Bioactive Bis(indole) Alkaloids from a Spongosorites sp. Sponge
by

Jae Sung Park
1, 
Eunji Cho
2,
Ji-Yeon Hwang
1,
Sung Chul Park
1,
Beomkoo Chung
2,
Oh-Seok Kwon
1,
Chung J. Sim
3,
Dong-Chan Oh
1,
Ki-Bong Oh
2,* and
Jongheon Shin
1,* 1
Natural Products Research Institute, College of Pharmacy, Seoul National University, San 56-1, Sillim, Gwanak, Seoul 151-742, Korea
2
Department of Agricultural Biotechnology, College of Agriculture and Life Sciences, Seoul National University, Seoul 08826, Korea
3
Department of Biological Sciences, College of Life Science and Nano Technology, Hannam University, 461-6 Jeonmin, Yuseong, Daejeon 305-811, Korea
*
Authors to whom correspondence should be addressed.
Mar. Drugs 2021, 19(1), 3; https://doi.org/10.3390/md19010003
Submission received: 23 November 2020
/
Revised: 15 December 2020
/
Accepted: 18 December 2020
/
Published: 23 December 2020
(This article belongs to the Special Issue Bioactive Compounds from Marine Sponges 2020)
Abstract
:Six new bis(indole) alkaloids (1–6) along with eight known ones of the topsentin class were isolated from a Spongosorites sp. sponge of Korea. Based on the results of combined spectroscopic analyses, the structures of spongosoritins A–D (1–4) were determined to possess a 2-methoxy-1-imidazole-5-one core connecting the indole moieties, and these were linked by a linear urea bridge for spongocarbamides A (5) and B (6). The absolute configurations of spongosoritins were assigned by electronic circular dichroism (ECD) computation. The new compounds exhibited moderate inhibition against transpeptidase sortase A and weak inhibition against human pathogenic bacteria and A549 and K562 cancer cell lines.
1. Introduction
Alkaloids are widely recognized to play a central role in drug development [1,2,3]. These nitrogen-containing compounds have been in the limelight of medical chemistry and pharmacology research, with high structural complexity and various types of often potent bioactivity [1,2]. The level of structural complexity remarkably increases in those from the marine environment [4,5]. For example, among the most representative marine alkaloids are makaluvamines, manzamines, triptans, veranamines, coscinamides, and aplysinopsins from diverse colonial animals including sponges [2,6,7,8,9,10]. Bioactivity evaluations have shown that marine alkaloids and/or their synthetic analogs have various drug-related activities, including anti-tumoral, anti-malarial, anti-bacterial, and anti-viral, as well as enzymatic inhibition [2,4,5,11,12,13,14]. Overall, the importance of searching for novel bioactive marine alkaloids is constant.
Among the numerous sponge-derived alkaloids, topsentins are one of the most well-known classes; thus, extensive research has been conducted with respect to their structural determination, total synthesis, and bioactivity [15,16,17]. The diverse types of bioactivity reported include anti-inflammatory, anti-bacterial, anti-cancer, DNA-intercalating, and skin photoprotective, as well as the inhibition of Gram-positive bacterial enzyme sortase A, from our previous study [17,18,19,20,21]. These bis(indole) alkaloids, typically bearing an imidazole ring as the central core, were originally isolated from the sponge Spongosorites genitrix, which belongs to the family Halichondriidae [21,22]. Experiencing wide structural variations at both the imidazole moiety and the indole functionality, the succeeding analogs of topsentins include dragmacidins, hamacanthins, hytinadine A, nortopsentins, and rhopaladins from the genus Spongosorites and even a Rhopalaea sp. ascidian [23,24,25,26,27]. In a recent example, two new bis(indole) alkaloids, calcicamides A and B, bearing imidazole-cleaved 1,2-dicarbonyl moiety and exhibiting weak cytotoxicity against HeLa cells, were reported from the Irish sponge Spongosorites calcicola [28].
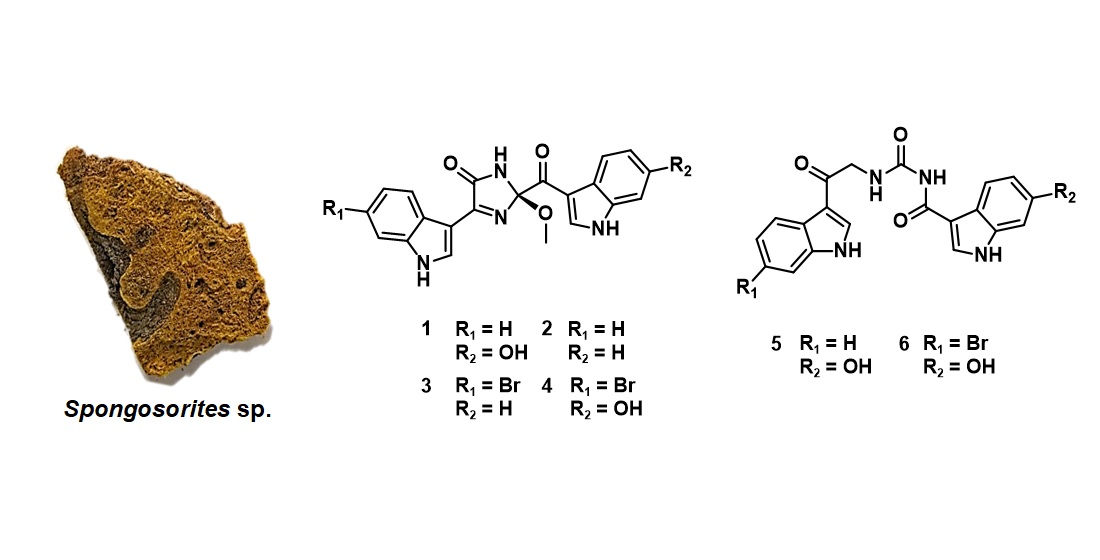
Herein, as a result of the continuing search for sponge-derived bioactive compounds, we report the structures of six new bis(indole) alkaloids related to the topsentin family, along with eight known ones from a Spongosorites sp. sponge, collected from offshore of Jeju Island, Korea (Figure 1). The new compounds, spongosoritins A–D (1–4), possess the 2-methoxy-1-imidazole-5-one moiety as a common structural core with different auxiliary substituents at the indole moieties. The other new compounds, spongocarbamides A (5) and B (6), also have a linear urea-type linkage connecting the indole groups. Both the 2-methoxy-1-imidazole-5-one and linear urea moieties of the new compounds are unprecedented in topsentin alkaloids. The eight known compounds (7–14) consist of four topsentins, two hamacanthins, and two monoindoles. Consistent with our previous results, these indole-bearing compounds exhibited moderate inhibition of Staphylococcus aureus-derived sortase A (SrtA) and weak antibacterial activity against human pathogenic strains. In addition, several compounds showed weak to moderate cytotoxicity against A549 and K562 cancer cell lines.
2. Results and Discussion
2.1. Structure Elucidation
The molecular formula of spongosoritin A (1) was deduced by high resolution fast atom bombardment mass spectrometry (HRFABMS) analysis to be C21H16N4O4 with an unsaturation degree of 16: [M + H]+ m/z 389.1248 (calcd for C21H17N4O4, 389.1250). In accordance with the high unsaturation degree, the 13C NMR data of this compound presented most of the carbon signals at the aromatic/olefinic zone (δC ca 150–100). The corresponding HSQC-based proton signals were also observed at the deshielded region (δH 8.67–6.70) in the 1H NMR data, indicating a polyaromatic nature for 1 (Table 1). Further examination of these data also revealed the presence of three exchangeable protons at the far deshielded region, δH 12.00, 11.80, and 10.10. Thus, 1 was thought to be an alkaloid bearing indoles or similar aromatic moieties.
