
Tools for Predicting the Nature and Magnitude of Magnetic Anisotropy in Transition Metal Complexes: Application to Co(II) Complexes


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Selection Rules and Computational Information
2.1. Selection Rules for the Spin-Orbit Coupling in Atoms and Molecules
2.2. Computational Information
3. Results and Discussion
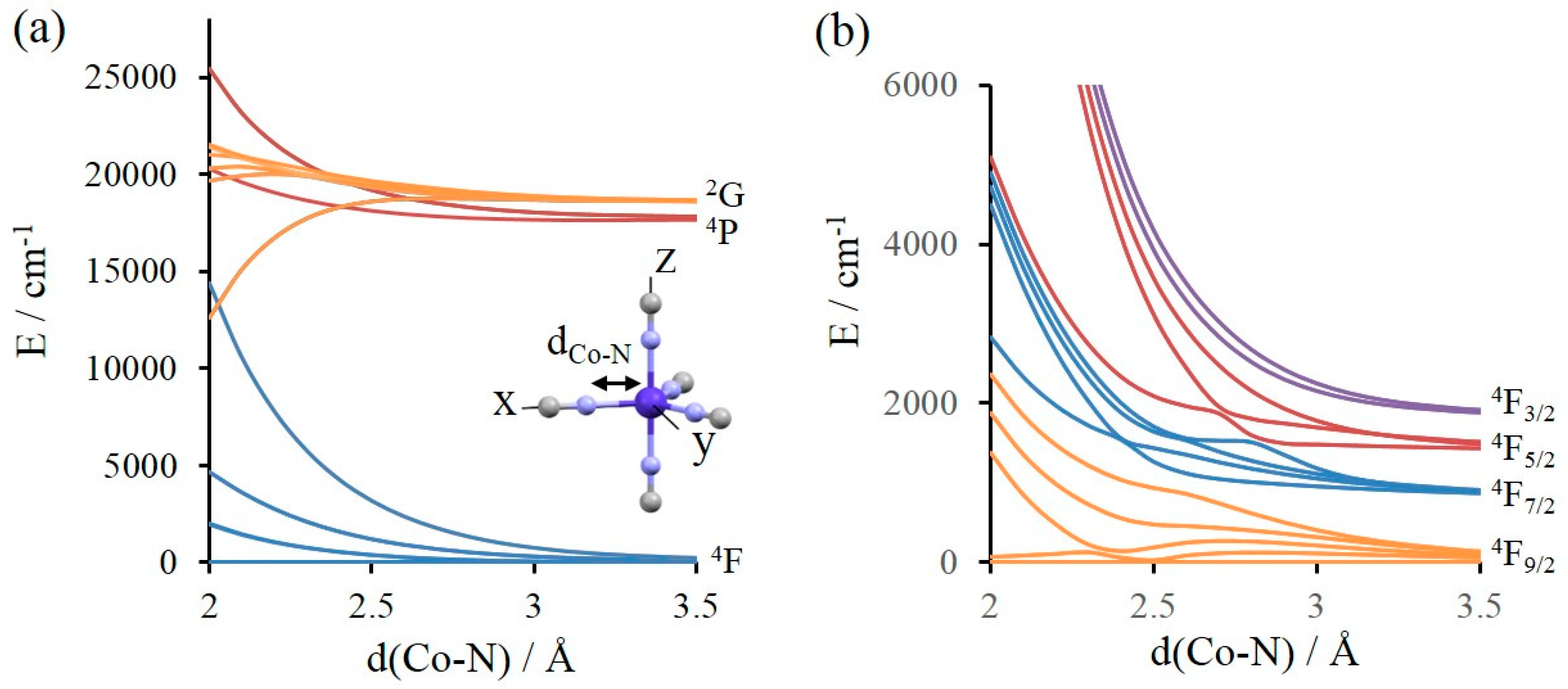
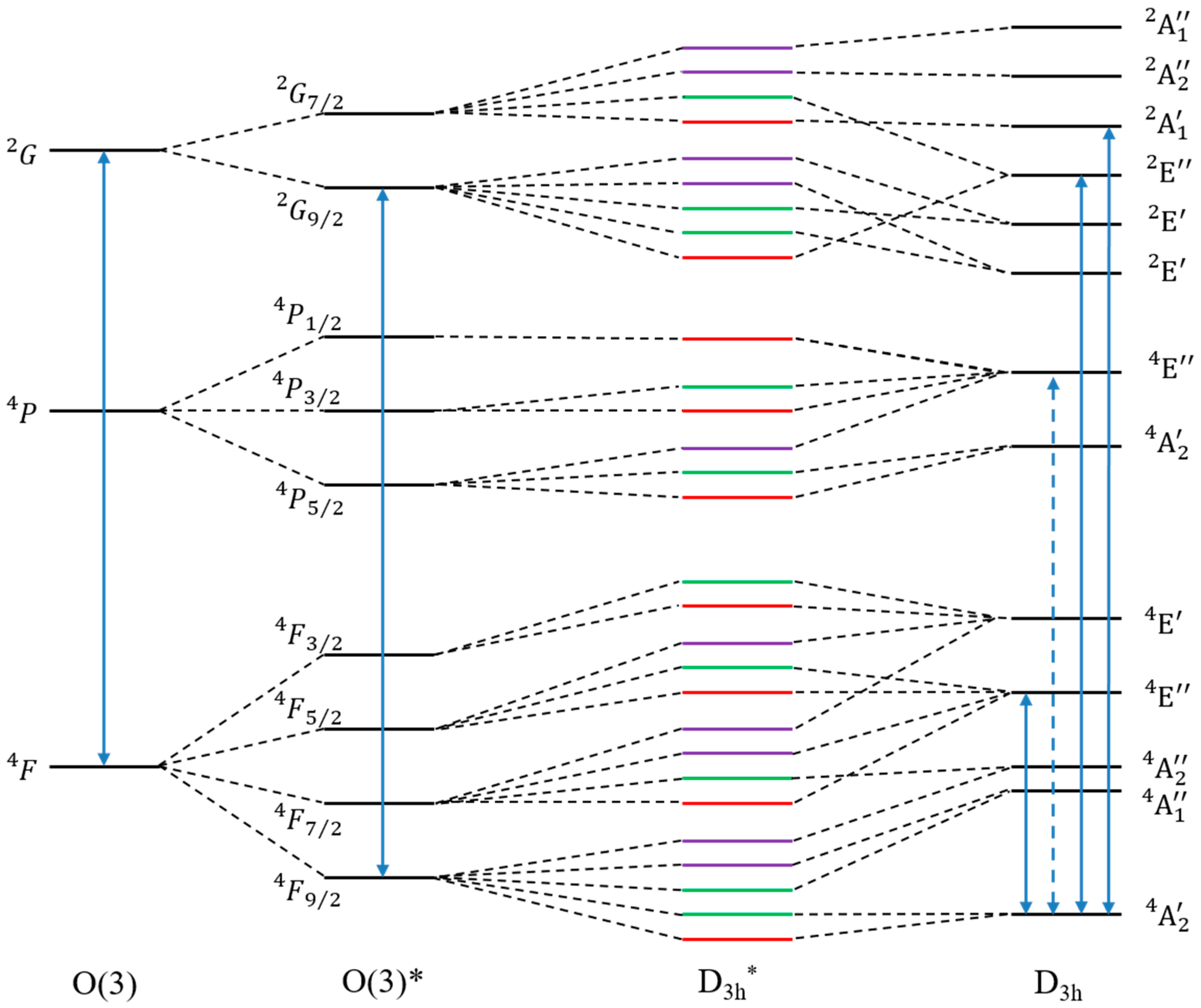
3.1. Reminiscence of the Physics of the Atom in the Molecular Spectrum
