Systemic Mastocytosis and Essential Thrombocythemia: Case Report and Literature Overview

 ,
, 
Abstract
:1. Introduction
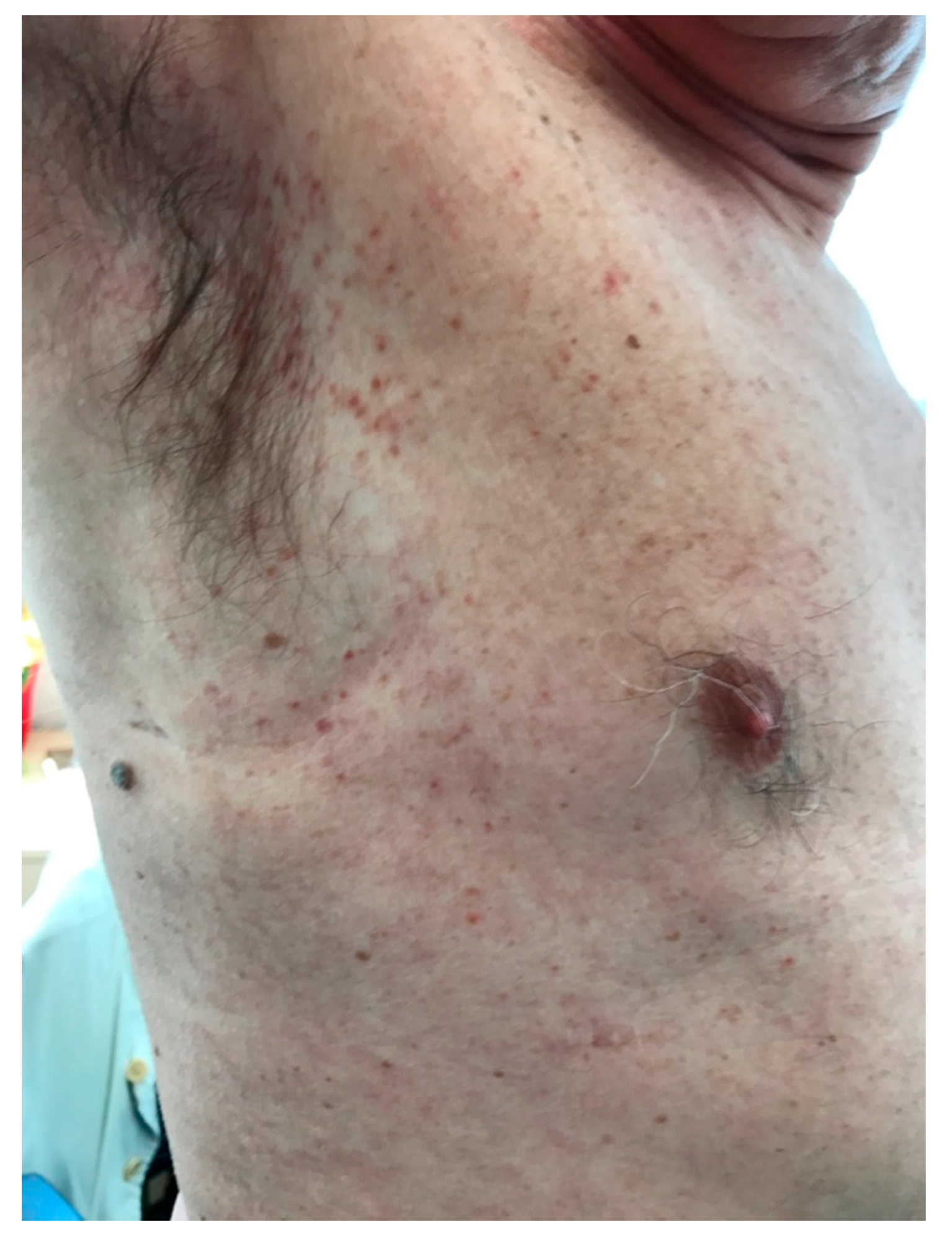
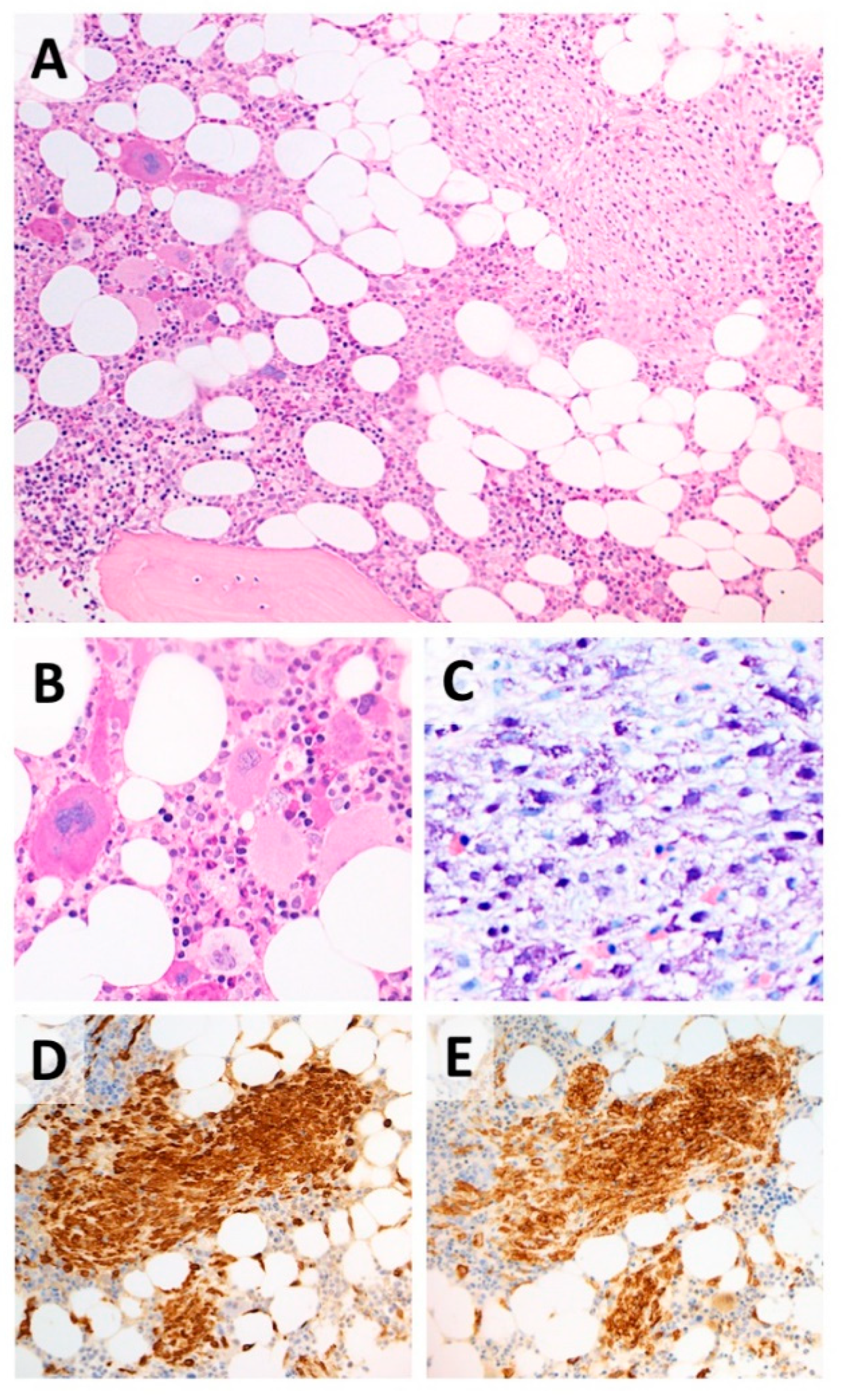
2. Case Report
3. Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Harris, N.L.; Stein, H.; Camp, E.; Swerdlow, S.H.; Jaffe, E.S.; Thiele, J.; Pileri, S.A.; Vardiman, J.W. Mastocytosis. In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; World Health Organization: Lyon, France, 2008; pp. 54–63. [Google Scholar]
- Van Doormaal, J.J.; Arends, S.; Brunekreeft, K.L.; van der Wal, V.B.; Sietsma, J.; van Voorst Vader, P.C.; Oude Elberink, J.N.; Kluin-Nelemans, J.C.; van der Veer, E.; de Monchy, J.G. Prevalence of indolent systemic mastocytosis in a Dutch region. J. Allergy Clin. Immunol. 2013, 131, 1429–1431. [Google Scholar] [CrossRef] [PubMed]
- Coltoff, A.; Mascarenhas, J. Relevant updates in systemic mastocytosis. Leuk. Res. 2019, 81, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A.; Lim, K.H.; Lasho, T.L.; Finke, C.; McClure, R.F.; Li, C.Y.; Tefferi, A. Prognostically relevant breackdown of 123 patients with systemic mastocytosis associated with other myeloid malignancies. Blood 2017, 114, 3769–3772. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Akin, C.; Gleixner, K.V.; Sperr, W.R.; Reiter, A.; Arock, M.; Triggiani, M. Multidisciplinary Challenges in Mastocytosis and How to Address with Personalized Medicine Approaches. Int. J. Mol. Sci. 2019, 20, 2976. [Google Scholar] [CrossRef] [PubMed]
- Jawhar, M.; Schwaab, J.; Horny, H.; Sotlar, K.; Naumann, N.; Fabarius, A.; Valent, P.; Cross, N.C.; Hofmann, W.; Metzgeroth, G.; et al. Impact of Centralized Evaluation of Bone Marrow Histology in Systemic Mastocytosis. Eur. J. Clin. Investig. 2016, 46, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Le Tourneau, A.; Gaulard, P.; D’Agay, M.F.; Vainchencker, W.; Cadiou, M.; Devidas, A.; Haioun, C.; Clauvel, J.P.; Audouin, J.; Diebold, J. Primary thrombocythemia associated with systemic mastocytosis: A report of five cases. Br. J. Haematol. 