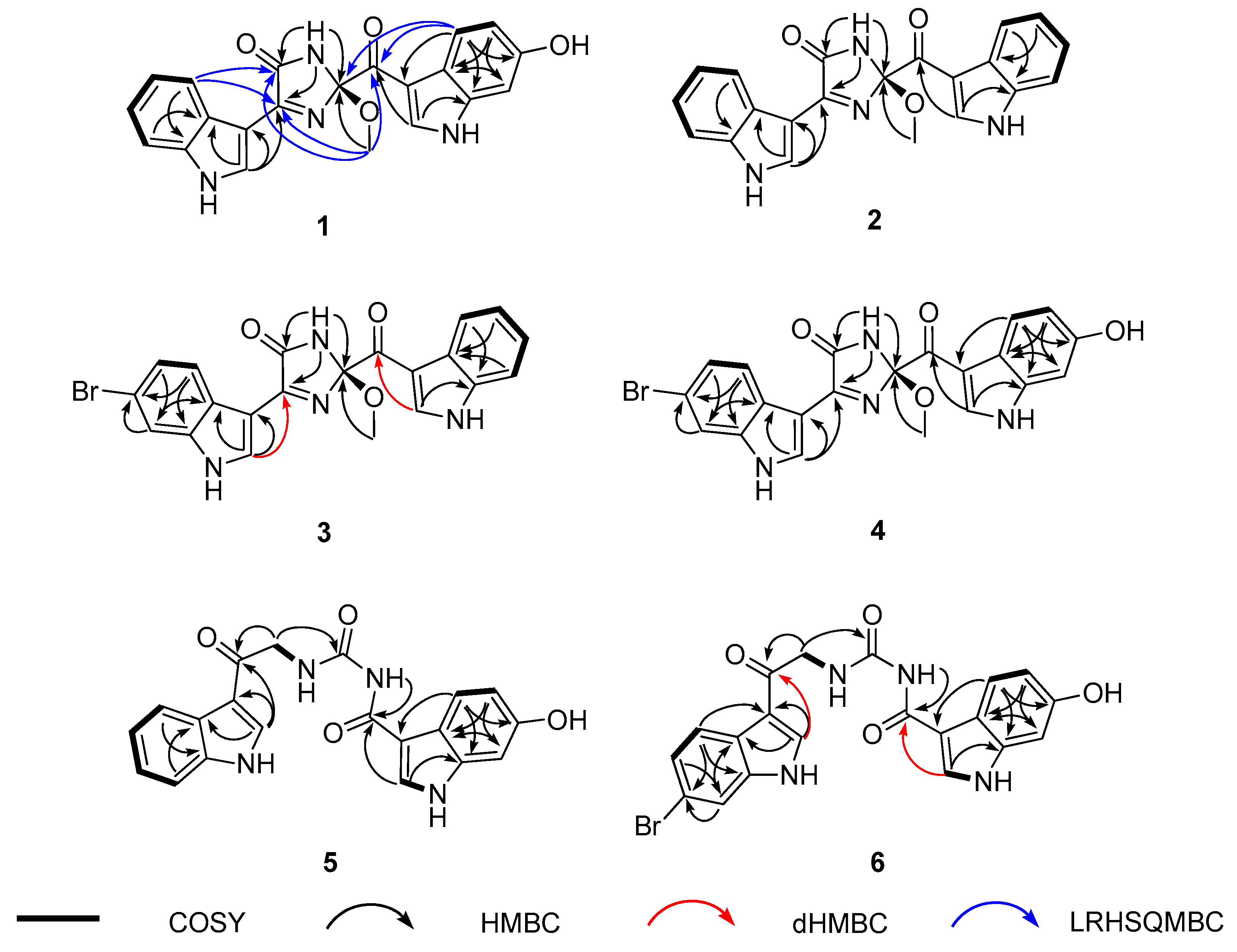
The preliminary spectroscopic interpretation was further investigated by combined 1H-1H COSY and HMBC experiments. First, the COSY data revealed a linear array of four aromatic protons, H-4′–H-7′ (δH 8.18, 7.16, 7.23, and 7.51, respectively) constructing an ortho-disubstituted benzene. Then the HMBC correlations of these and an isolated H-2′ methine proton (δH 8.67) with neighboring carbons readily deduced an indole moiety (N-1′ and C-2′-C-7a′) (Figure 2). Subsequently, a dHMBC correlation with H-2′ attached a nonprotonated carbon (C-4, δC 160.1) at C-3′ (δC 105.9) of this indole, whose functionality was clarified later. Another indole moiety (N-1′′ and C-2′′–C-8′′) was also constructed by the COSY-based ABX-type spin-coupling pattern of three protons, H-4′′ (δH 7.92), H-5′′ (δH 6.70), and H-7′′ (δH 6.87), as well as the HMBC correlations of these and an isolated H-2′′ (δH 8.55) with aromatic carbons. A hydroxy group was attached at C-6′′ (δC 154.2) from the significantly deshielded chemical shift of this carbon. Similarly, a ketone was placed at C-8′′ (δC 186.2) from its chemical shift as well as a three-bond dHMBC correlation with H-2′′.
The 2D NMR interpretation of 1 remained a partial structure with the formula C4H4N2O2 bearing an unsaturation degree of 3 and connecting two indole moieties. A methoxy group (δC/δH 49.3/3.28) was readily evidenced by the HSQC data. Then the other elements were thought to form a cyclic moiety with two double bonds from the chemical shifts of three non-protonated carbons at δC 166.4, 160.1, and 106.9. This was fulfilled by sequential interpretation of the HMBC data, and then extended by the long-range heteronuclear single quantum multiple bond correlation (LRHSQMBC) data for long-range couplings (Figure 2) [29]. First, the methoxy protons (2-OCH3) showed a three-bond HMBC correlation with C-2 (δC 106.9). This oxygenated carbon was directly attached at C-8′′ by LRHSQMBC couplings at H-4′′/C-2 and C-8′′ and 2-OCH3/C-8′′.
Meanwhile the exchangeable NH-1 proton (δH 10.1) was linked with all three carbons at δC 166.4, 160.1, and 106.9 (C-2) by the HMBC data. Of these, the carbon at δC 160.1 was preliminarily found to be connected at C-3′ of the C-2′ indole moiety. Subsequently, the LRHSQMBC-based extended correlations of the former two carbons with H-5′ placed these at C-5 and C-4, respectively. Their deshielded chemical shifts, in conjunction with the molecular formula and unsaturation degree, defined them as an amide carbonyl (C-5) and an imine (C-4) with sp2 nitrogen, together forming a cyclic moiety. This was confirmed by the LRHSQMBC data, in which both carbons showed long-range correlations with the 2-OCH3 protons, constructing a 2-methoxy-1-imidazole-5-one moiety (Figure 2). Alternatively, the switch of functionality between C-4 and C-5 formed a four-member cyclic diazete moiety (N = C–N) consisting of C-2, C-5, and two nitrogen atoms, while the C-4 carbon was a ketone. This interpretation was readily erased by a far shielded chemical shift of C-4 (δC 160.1) as a ketone. Thus, the structure of spongosoritin A (1) was determined to be an imidazolone-bearing bis(indole) alkaloid related to the topsentin class. The presence of imidazolone core is unprecedented among the topsentins, to the best of our knowledge. The absolute configuration at the C-2 stereogenic center was discussed later with congeners.
The 13C and 1H NMR data of spongosoritin B-D (2–4) were very similar to those of 1 at the imidazolone moiety (Table 1). Therefore, the structural difference must occur at the substitutions at the indole moieties, as commonly found in the topsentin alkaloids. Bearing this in mind, the molecular formula of 2 was analyzed for C21H16N4O3 by HRFABMS data: [M + H]+ m/z 373.1303 (calcd for C21H17N4O3, 373.1301). The combined 1D and 2D NMR data readily revealed the presence of a fully protonated indole replacing the hydroxy-bearing one (N-1′′and C-2′′-C-7a′′) in 1 (Figure 2), defining spongosoritin B (2) as a dehydroxy derivative of spongosoritin A (1).
Spongosoritin C (3) exhibited a 1:1 peak ratio of [M]+ and [M + 2]+ in low-resolution electrospray ionization mass spectrometry (LRESIMS) analysis, which is a characteristic pattern attributed to the presence of a bromine. Indeed, the molecular formula of 3 was established to be C21H15BrN4O3 by the HRFABMS data: [M + H]+ m/z 451.0413 (calcd for C21H16Br79N4O3, 451.0406). The position of bromine on an indole group was verified at C-6′ (δC 115.7) by its shielded carbon chemical shift and by the HMBC correlations with the neighboring H-4′ (δH 8.17), H-5′ (δH 7.30), and H-7′ (δH 7.72). Then this bromoindole moiety was directly connected to C-4 of the imidazolone ring by the crucial HMBC correlation at H-2′ (δH 8.69)/ C-4 (δC 161.1) (Figure 2). Based on the same 2D NMR analysis, the other indole moiety was found to be a fully protonated one, and was assigned as a brominated derivative of 2.
The molecular formula of spongosoritin D (4) was analyzed for C21H15BrN4O4 by HRFABMS analysis: [M + H]+ m/z 467.0351 (calcd for C21H16Br79N4O4, 467.0355). As indicated by the MS and 13C and 1H NMR data (Table 1), this compound was anticipated to possess one bromoindole and one hydroxyindole. This interpretation was confirmed by combined 2D NMR analysis, in which these substituents were first secured at C-6′ (δC 115.1) and C-6′′ (δC 154.3) by their HMBC correlation with neighboring ring protons. Subsequently, the indole units were connected to the imidazolone core by diagnostic 3-bond dHMBC correlations at H-2′ (δH 8.67)/C-4 (δC 160.9) and H-2′′ (δH 8.53)/C-8′′ (δC 185.9), respectively. Thus, the structure of spongosoritin D (4) showed that it was a member of the spongosoritin class possessing two substituted indoles.