3.2. Selection Rules for the Calculation of the ZFS in Molecular Complexes
3.2.1. Molecular Selection Rules Based on the Symmetry Point Group
3.2.2. Molecular Selection Rules Based on the Double Group Theory
3.2.3. Atomic Selection Rules
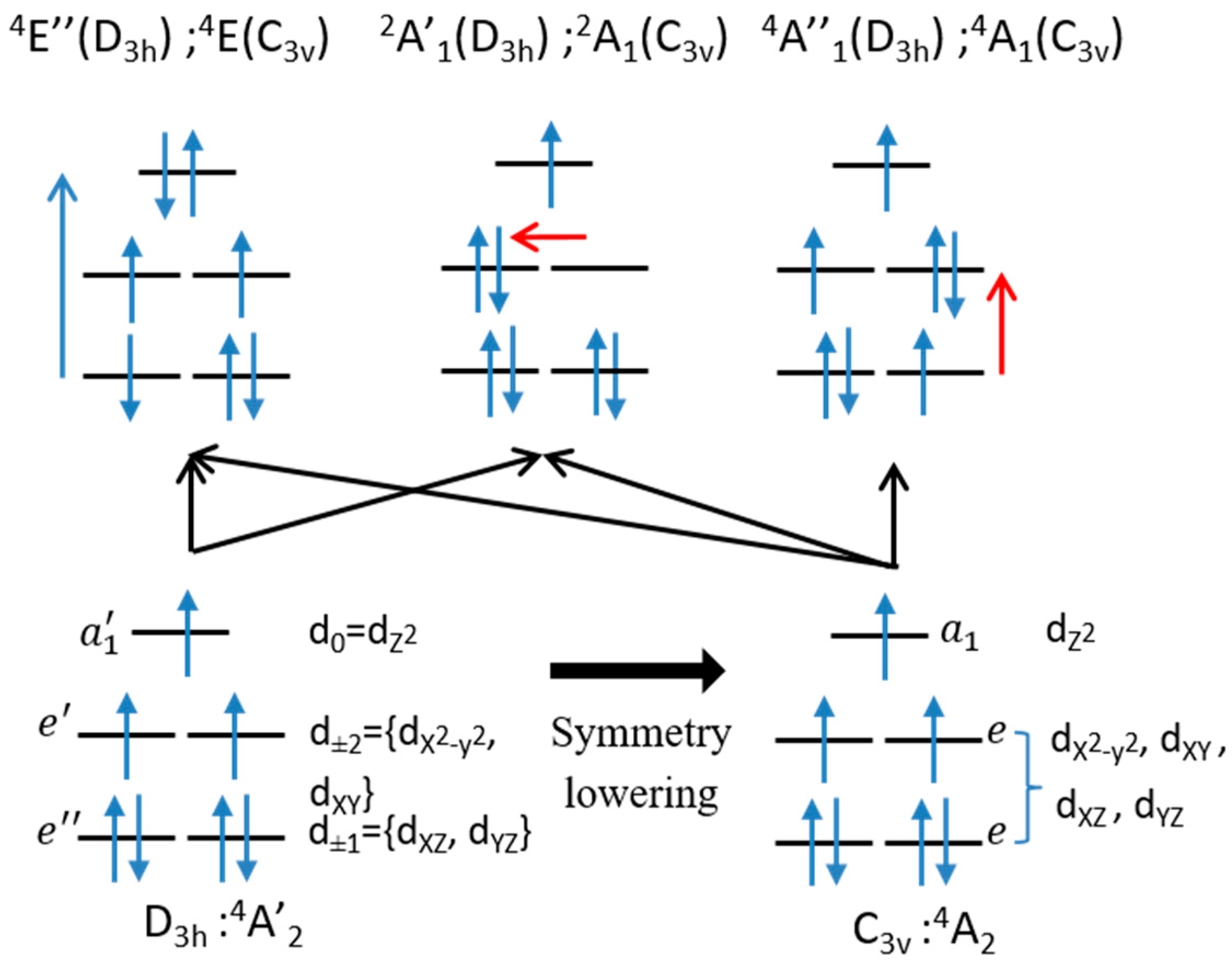
3.3. Rationalization of the ZFS Nature