1991, 79, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Krsnik, I.; Ricard, M.P.; Escribano, L.M.; Calero, M.A.; Perez, R.G.; Garcia, S.J.; Perera, F.; Merino, J.L. Systemic mastocytosis and primary thrombocythemia. Br. J. Haematol. 1991, 77, 437. [Google Scholar] [CrossRef] [PubMed]
- Dobrea, C.; Ciochinaru, M.; Găman, A.; Dănăilă, E.; Coriu, D. Systemic mastocytosis associated with essential thrombocythemia. Rom. J. Morphol. Embryol. Rev. Roum. Morphol. Embryol. 2012, 53, 197–202. [Google Scholar]
- Pieri, L.; Bonadonna, P.; Elena, C.; Papayannidis, C.; Grifoni, F.I.; Rondoni, M.; Girlanda, S.; Mauro, M.; Magliacane, D.; Elli, E.M.; et al. Clinical presentation and management practice of systemic mastocytosis. A survey on 460 Italian patients. Am. J. Hematol. 2016, 91, 692–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbui, T.; Thiele, J.; Passamonti, F.; Rumi, E.; Boveri, E.; Ruggeri, M.; Rodeghiero, F.; D’Amore, E.S.; Randi, M.L.; Bertozzi, I.; et al. Survival and Disease Progression in Essential Thrombocythemia Are Significantly Influenced by Accurate Morphologic Diagnosis: An International Study. J. Clin. Oncol. 2011, 29, 3179–3184. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, D.; Carreau, N.; Kremyanskaya, M.; Mascarenhas, J. Systemic Mastocytosis: Clinical Update and Future Directions. Clin. Lymphoma Myeloma Leuk. 2015, 15, 728–738. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Pardanani, A. Myeloproliferative neoplasms: A contemporary review. JAMA Oncol. 2015, 1, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Greene, L.W.; Asadipooya, K.; Corradi, P.F.; Akin, C. Endocrine manifestations of systemic mastocytosis in bone. Rev. Endocr. Metab. Disord. 