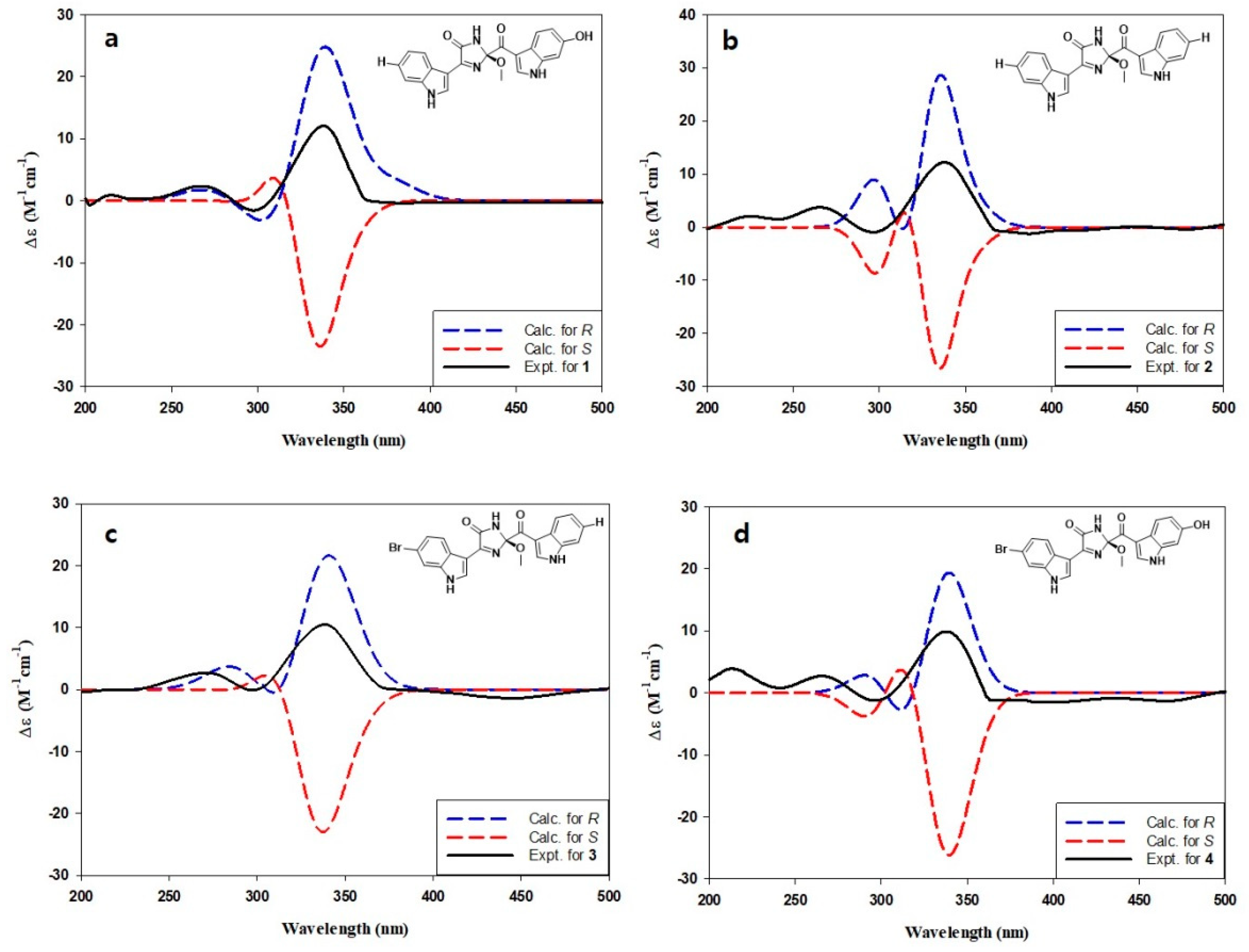
Compounds 1–4 possessed a common stereogenic center at the methoxy-bearing C-2 of the imidazolone moiety. In order to determine the absolute configuration at this position, a density functional theory (DFT)-based computational method was carried out. That is, the initial approach was to measure the specific rotations for 1–4 to verify the same absolute configuration at the C-2 center. However, the obtained rotations were severely irregular and resulted in alternating signs, possibly due to the presence of significantly different conformers (Supplementary Figures S40–S43) [30,31]. Notable examples of this phenomenon among marine natural products would be the sponge-derived discorhabdins, fungus-derived herqueinones, and ascidian-derived isocadiolides [32,33,34,35,36]. This problem was solved by the ECD measurements. Unlike the optical rotations measured at the fixed wavelength of 589 nm, the ECD profiles of 1–4 were stable in the range of 200–400 nm and showed very similar profiles to each other (Figure 3) [32]. Subsequently, the measured data were compared with the calculated ECD profiles, thus assigning the 2R configuration for spongosoritins A–D.
In addition to 1–4, two structurally related but remarkably distinct congeners were isolated. The molecular formula of spongocarbamide A (5) was deduced to be C20H16N4O4 by HRFABMS data: [M + H]+ m/z 377.1249 (calcd for C20H17N4O4, 377.1250). The 13C and 1H NMR data of this compound were highly reminiscent of those of 1–4, indicating the presence of two indole moieties (Table 2). Based on the results of combined 2D NMR analyses, these were identified to be each one of fully protonated and hydroxy-bearing indole as 1 (Figure 2). The remaining signals in the NMR data were three non-protonated carbons (δC 189.7, 165.5 and 154.5), one methylene (δC/δH 46.5/4.71 (2H)), and two exchangeable NH protons (δH 10.20 and 9.35). Of the non-protonated carbons, two carbonyls were secured at C-1 (δC 189.7) and C-8′′ (δC 165.5) by their diagnostic 3-bond HMBC correlation with H-2′ (δH 8.47) and H-2′′ (δH 8.34), respectively.
The COSY data showed a spin correlation between NH (δH 9.35) and methylene protons, which were directly connected to each other, forming an amino-methylene group by the vicinal coupling constant (J = 5.2 Hz). This group was placed between the C-1 carbonyl and another nonprotonated carbon (δC 154.5) by their HMBC correlation with the methylene protons. Based on the chemical shift of C-1 carbonyl (δC 189.7), the amino-methylene group was unambiguously designated as 3-NH and 2-CH2, leaving the other carbon at C-4. Additionally, the HMBC data showed a long-range correlation between an NH (δH 10.20) and C-8′′, assigning this amide nitrogen at 5-NH and functionalizing the C-4 carbon as a urea. Although it was not directly proven by either 2D NMR or MS fragmentation analysis, the chemical shift of C-4 (δC 154.5) was decisive for the assignment of the urea group. Thus, the structure of spongocarbamide A (5) was determined to be a linear bis(indole) alkaloid bearing a urea group.
The molecular formula of spongocarbamide B (6) was deduced to be C20H15BrN4O4 by HRFABMS analysis: [M + H]+ m/z 455.0356 (calcd for C20H16Br79N4O4, 455.0355). The spectroscopic data of this compound were similar to those of 5, revealing the same urea and linear portion between them (Table 2). Therefore, the structural difference was anticipated to exist at the bis(indole) moieties. As for the topsentin congeners, the indole groups were identified by combined 1D and 2D NMR analyses followed by their HMBC- and dHMBC-based connectivity with the linear portion (Figure 2). Conclusively, the bromoindole and hydroxyindole were placed at C-2′ and C-2′′, respectively, identical to 4, defining spongocarbamide B (6) as a brominated derivative of spongocarbamide A (5).