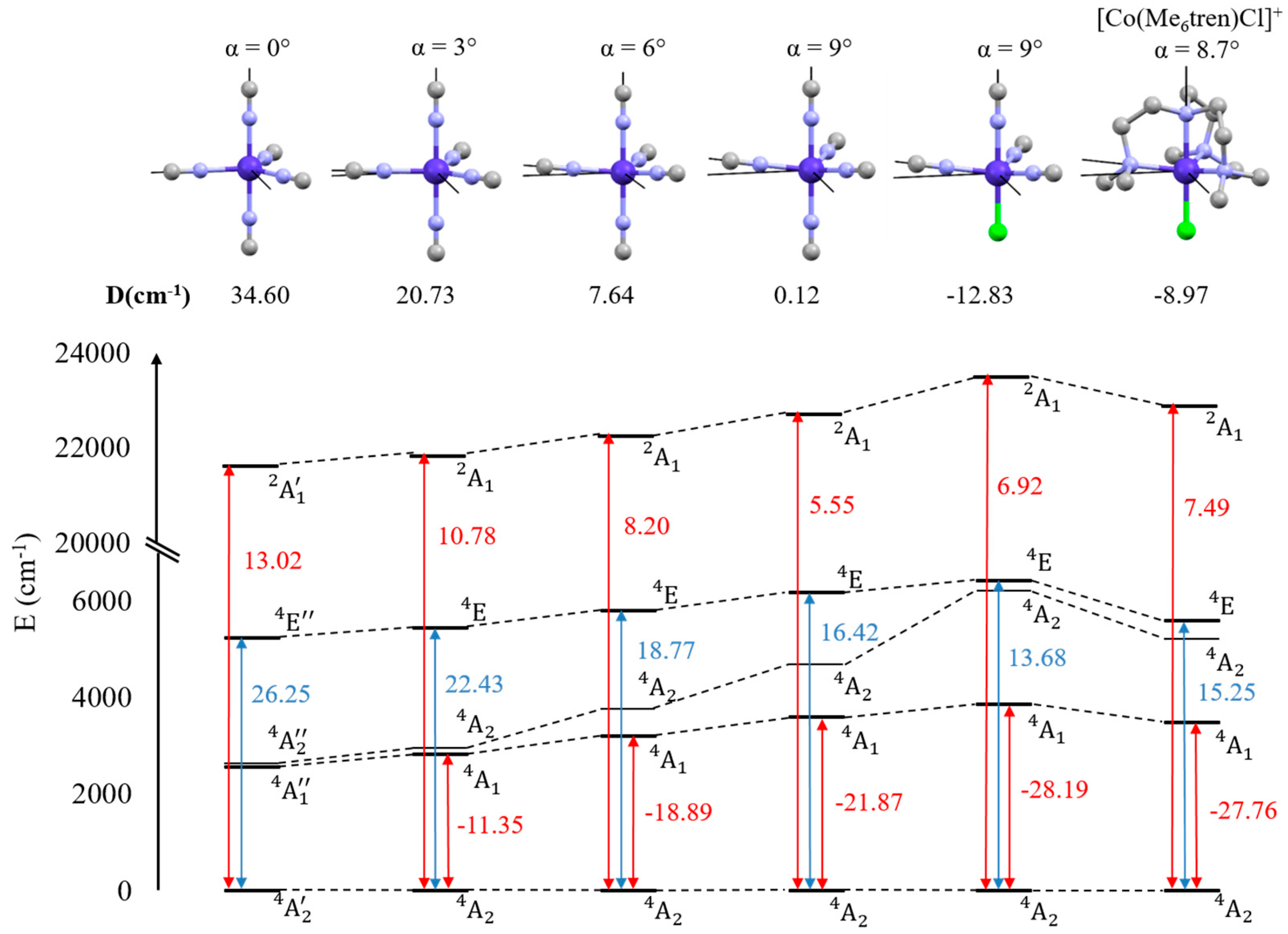
- (i)
- If an easy axis of magnetization is needed (negative D value), the symmetry lowering from D3h to C3v is beneficial because it allows a coupling with the first excited state that brings a negative contribution to D.
