 1p36.33-1p36.32 containing the
HES5 gene were also identified in the current study. Our newly-identified CNA containing this gene on 7p22.2 was also observed in the recurrent pre-cancer and cancer subjects (
Table 3).
Moreover, we also examined deletion burden observed to be similar to those for other cancers and found more deletions identified than amplification in most of the cancer studies, which supports our findings. A recent study using the TCGA data identified nine regions of deletion that were unique to estrogen receptor positive (ER+) post menopause tumors in patients with breast cancer [
45], including deletion in 7p22.3, where our newly-identified deletion in cases only was located, and it contains a known tumor suppressor gene.
To analyze the possible effect of genomic alterations, to further capture cancer-causing gene information and to see whether any GO categories are overrepresented among CNA regions, we searched the KEGG pathway database and GO categories and identified a number of pathways and functions where CNAs occur in the SCC group, but not in the reference group (
Table 6 and
Table 7). Furthermore, our results were partly in agreement with previous reports about cervical cancer. For example, using the functional enrichment of KEGG pathway analysis, we discovered the insulin signaling pathway in the case group, and it was evidenced that HPV 16 E6 oncoprotein interferes with the insulin signaling pathway by binding to tuberin [
46]. As is already known, high-risk HPV infection is a causal agent for cervical cancer. In addition, two pathways of interest are endometrial cancer and the estrogen signaling pathway observed in the current study. The NF-κB signaling pathway was also identified in our cancer group (SCC), but not in the reference group, which supports a previous study where bisphenol A (BPA) stimulated the cervical cancer migration via IKK-β/NF-κB signals [
47]. The estrogen signaling pathway was also shown in our KEGG analysis. A previous study was performed to evaluate the potential of miRNAs as novel markers for the post-therapeutic monitoring of cervical SCC patients. A regulatory network of differentially-expressed serum miRNAs was identified, and a number of target genes was predicted in the estrogen-mediated signal pathways [
48]. Ours and others’ findings support that cervical cancer is a hormone-associated gynecological disease. For example, HPV infection has been associated with the deregulation of the PI3K-Akt-mTOR pathway in invasive cervical carcinomas [
49]. The PI3K-Akt signaling pathway was also found in our current study, and 14 genes in this pathway were found in two or more SCC cases (
Table 6).
Moreover, signal transducer activity with an enrichment score of 4.2 and a
p value of 0.01 containing 11 genes was also identified in a previous report on cervical cancer [
50] using GO analysis. Signal transduction was identified in our current study in cervical cancer (SCC), but not in the reference group; a recent study also supports that a signal transduction cascade and mitogen-activated protein kinase kinase kinase 3 (MEKK3) serve as key integration points and are important factors in regulating cellular responses to environmental stress. This signal transduction and MEKK3 not only link diverse extracellular stimuli to subsequent signaling molecules, but they also amplify the initiating signals to ultimately activate effector molecules and induce cell proliferation, differentiation and survival of cervical cancer [
51].
These pathways and functions were identified as overlapping biological themes, and these data may provide useful information on the molecular mechanisms for cervical cancer, its prognosis and treatment responses.
There are a number of novelties in this study: (1) this is the first report of CNAs in the relatively ethnically homogeneous group of Mexican Americans using high density mapping (2.5 million in number); previous studies using less density markers may result in an underestimation of the genetic changes that take place in cervical cancer; (2) newly-identified CNAs in cases together with results in the in silico analysis using KEGG and GO function provide insight into how multiple CNAs might contribute to cervical cancer development. We also are aware of some limitations in our study. (1) Our small sample size (14 cases and 125 references) is a major limitation for this type of study. Due to a small number of samples, the nature of genome-wide alteration of copy number may not be fully explained in pre-cancer and SCC. Future confirmation studies using an independent sample will provide an opportunity to more accurately dissect the genetic complexity of somatic CNAs for cervical cancer. (2) There might be a bias of the CNA identified in cervical cancer for the Mexican Americans, since we used the 1000 Genome admixed populations, not Mexican Americans; thus, we currently are recruiting more subjects with cervical cancer from the same population and plan to validate the findings in more samples. (3) Other biomarkers, such as RNA-Seq or DNA methylation profile and sequencing data using next generation sequencing technologies (such as target gene sequencing in these CNA regions), will provide an opportunity for in-depth molecular profiling of the fundamental biological processes of cervical cancer.
We expect that with future validation and confirmation, newly-discovered CNAs can be used in cancer classification, diagnosis, prognosis and treatment responses.





{kind=link}
{kind=link}