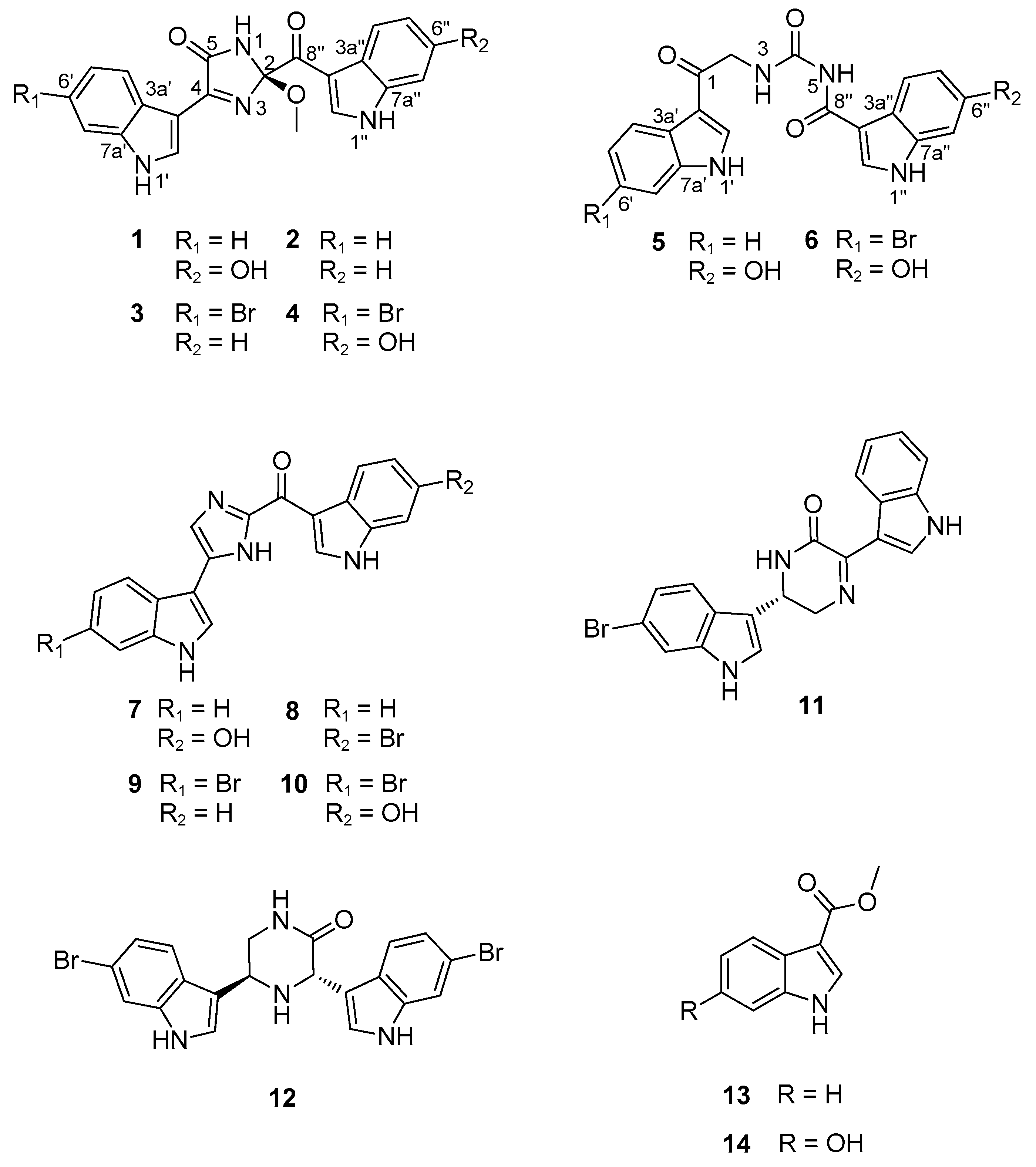
In addition to new compounds 1–6, the crude extract of Spongosorites sp. contained several indole-bearing congeners. Extensive chromatographic separation of these followed by combined spectroscopic analyses led to the identification of eight previously reported compounds. Categorized as four topsentins, two hamacanthins, and two monoindoles, these were topsentin (7) [37], isobromodeoxytopsentin (8) [14], bromodeoxytopsentin (9) [14], bromotopsentin (10) [14], (R)-6′′-debromohamacanthin A (11) [38], (3S, 5R)-6-dibromo-trans-3,4-dihydrohamacanthin B (12) [39], 3-carbomethoxyindole (13) [40], and 6-hydroxy-3-carbomethoxyindole (14) [40]. The spectroscopic data of these were in good accordance with those in the literature.
New compounds 1–6 from Spongosorites sp. showed remarkable structural similarity with topsentins by the presence of not only bis(indole) moieties but also two nitrogen atoms associated with carbonyl group(s). The diversity of indole functionalities and the unanimously presented C-8′′ keto group are further supportive of the structural relation. On the contrary, the new compounds possessed either imidazolone core (1–4) or linear urea linkage (5 and 6), which is unprecedented among topsentin alkaloids. Regarding this, the high oxidant state of the new compounds raises the possibility that these are artifacts derived from topsentins or structurally related precursors during the storage or isolation process. Despite the significant differences in both chemical structure and reaction type, dye-sensitized photooxidation of mono- and bis-phenylimidazoles to corresponding imidazolones would increase the possibility that the new compounds are artifacts derived from natural alkaloids [41]. However, the significant structural deviations of new compounds from topsentins prevented us from postulating plausible mechanisms for either their biotic or abiotic production. Although their origin remains unclear, the structural novelty of these new compounds and their bioactivity, described later, contribute to the value of sponge-derived topsentin alkaloids.
2.2. Biological Activity
In our previous studies, topsentin alkaloids showed antibacterial and cytotoxic activities and SrtA inhibition [17,19]. The same bioassays were pursued with the new compounds and the known congeners, with a particular emphasis on SrtA inhibition. The rationale of SrtA inhibition is that this transpeptidase decorates the surfaces of Gram-positive bacteria with a diverse array of proteins that enable microbes to effectively interact with their environment, and it is not required for bacterial growth or viability [42,43]. It is thus considered a promising target for the development of anti-virulence drugs aimed at interfering with important virulence mechanisms such as adhesion to host tissues. To assess the SrtA inhibitory activity of compounds 1–14, recombinant SrtA derived from S. aureus ATCC6538p was purified from Escherichia coli extracts using metal chelate-affinity chromatography [44]. The enzyme activity was determined from the fluorescence intensity upon cleavage of a peptide substrate containing the LPETG motif [45,46]. The inhibitory potency of the isolated compounds against recombinant SrtA, expressed as IC50 values, is shown in Table 1 and was compared to that of known SrtA inhibitors berberine chloride (IC50 = 87.4 μM) and triphasiol (IC50 = 37.9 μM) [47]. Compounds 2, 3, 5, 6, and 8–11 displayed moderately inhibited SrtA activity (IC50 values of 79.4–34.0 μM). Among them, bromodeoxytopsentin (9) exhibited the most potent inhibitory activity (IC50 = 34.0 μM). The noticeably weak inhibition of monoindoles (13 and 14) emphasizes the role of bis(indole) in SrtA activity inhibition (Table 3).
The antimicrobial activity of compounds 1–14 was also evaluated against phylogenetically diverse pathogenic bacterial and fungal strains [48]. Among these compounds, compounds 7 and 10–12 displayed moderate to significant antibacterial activity (minimum inhibitory concentration (MIC) values of 64–8 μg/mL) against Gram-positive Staphylococcus aureus strain Newman, Enterococcus faecalis ATCC19433, and Enterococcus faecium ATCC19434. Compounds 7 and 10–12 also showed inhibitory activity against Gram-negative Klebsiella pneumoniae ATCC10031 (MIC = 64–8 μg/mL) and Salmonella enterica ATCC14028 (MIC = 64–8 μg/mL), but not Escherichia coli ATCC25922 (MIC > 128 μg/mL), using ampicillin and tetracycline as positive control compounds. However, new compounds 1–6 showed far weaker antibacterial activity than topsentin congeners. The antifungal activity of compounds 1–14 was also evaluated against pathogenic fungal strains Candida albicans ATCC10231, Aspergillus fumigatus HIC6094, Trichophyton rubrum NBRC9185, and Trichophyton mentagrophytes IFM40996 using amphotericin B as a positive control compound. However, none of these compounds exhibited inhibitory activity (MIC > 128 μg/mL) against the tested strains. In addition to cytotoxicity, several compounds showed weak inhibition against A549 and K562 cancer cell lines. Consistent with the antibacterial activity, the cytotoxicity of new compounds 1–6 was remarkably weaker than that of topsentins 7–9 and 12, suggesting the contribution of imidazole or a similar ring moiety to the bioactivity.
3. Materials and Methods
3.1. General Experimental Procedures
Optical rotations were measured using a JASCO P-1020 polarimeter (JASCO Inc., Easton, MD, USA) with a 1 cm cell. UV spectra were recorded by a Hitachi U-3010 spectrophotometer (Hitachi, Fukuoka, Japan). CD spectra were obtained with an Applied Photophysics Chirascan Plus CD spectrometer (Applied Photophysics, Leatherhead, Surrey, UK) using a 0.2 mm cell. IR spectra were acquired on a JASCO 4200 FT-IR spectrometer (JASCO Inc., Easton, MD, USA) using a ZnSe cell. High-resolution FABMS spectrometric data were obtained using a Jeol JMS 700 mass spectrometer (JEOL Ltd., Tokyo, Japan) with meta-nitrobenzyl alcohol (NBA) as a matrix at the National Center for Inter-University Research Facilities (Seoul, Korea). NMR spectra were recorded using Bruker Avance 600 and 800 MHz (Bruker, Billerica, MA, USA) in DMSO-d6 with solvent peaks at δH 2.50/δc 39.50 as the internal standards. The dHMBC NMR experiments were performed on a Bruker Avance 800 MHz utilizing the standard Bruker pulse sequence code (hmqcgpgf) with data points of 2048 × 570 and optimized for JCH measurement at 2Hz. The LRHSQMBC NMR experiment was measured on a Bruker Avance 800 MHz using the same pulse program described in a previous study, with data points of 2048 × 620 and optimized for JCH measurement at 2 Hz [28]. HPLC was performed using a SpectraSYSTEM p2000 (Thermo Fisher Scientific, Waltham, MA, USA) equipped with a refractive index detector (SpectraSYSTEM RI-150 (Thermo Fisher Scientific, Waltham, MA, USA) and a UV-Vis detector (Gilson UV-Vis-151 (Gilson Inc., Middleton, WI, USA). All solvents were of spectroscopy grade or distilled prior to use.