2016, 17, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Rossini, M.; Zanotti, R.; Viapiana, O.; Tripi, G.; Idolazzi, L.; Biondan, M.; Orsolini, G.; Bonadonna, P.; Adami, S.; Gatti, D. Zoledronic Acid in Osteoporosis Secondary to Mastocytosis. Am. J. Med. 2014, 127, 1127.e1–1127.e4. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Sex/Age | Platelets (× 109/L) | Clinical Findings at Presentation | Molecular Biology | Treatment | Outcome | Ref. |
|---|---|---|---|---|---|---|
| F/55 | 900 | Bone lesions | - | None | Stable | [8] |
| M/64 | 900 | Superficial adenopathy | - | HU | No SM (3 years) | |
| F/22 | 1000 | Maculo-papular skin lesions | - | HU | No SM (3 years) | |
| M/60 | 1000 | Bone lesions | - | HU | Normal Plts. Stable (6 years) | |
| M/68 | 940 | Mild SM | - | HU | Normal Plts. Stable (3 years) | |
| F/65 | 1373 | Maculo-papular skin lesions Bone lesions | - | HU | Normal Plts. | [9] |
| F/31 | 820 | Mild spleen enlargement | JAK2 V617F cKIT D816V | IFN-a | Normal Plts. Stable BM (3 years) | [10] |
| M/62 | 640 | Maculo-papular skin lesions Bone lesions Mild spleen enlargement | JAK2 V617F cKIT D816V | HU zoledronic acid | Normal Plts. Stable (3 years) | Present case |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cancian, M.; Cosi, E.; Pizzi, M.; Giannini, S.; Bertozzi, I.; Fabris, F.; Randi, M.L. Systemic Mastocytosis and Essential Thrombocythemia: Case Report and Literature Overview. Medicina 2019, 55, 528. https://doi.org/10.3390/medicina55090528
Cancian M, Cosi E, Pizzi M, Giannini S, Bertozzi I, Fabris F, Randi ML. Systemic Mastocytosis and Essential Thrombocythemia: Case Report and Literature Overview. Medicina. 2019; 55(9):528. https://doi.org/10.3390/medicina55090528
Chicago/Turabian StyleCancian, Mauro, Elisabetta Cosi, Marco Pizzi, Sandro Giannini, Irene Bertozzi, Fabrizio Fabris, and Maria Luigia Randi. 2019. "Systemic Mastocytosis and Essential Thrombocythemia: Case Report and Literature Overview" Medicina 55, no. 9: 528. https://doi.org/10.3390/medicina55090528