3.2. Animal Material
A specimen of Spongosorites sp. sponge (sample number 14J-17) was collected by hand using SCUBA offshore of Seogwipo, Jeju Island, Korea, at a depth of 25–30 m on 1 October 2014. The specimen was a small piece (5.5 × 3.5 × 1.5 cm). The surface was smooth and covered with other sponge. Oscules were 0.5–1.5 cm in diameter. The texture was firm and compressible. The live color was yellowish brown, turning to dark brown after collection. The skeleton was composed of small and large oxeas (70–200 × 3–10 μm, 900–1200 × 30–40 μm), styles (rare, 760–1100 × 18–36 μm), and strongyles (rare, 420 × 1020 × 24–42 μm). A voucher specimen (registry no. Spo. 83) is deposited in the Natural History Museum, Hannam University, Daejeon, Korea.
3.3. Extraction and Isolation
The sponge specimen was immediately frozen at −25 °C until use. Lyophilized and macerated specimens (120 g) were repeatedly extracted with MeOH (3 × 2 L) and CH2Cl2 (3 × 2 L). The combined yellow extracts (29.8 g) were dried in vacuo and partitioned between H2O (18.0 g) and n-BuOH (10.7 g), then the latter layer was repartitioned between H2O-MeOH (15:85, 7.1 g) and n-hexane (2.6 g). The H2O-MeOH layer was separated by vacuum flash chromatography with ODS resin using sequential mixtures of MeOH and H2O as the eluents (6 fractions in H2O-MeOH gradient, from 50:50 to 0:100), followed by acetone and finally EtOAc. Based on the aromatic proton signals observed in 1H-NMR analysis, 3 fractions were chosen for HPLC separation: 40:60 H2O-MeOH (0.51 g), 30:70 H2O-MeOH (1.08 g), and 20:80 H2O-MeOH (0.40 g).
The first fraction, 50:50 H2O-MeOH, was separated by semi-preparative reversed-phase HPLC (YMC-ODS column, 10 × 250 mm; H2O-MeOH (70:30); 2.0 mL/min) to yield 14 (tR = 60.1 min). The 40:60 H2O-MeOH fraction was separated by semi-preparative HPLC (H2O-MeOH (54:46); 2.0 mL/min) to yield 4 peaks at tR = 35.1, 46.8, 62.4, and 66.3 min. The first peak was purified by reversed-phase analytical HPLC (YMC-ODS column, 4.6 × 250 mm; H2O-MeCN (70:30); 0.7 mL/min) to afford 1 (tR = 26.8 min). The third peak obtained at tR = 62.4 min was purified by similar HPLC conditions (H2O-MeCN (72:28); 0.7 mL/min) to afford 5 (tR = 34.1 min). Compounds 13 and 7 from the original HPLC peaks at tR = 46.8 and 66.3 min, respectively, were pure by 1H NMR analysis, so were not purified further.
The 30:70 H2O-MeOH fraction from vacuum flash chromatography was separated by semi-preparative HPLC (H2O-MeOH (43:57); 2.0 mL/min) to afford 4 peaks at tR = 34.7, 46.2, 52.9, and 60.1 min. The first peak was separated by analytical HPLC (H2O-MeCN gradient (from 75:25 to 60:40 in 80 min); 0.7 mL/min) to yield compounds 4 (tR = 72.0 min) and 2 (tR = 78.3 min). The third peak at tR = 52.9 min was purified by the same gradient HPLC condition to afford 6 (tR = 54.1 min). Compounds 8 and 10 from the original HPLC peaks at tR = 46.2 and 60.1 min, respectively, were pure by 1H NMR analysis, so were not purified further. Finally, the 20:80 H2O-MeOH fraction was separated by semi-preparative HPLC (H2O-MeOH, (36:64); 2.0 mL/min) to yield 4 peaks at tR = 48.7, 52.6, 54.1, and 60.3 min. The peak at tR = 54.1 min was then purified by analytical HPLC (0.7 mL/min; H2O-MeCN (62:38)) to afford 3 (tR = 54.7 min). The other peaks obtained at tR = 48.7, 52.6, and 60.3 min were determined to be 9, 11, and 12, respectively. Overall, the amounts of isolated materials were 7.8, 2.1, 1.1, 4.8, 0.8, 0.7, 28.1, 33.1, 2.3, 1.8, 2.8, 2.1, 1.8, and 1.4 mg for 1–14, respectively.
Spongosoritin A (1). yellow, amorphous solid; ± 40 (c 1.0, MeOH); UV (MeOH) λmax (logε) 280 (3.40), 330 (3.31) nm; ECD (c 1.3 × 10−3 M, MeOH) λmax (Δε) 338 (+12.0), 298 (−1.6) nm; IR (ZnSe) νmax 3289, 2353, 1717, 1599, 1516, 1431 cm−1; 1H and 13C NMR data, see Table 1; HRFABMS m/z 389.1248 [M + H]+ (calcd for C21H17N4O4, 389.1250).
Spongosoritin B (2). yellow, amorphous solid; ± 7 (c 0.8, MeOH); UV (MeOH) λmax (logε) 280 (3.15), 330 (3.09) nm; ECD (c 1.1 × 10−3 M, MeOH) λmax (Δε) 336 (+12.2), 296 (−0.9) nm; IR (ZnSe) νmax 3278, 2354, 1718, 1598, 1516 cm−1; 1H and 13C NMR data, see Table 1; HRFABMS m/z 373.1303 [M + H]+ (calcd for C21H17N4O3, 373.1301).
Spongosoritin C (3). yellow, amorphous solid; ± 10 (c 0.8, MeOH); UV (MeOH) λmax (logε) 280 (3.36), 330 (3.33) nm; ECD (c 1.0 × 10−3 M, MeOH) λmax (Δε) 343 (+12.0), 300 (−1.5) nm; IR (ZnSe) νmax 3312, 2354, 1712, 1599, 1515, 1425 cm−1; 1H and 13C NMR data, see Table 1; HRFABMS m/z 451.0413 [M + H]+ (calcd for C21H16Br79N4O3, 451.0406).
Spongosoritin D (4). yellow, amorphous solid; ± 20 (c 1.0, MeOH); UV (MeOH) λmax (logε) 280 (3.50), 330 (3.40) nm; ECD (c 0.8 × 10−3 M, MeOH) λmax (Δε) 338 (+9.9), 296 (−1.3) nm; IR (ZnSe) νmax 3278, 2354, 1750, 1711, 1600, 1515, 1424 cm−1; 1H and 13C NMR data, see Table 1; HRFABMS m/z 467.0351 [M + H]+ (calcd for C21H16Br79N4O4, 467.0355).
Spongocarbamide A (5). yellow, amorphous solid; UV (MeOH) λmax (logε) 290 (3.34) nm; IR (ZnSe) νmax 3278, 2354, 1684, 1640 cm−1; 1H and 13C NMR data, see Table 2; HRFABMS m/z 377.1249 [M + H]+ (calcd for C20H17N4O4, 377.1250).
Spongocarbamide B (6). yellow, amorphous solid; UV (MeOH) λmax (logε) 280 (3.34) nm; IR (ZnSe) νmax 3290, 2354, 1684, 1641, 1516, 1444 cm−1; 1H and 13C NMR data, see Table 2; HRFABMS m/z 455.0356 [M + H]+ (calcd for C20H16Br79N4O4, 455.0355).
3.4. ECD Calculations
Based on the density functional theory (DFT) calculations, the geometries were optimized to the ground-state level by Turbomole computer software. The basis parameter sets used were def-SVP and the B3-LYP functional for all atoms. The stabilized ground state of each model compound was further confirmed through the harmonic frequency calculation. The calculated ECD data were derived from the optimized structures obtained with TD-DFT at the B3-LYP functional. The ECD spectra were simulated by overlapping Gaussian functions for each transition, where σ is the width of the band at height 1/e. Values ΔEi and Ri are the excitation energy and rotatory strength for transition i, respectively. In the current work, the value of σ was 0.10 eV.
3.5. SrtA Inhibition Assay
Before evaluating the SrtA inhibitory activity of the test compounds, recombinant SrtA from S. aureus ATCC6538p was prepared according to methods described in a previous paper [44]. The SrtA inhibitory activity of the test compounds was determined according to previously described methods [45,46] with minor modifications. The reaction was carried out with 100 μL buffer (50 mM Tris-HCl, 5 mM CaCl2, and 150 mM NaCl, pH 7.5), 7.5 µM synthetic peptide dabcyl-LPETG-edans (AnaSpec, Inc., Fremont, CA, USA), 7.5 µM purified SrtA, and test samples at various concentrations. Each sample was dissolved in dimethyl sulfoxide (DMSO) and diluted with reaction buffer to obtain a final concentration of 1% DMSO, which did not influence enzyme activity. The SrtA inhibition assay was conducted at 37 °C for 1 h, and inhibition was quantified fluorometrically using a microplate reader (FLx800, BioTek Instruments, Winooski, VT, USA) at excitation and emission wavelengths of 350 and 495 nm, respectively. Berberine chloride and triphasiol [47] were used as reference inhibitors of SrtA.
3.6. Antibacterial, Antifungal, and Cytotoxic Assays
The antibacterial activity of compounds 1–14 was evaluated against Gram-positive bacteria (S. aureus strain Newman, E. faecalis ATCC19433, and E. faecium ATCC19434) and Gram-negative bacteria (K. pneumoniae ATCC10031, S. enterica ATCC14028, and E. coli ATCC25922) using a previously reported method [48]. For antifungal activity, C. albicans ATCC10231, A. fumigatus HIC6094, T. mentagrophytes IFM40996, and T. rubrum NBRC9185 strains were used to measure the inhibition of compounds 1–14 by following previously reported procedures [48]. An MTT assay was performed as previously described to determine the cytotoxicity against A549 and K562 cell lines [19]. These cancer cell lines were purchased from the Korean Cell Line Bank (KCLB), Seoul, Republic of Korea.
4. Conclusions
Six new bis(indole) alkaloids along with eight known ones of the topsentin class were isolated from a Spongosorites sp. sponge collected from Jeju Island, Korea. Based on the results of combined spectroscopic analyses, new compounds spongosoritin A–D (1–4) were defined to possess 2-methoxy-1-imidazole-5-one as a common structural core, while spongocarbamides A (5) and B (6) had a linear urea-type linkage connecting the indole groups. Both the 2-methoxy-1-imidazole-5-one and linear urea moieties of the new compounds were unprecedented among the topsentins and related indole alkaloids. These indole-bearing compounds exhibited moderate inhibition of sortase A and weak antibacterial activity against human pathogenic strains. In addition, several compounds were weakly cytotoxic against A549 and K562 cancer cell lines. Thus, the new compounds could contribute significantly to both the structural diversity and bioactivity of sponge-derived topsentin alkaloids.
Supplementary Materials
The following are available online at https://www.mdpi.com/1660-3397/19/1/3/s1, Figures S1–S43: 1D and 2D NMR spectra and HRFABMS data of 1–6.
Author Contributions
J.S.P. carried out the isolation and spectroscopic analyses; J.S.P. and O.-S.K. elucidated DFT models and calculated ECD data; J.-Y.H. and S.C.P. performed MTT assay of A549 and K562; E.C., B.C. and K.-B.O. performed antimicrobial and sortase A inhibition assays; C.J.S. identified the sponge specimen; J.S. and D.-C.O. reviewed and evaluated all data; J.S. and K.-B.O. supervised the research work and prepared the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the National Research Foundation (NRF, grant no. 2018R1A4A1021703) funded by the Ministry of Science, ICT and Future Planning, Korea.
Data Availability Statement
The data presented in this study are available in the supplementary material.
Acknowledgments
We thank the National Center for Inter-university Research Facilities (NCIRF), Korea, for providing mass spectrometric data.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cordell, G.A.; Quinn-Beattie, M.L.; Farnsworth, N.R. The potential of alkaloids in drug discovery. Phytother. Res. 2001, 15, 183–205. [Google Scholar] [CrossRef] [PubMed]
- Kochanowska-Karamyan, A.J.; Hamann, M.T. Marine Indole Alkaloids: Potential New Drug Leads for the Control of Depression and Anxiety. Chem. Rev. 2010, 110, 4489–4497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, Y.P.; Or, T.C.T.; Ip, N.Y.; Cho, T.; Or, T. Plant alkaloids as drug leads for Alzheimer’s disease. Neurochem. Int. 2015, 89, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Huang, G. The synthesis and biological activity of marine alkaloid derivatives and analogues. RSC Adv. 2020, 10, 31909–31935. [Google Scholar] [CrossRef]
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2020, 37, 175. [Google Scholar] [CrossRef]
- Schmidt, E.W.; Harper, M.K.; Faulkner, D.J. Makaluvamines H-M and Damirone C from the Pohnpeian Sponge Zyzzya fuliginosa. J. Nat. Prod. 1995, 58, 1861–1867. [Google Scholar] [CrossRef]
- Sakai, R.; Higa, T.; Jefford, C.W.; Bernardinelli, G. Manzamine A, a novel antitumor alkaloid from a sponge. J. Am. Chem. Soc. 1986, 108, 6404–6405. [Google Scholar] [CrossRef]
- Kochanowska-Karamyan, A.J.; Araujo, H.C.; Zhang, X.; El-Alfy, A.; Carvalho, P.; Avery, M.A.; Holmbo, S.D.; Magolan, J.; Hamann, M.T. Isolation and Synthesis of Veranamine, an Antidepressant Lead from the Marine Sponge Verongula rigida. J. Nat. Prod. 2020, 83, 1092–1098. [Google Scholar] [CrossRef]
- Ma, Y.; Yakushijin, K.; Miyake, F.; Horne, D. A concise synthesis of indolic enamides: Coscinamide A, coscinamide B, and igzamide. Tetrahedron Lett. 2009, 50, 4343–4345. [Google Scholar] [CrossRef]
- Guella, G.; Mancini, I.; Zibrowius, H.; Pietra, F. Aplysinopsin-type alkaloids fromDendrophyllia sp., a scleractinian coral of the family dendrophylliidae of the philippines, facile photochemical (Z/E) photoisomerization and thermal reversal. Helvetica Chim. Acta 1989, 72, 1444–1450. [Google Scholar] [CrossRef]
- Gul, W.; Hamann, M.T. Indole alkaloid marine natural products: An established source of cancer drug leads with considerable promise for the control of parasitic, neurological and other diseases. Life Sci. 2005, 78, 442–453. [Google Scholar] [CrossRef]
- Ang, K.K.H.; Holmes, M.J.; Higa, T.; Hamann, M.T.; Kara, U.A.K. In Vivo Antimalarial Activity of the Beta-Carboline Alkaloid Manzamine A. Antimicrob. Agents Chemother. 2000, 44, 1645–1649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamanokuchi, R.; Imada, K.; Miyazaki, M.; Kato, H.; Watanabe, T.; Fujimuro, M.; Saeki, Y.; Yoshinaga, S.; Terasawa, H.; Iwasaki, N.; et al. Hyrtioreticulins A–E, indole alkaloids inhibiting the ubiquitin-activating enzyme, from the marine sponge Hyrtios reticulatus. Bioorganic Med. Chem. 2012, 20, 4437–4442. [Google Scholar] [CrossRef] [PubMed]
- Shinkre, B.A.; Raisch, K.P.; Fan, L.; Velu, S.E. Analogs of the marine alkaloid makaluvamines: Synthesis, topoisomerase II inhibition, and anticancer activity. Bioorganic Med. Chem. Lett. 2007, 17, 2890–2893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, J.; Seo, Y.; Cho, K.W.; Rho, J.-R.; Sim, C.J. New Bis(Indole) Alkaloids of the Topsentin Class from the SpongeSpongosorites genitrix. J. Nat. Prod. 1999, 62, 647–649. [Google Scholar] [CrossRef]
- Guinchard, X.; Vallee, Y.; Denis, J.-N. Total Synthesis of Marine Sponge Bis(indole) Alkaloids of the Topsentin Class. J. Org. Chem. 2007, 72, 3972–3975. [Google Scholar] [CrossRef]
- Oh, K.-B.; Mar, W.; Kim, S.; Kim, J.-Y.; Oh, M.-N.; Kim, J.-G.; Shin, D.; Sim, C.J.; Shin, J. Bis(indole) alkaloids as sortase A inhibitors from the sponge Spongosorites sp. Bioorganic Med. Chem. Lett. 2005, 15, 4927–4931. [Google Scholar] [CrossRef]
- Keyzers, R.A.; Davies-Coleman, M.T. Anti-inflammatory metabolites from marine sponges. Chem. Soc. Rev. 2005, 34, 355–365. [Google Scholar] [CrossRef]
- Oh, K.-B.; Mar, W.; Kim, S.; Kim, J.-Y.; Lee, T.-H.; Kim, J.-G.; Shin, D.; Sim, C.J.; Shin, J. Antimicrobial Activity and Cytotoxicity of Bis(indole) Alkaloids from the Sponge Spongosorites sp. Biol. Pharm. Bull. 2006, 29, 570–573. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.-G.; Huang, H.; Jiang, B. Progress in Studies of Novel Marine Bis(indole) Alkaloids. Curr. Org. Chem. 2004, 8, 1691–1720. [Google Scholar] [CrossRef]
- Hwang, J.; Kim, D.; Park, J.S.; Park, H.J.; Shin, J.; Lee, S.K. Park Photoprotective Activity of Topsentin, A Bis(Indole) Alkaloid from the Marine Sponge Spongosorites genitrix, by Regulation of COX-2 and Mir-4485 Expression in UVB-Irradiated Human Keratinocyte Cells. Mar. Drugs 2020, 18, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, M.S.; Pandita, N.S.; Pal, A.K. LC-MS/MS as a tool for identification of bioactive compounds in marine sponge Spongosorites halichondriodes (Dendy 1905). Toxicon 2012, 60, 1135–1147. [Google Scholar] [CrossRef] [PubMed]
- Garg, N.K.; Sarpong, R.; Stoltz, B.M. The First Total Synthesis of Dragmacidin D. J. Am. Chem. Soc. 2002, 124, 13179–13184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunasekera, S.P.; McCarthy, P.J.; Kelly-Borges, M. Hamacanthins A and B, New Antifungal Bis Indole Alkaloids from the Deep-Water Marine Sponge, Hamacantha Sp. J. Nat. Prod. 1994, 57, 1437–1441. [Google Scholar] [CrossRef] [PubMed]
- Endo, T.; Tsuda, M.; Fromont, J.; Kobayashi, J. Hyrtinadine A, a Bis-indole Alkaloid from a Marine Sponge. J. Nat. Prod. 2007, 70, 423–424. [Google Scholar] [CrossRef]
- Diana, P.; Carbone, A.; Barraja, P.; Kelter, G.; Fiebig, H.-H.; Cirrincione, G. Synthesis and antitumor activity of 2,5-bis(3′-indolyl)-furans and 3,5-bis(3′-indolyl)-isoxazoles, nortopsentin analogues. Bioorganic Med. Chem. 2010, 18, 4524–4529. [Google Scholar] [CrossRef]
- Sato, H.; Tsuda, M.; Watanabe, K.; Kobayashi, J. Rhopaladins A ∼ D, new indole alkaloids from marine tunicate Rhopalaea sp. Tetrahedron 1998, 54, 8687–8690. [Google Scholar] [CrossRef]
- Jennings, L.K.; Khan, N.M.D.; Kaur, N.; Rodrigues, D.; Morrow, C.; Boyd, A.; Thomas, O.P. Brominated Bisindole Alkaloids from the Celtic Sea Sponge Spongosorites calcicola. Molecules 2019, 24, 3890. [Google Scholar] [CrossRef] [Green Version]
- Williamson, R.T.; Buevich, A.V.; Martin, G.E.; Parella, T. LR-HSQMBC: A Sensitive NMR Technique To Probe Very Long-Range Heteronuclear Coupling Pathways. J. Org. Chem. 2014, 79, 3887–3894. [Google Scholar] [CrossRef]
- Pecul, M.; Ruud, K.; Rizzo, A.; Helgaker, T. Conformational Effects on the Optical Rotation of Alanine and Proline. J. Phys. Chem. A 2004, 108, 4269–4276. [Google Scholar] [CrossRef]
- Wiberg, K.B.; Vaccaro, P.H.; Cheeseman, J.R. Conformational Effects on Optical Rotation. 3-Substituted 1-Butenes. J. Am. Chem. Soc. 2003, 125, 1888–1896. [Google Scholar] [CrossRef] [PubMed]
- Grkovic, T.; Ding, Y.; Li, X.-C.; Webb, V.L.; Ferreira, D.; Copp, B.R. Enantiomeric Discorhabdin Alkaloids and Establishment of Their Absolute Configurations Using Theoretical Calculations of Electronic Circular Dichroism Spectra. J. Org. Chem. 2008, 73, 9133–9136. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.-E.; Na, Z.; Jung, M.; Lee, H.-S.; Sim, C.J.; Nahm, K.; Oh, K.-B.; Shin, J. Discorhabdins from the Korean Marine SpongeSceptrellasp. J. Nat. Prod. 2010, 73, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Cason, J.; Koch, C.W.; Correia, J.S. Structures of herqueinone, isoherqueinone and norherqueinone. J. Org. Chem. 1970, 35, 179–186. [Google Scholar] [CrossRef]
- Park, S.C.; Julianti, E.; Ahn, S.; Kim, D.; Lee, S.K.; Noh, M.; Oh, D.-C.; Oh, K.-B.; Shin, A.J.; Shin, J. Phenalenones from a Marine-Derived Fungus Penicillium Sp. Mar. Drugs 2019, 17, 176. [Google Scholar] [CrossRef] [Green Version]
- Bae, J.; Cho, E.; Park, J.S.; Won, T.H.; Seo, S.-Y.; Oh, D.-C.; Oh, K.-B.; Shin, J. Isocadiolides A–H: Polybrominated Aromatics from a Synoicum sp. Ascidian. J. Nat. Prod. 2020, 83, 429–437. [Google Scholar] [CrossRef]
- Zhang, Z.-W.; Xue, H.; Li, H.; Kang, H.; Feng, J.; Lin, A.; Liu, S. Collective Synthesis of 3-Acylindoles, Indole-3-carboxylic Esters, Indole-3-sulfinic Acids, and 3-(Methylsulfonyl)indoles from Free (N–H) Indoles via Common N-Indolyl Triethylborate. Org. Lett. 2016, 18, 3918–3921. [Google Scholar] [CrossRef]
- Casapullo, A.; Bifulco, G.; Bruno, I.; Riccio, R. New Bisindole Alkaloids of the Topsentin and Hamacanthin Classes from the Mediterranean Marine SpongeRhaphisia lacazei. J. Nat. Prod. 2000, 63, 447–451. [Google Scholar] [CrossRef]
- Bao, B.; Sun, Q.; Yao, X.; Hong, J.; Lee, C.-O.; Cho, H.Y.; Jung, J.H. Bisindole Alkaloids of the Topsentin and Hamacanthin Classes from a Marine SpongeSpongosoritessp. J. Nat. Prod. 2007, 70, 2–8. [Google Scholar] [CrossRef]
- Bao, B.; Zhang, P.; Lee, Y.; Hong, J.; Lee, C.-O.; Jung, J.H. Monoindole Alkaloids from a Marine Sponge Spongosorites sp. Mar. Drugs 2007, 5, 31–39. [Google Scholar] [CrossRef]
- Kai, S.; Suzuki, M. ChemInform Abstract: Dye-Sensitized Photooxidation of 2,4-Disubstituted Imidazoles: The Formation of Isomeric Imidazolinones. ChemInform 2010, 27, 1185–1188. [Google Scholar] [CrossRef]
- Maresso, A.W.; Schneewind, O. Sortase as a Target of Anti-Infective Therapy. Pharmacol. Rev. 2008, 60, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Cascioferro, S.; Totsika, M.; Schillaci, D. Sortase A: An ideal target for anti-virulence drug development. Microb. Pathog. 2014, 77, 105–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, K.-B.; Kim, S.-H.; Lee, J.; Cho, W.-J.; Lee, T.; Kim, S. Discovery of Diarylacrylonitriles as a Novel Series of Small Molecule Sortase A Inhibitors. J. Med. Chem. 2004, 47, 2418–2421. [Google Scholar] [CrossRef] [PubMed]
- Park, S.C.; Chung, B.; Lee, J.; Cho, E.; Hwang, J.-Y.; Oh, D.-C.; Shin, J.; Oh, K.-B. Sortase A-Inhibitory Metabolites from a Marine-Derived Fungus Aspergillus sp. Mar. Drugs 2020, 18, 359. [Google Scholar] [CrossRef]
- Mazmanian, S.K.; Ton-That, H.; Su, K.; Schneewind, O. An iron-regulated sortase anchors a class of surface protein during Staphylococcus aureus pathogenesis. Proc. Natl. Acad. Sci. USA 2002, 99, 2293–2298. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-S.; Chung, B.; Lee, W.-H.; Lee, J.; Suh, Y.; Oh, D.-C.; Oh, K.-B.; Shin, J. Sortase A-Inhibitory Coumarins from the Folk Medicinal Plant Poncirus trifoliata. J. Nat. Prod. 2020, 83, 3004–3011. [Google Scholar] [CrossRef]
- Cho, E.; Kwon, O.-S.; Chung, B.; Lee, J.; Sun, J.; Shin, J.; Oh, K.-B. Antibacterial Activity of Chromomycins from a Marine-Derived Streptomyces microflavus. Mar. Drugs 2020, 18, 522. [Google Scholar] [CrossRef]
Figure 1.
Chemical structures of compounds 1–14.

Figure 2.
Key correlations of 1H-1H COSY (bold line), HMBC (arrow), dHMBC (red arrow), and LRHSQMBC (blue arrow) experiments for compounds 1–6.
Figure 2.
Key correlations of 1H-1H COSY (bold line), HMBC (arrow), dHMBC (red arrow), and LRHSQMBC (blue arrow) experiments for compounds 1–6.

Figure 3.
(a–d) Comparison of measured and calculated ECD profiles of compounds 1–4, respectively.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
13C and 1H NMR assignments for compounds 1–4a.
| No. | 1 b | 2 b | 3 b | 4 b | ||||
|---|---|---|---|---|---|---|---|---|
| ppm, Type | δ, mult (J in Hz) | ppm, Type | δ, mult (J in Hz) | ppm, Type | δ, mult (J in Hz) | ppm, Type | δ, mult (J in Hz) | |
| 1 | 10.10, s | 10.20, s | 10.20, s | 10.10, s | ||||
| 2 | 106.9, C | 106.9, C | 107.0, C | 107.0, C | ||||
| 4 | 160.1, C | 161.1, C | 161.1, C | 160.9, C | ||||
| 5 | 166.4, C | 166.4, C | 166.1, C | 166.2, C | ||||
| 1′ | 12.00, s | ND c | 12.10, s | 11.90, s | ||||
| 2′ | 132.9, CH | 8.67, s | 133.0, CH | 8.68, s | 133.9, CH | 8.69, d (3.0) | 132.9, CH | 8.67, s |
| 3′ | 105.9, C | 105.9, C | 106.0, C | 106.0, C | ||||
| 3a′ | 125.6, C | 125.6, C | 124.6, C | 124.4, C | ||||
| 4′ | 121.5, CH | 8.18, d (7.8) | 121.5, CH | 8.18, d (7.8) | 123.1, CH | 8.17, d (7.8) | 123.1, CH | 8.09, d (8.2) |
| 5′ | 121.5, CH | 7.16, dd (8.0, 7.8) | 121.6, CH | 7.16, dd (7.8, 7.0) | 124.4, CH | 7.30, dd (7.8, 1.5) | 124.6, CH | 7.30, dd (8.2, 1.9) |
| 6′ | 123.1, CH | 7.23, dd (8.0, 7.4) | 123.1, CH | 7.23, dd (8.0, 7.0) | 115.7, C | 115.1, C | ||
| 7′ | 112.2, CH | 7.51, d (8.0) | 112.4, CH | 7.51, d (8.0) | 115.1, CH | 7.72, d (1.5) | 115.7, CH | 7.72, d (1.9) |
| 7a′ | 136.4, C | 136.5, C | 137.4, C | 137.4, C | ||||
| 1′′ | 11.80, s | ND c | 12.20, s | ND c | ||||
| 2′′ | 136.4, CH | 8.55, s | 137.7, CH | 8.74, s | 137.7, CH | 8.73, d (3.0) | 123.1, CH | 8.53, s |
| 3′′ | 112.3, C | 112.1, C | 112.1, C | 112.1, C | ||||
| 3a′′ | 119.5, C | 126.7, C | 126.7, C | 119.5, C | ||||
| 4′′ | 121.7, CH | 7.92, d (8.6) | 121.2, CH | 8.14, d (7.8) | 121.2, CH | 8.09, d (8.2) | 121.7, CH | 7.91, d (8.4) |
| 5′′ | 112.2, CH | 6.70, dd (8.6, 2.0) | 122.2, CH | 7.19, dd (7.8, 7.6) | 122.3, CH | 7.22, dd (8.2, 7.7) | 112.7, CH | 6.69, dd (8.4, 2.1) |
| 6′′ | 154.2, C | 123.0, CH | 7.24, dd (8.0, 7.6) | 123.0, CH | 7.22, dd (7.7, 7.6) | 154.3, C | ||
| 7′′ | 97.5, CH | 6.87, d (2.0) | 112.4, CH | 7.55, d (8.0) | 112.4, CH | 7.54, d (7.6) | 97.5, CH | 6.88, d (2.1) |
| 7a′′ | 137.1, C | 136.0, C | 135.9, C | 137.1, C | ||||
| 8′′ | 186.2, C | 186.5, C | 186.3, C | 185.9, C | ||||
| 2-OMe | 49.3, CH3 | 3.28, s | 49.3, CH3 | 3.30, s | 49.3, CH3 | 3.30, s | 49.4, CH3 | 3.28, s |
a Data were obtained in DMSO-d6. b Data were measured at 500/125 (1 and 2) and 800/200 (3 and 4) MHz for 1H/13C, respectively. c Not detected.
Table 2.
13C and 1H NMR assignments for compounds 5 and 6 a.
| No. | 5 b | 6 b | ||
|---|---|---|---|---|
| ppm, Type | δ, mult (J in Hz) | ppm, Type | δ, mult (J in Hz) | |
| 1 | 189.7, C | 189.7, C | ||
| 2 | 46.5, CH2 | 4.71, d (5.2) | 46.6, CH2 | 4.74, d (5.2) |
| 3 | 9.35, t (5.2) | 9.33, t (5.2) | ||
| 4 | 154.5, C | 154.5, C | ||
| 5 | 10.20, s | 10.20, s | ||
| 1′ | 12.00, s | NDc | ||
| 2′ | 133.7, CH | 8.47, s | 134.6, CH | 8.50, s |
| 3′ | 113.8, C | 113.9, C | ||
| 3a′ | 125.4, C | 124.4, C | ||
| 4′ | 121.1, CH | 8.18, d (7.8) | 122.8, CH | 8.11, d (8.4) |
| 5′ | 121.9, CH | 7.22, dd (7.8, 7.0) | 128.4, CH | 7.36, dd (8.4, 1.6) |
| 6′ | 122.9, CH | 7.23, dd (8.0, 7.0) | 114.9, C | |
| 7′ | 112.2, CH | 7.49, d (8.0) | 115.5, CH | 7.69, d (1.6) |
| 7a′ | 136.5, C | 137.4, C | ||
| 1′′ | 11.50, s | 11.60, s | ||
| 2′′ | 129.3, CH | 8.34, d (2.2) | 129.2, CH | 8.33, s |
| 3′′ | 108.4, C | 108.4, C | ||
| 3a′′ | 119.5, C | 119.4, C | ||
| 4′′ | 121.5, CH | 7.96, d (8.5) | 121.5, CH | 7.95, d (8.6) |
| 5′′ | 111.8, CH | 6.70, dd (8.5, 2.0) | 111.8, CH | 6.74, dd (8.6, 2.0) |
| 6′′ | 154.0, C | 154.0, C | ||
| 7′′ | 97.1, CH | 6.80, d (2.0) | 97.1, CH | 6.84, d (2.0) |
| 7a′′ | 137.6, C | 137.6, C | ||
| 8′′ | 165.5, C | 165.5, C | ||
a Data were obtained in DMSO-d6. b Data were measured at 600/150 (5) and 800/200 (6) MHz. c Not detected.
Table 3.
Results of antibacterial, sortase A inhibitory, and cytotoxic assays of compounds 1–14.
| Antibacterial Activity MIC (μg/mL) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Gram (+) Positive | Gram (−) Negative | IC50 (μM) | |||||||
| No. | S. aureus | Enterococcus faecalis | Enterococcus faecium | Klebsiella pneumonia | Salmonella enterica | E. coli | Srt A | A549 | K562 |
| 1 | >128 | >128 | >128 | >128 | >128 | >128 | >329.8 | 77.3 | 24.2 |
| 2 | 64 | >128 | >128 | 128 | 128 | >128 | 62.7 | 55.7 | 28.5 |
| 3 | 32 | 128 | >128 | >128 | 64 | >128 | 43.9 | 61.2 | 37.7 |
| 4 | 16 | 128 | 128 | >128 | 64 | >128 | >274.7 | 70.9 | 54.2 |
| 5 | >128 | >128 | >128 | >128 | >128 | >128 | 79.4 | >100 | 92.8 |
| 6 | 64 | >128 | >128 | >128 | 128 | >128 | 52.4 | >100 | >100 |
| 7 | 32 | 64 | 64 | 64 | 64 | >128 | >374.1 | 25.3 | 15.3 |
| 8 | 32 | >128 | >128 | >128 | >128 | >128 | 41.3 | 22.7 | 12.5 |
| 9 | 32 | >128 | >128 | >128 | >128 | >128 | 34.0 | 29.5 | 14.4 |
| 10 | 8 | 32 | 16 | 64 | 16 | >128 | 53.2 | 38.9 | 43.2 |
| 11 | 8 | 32 | 32 | 8 | 16 | >128 | 47.5 | 39.6 | >100 |
| 12 | 8 | 16 | >128 | 8 | 8 | >128 | >263.4 | 34.3 | 9.7 |
| 13 | 128 | >128 | >128 | >128 | >128 | >128 | 164.4 | 59.2 | >100 |
| 14 | 128 | >128 | >128 | >128 | >128 | >128 | 191.1 | 66.8 | >100 |
| ampicillin | 0.13 | 0.5 | 1 | ND a | 0.25 | 8 | |||
| tetracycline | ND a | ND a | ND a | 0.25 | ND a | ND a | |||
| triphasiol | 37.9 | ||||||||
| berberine chloride | 87.4 | ||||||||
| doxorubicin | 0.64 | 0.92 | |||||||
a Not detected.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Park, J.S.; Cho, E.; Hwang, J.-Y.; Park, S.C.; Chung, B.; Kwon, O.-S.; Sim, C.J.; Oh, D.-C.; Oh, K.-B.; Shin, J. Bioactive Bis(indole) Alkaloids from a Spongosorites sp. Sponge. Mar. Drugs 2021, 19, 3. https://doi.org/10.3390/md19010003
AMA Style
Park JS, Cho E, Hwang J-Y, Park SC, Chung B, Kwon O-S, Sim CJ, Oh D-C, Oh K-B, Shin J. Bioactive Bis(indole) Alkaloids from a Spongosorites sp. Sponge. Marine Drugs. 2021; 19(1):3. https://doi.org/10.3390/md19010003
Chicago/Turabian StylePark, Jae Sung, Eunji Cho, Ji-Yeon Hwang, Sung Chul Park, Beomkoo Chung, Oh-Seok Kwon, Chung J. Sim, Dong-Chan Oh, Ki-Bong Oh, and Jongheon Shin. 2021. "Bioactive Bis(indole) Alkaloids from a Spongosorites sp. Sponge" Marine Drugs 19, no. 1: 3. https://doi.org/10.3390/md19010003
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.