- (ii)
- In order to obtain a more negative overall D value, one can decrease its positive contribution by destabilizing the 4E(C3v) excited state. As the main determinant of this state coupled to the ground state has a double occupancy in the dz2 orbital, ligands with a strong field in axial positions would be appropriate to do so.
- (iii)
- Finally, one may question the role of the chemical substitution by chlorine. To answer this query and separate the role of the distortion brought by the change of the value of the angle α from that of the electronic effect of chlorine, we have also substituted an axial NCH ligand in a slightly distorted (α = 3°) complex. In both cases (α = 3° and α = 9°), the main effect comes from the first excited state that for chlorine has a two-fold stronger coupling (see Figure S2) to the ground state, resulting in a much larger negative contribution to D. From this last observation, one may learn that not only the energy differences between the states (diagonal elements of the state interaction matrix) but also the magnitude of the coupling (off diagonal elements which are proportional to the fine structure constant and depend on the chemical nature of the ligand, i.e., electronic effect) can be tuned with the choice of appropriate ligands.
4. Summary
- (i)
- The atomic selection rules are essentially fulfilled. The contribution to D of the single excited state that is coupled to the ground state in the molecular complex but not in the atom is only 0.58 cm−1 and can be safely neglected in any attempt of rationalization of the SOC nature.
- (ii)
- The double group selection rule is less restrictive than the symmetry point group ones.
- (iii)
- From the symmetry point group selection rules, one may not only determine the excited states that are coupled to the ground state but also identify which part of the SOC operator, or , is responsible for the coupling and therefore the sign of their contribution to D.
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ZFS | Zero-Field Splitting |
| CASSCF | Complete Active Space Self-Consistent Field |
| SO–SI | Spin-Orbit State Interaction |
| SOC | Spin-orbit coupling |
References
- Barra, A.L.; Caneschi, A.; Cornia, A.; de Biani, F.F.; Gatteschi, D.; Sangregorio, C.; Sessoli, R.; Sorace, L. Single-molecule magnet behavior of a tetranuclear iron(iii) complex. The origin of slow magnetic relaxation in iron(III) clusters. J. Am. Chem. Soc. 1999, 121, 5302–5310. [Google Scholar] [CrossRef]
- Brechin, E.K.; Yoo, J.; Nakano, M.; Huffman, J.C.; Hendrickson, D.N.; Christou, G. A new class of single-molecule magnets: Mixed-valent [Mn4(O2CMe)2(Hpdm)6][ClO4]2 with an S = 8 ground state. Chem. Commun. 1999, 783–784. [Google Scholar] [CrossRef]
- Caneschi, A.; Gatteschi, D.; Sessoli, R.; Barra, A.L.; Brunel, L.C.; Guillot, M. Alternating-current susceptibility, high-field magnetization, and millimeter band EPR evidence for a ground S = 10 state in [Mn12O12(CH3COO)16(H2O)4].2CH3COOH.4H2O. J. Am. Chem. Soc. 1991, 113, 5873–5874. [Google Scholar] [CrossRef]
- Delfs, C.; Gatteschi, D.; Pardi, L.; Sessoli, R.; Wieghardt, K.; Hanke, D. Magnetic-properties of an octanuclear iron(III) cation. Inorg. Chem. 1993, 32, 3099–3103. [Google Scholar] [CrossRef]
- Gatteschi, D.; Caneschi, A.; Pardi, L.; Sessoli, R. Large clusters of metal-ions: The transition from molecular to bulk magnets. Science 1994, 265, 1054–1058. [Google Scholar] [CrossRef] [PubMed]
- Maheswaran, S.; Chastanet, G.; Teat, S.J.; Mallah, T.; Sessoli, R.; Wernsdorfer, W.; Winpenny, R.E. Phosphonate ligands stabilize mixed-valent {MnIII20−xMnIIx} clusters with large spin and coercivity. Angew. Chem. Int. Ed. Engl. 2005, 44, 5044–5048. [Google Scholar] [CrossRef] [PubMed]
- Sessoli, R.; Gatteschi, D.; Caneschi, A.; Novak, M.A. Magnetic bistability in a metal-ion cluster. Nature 1993, 365, 141–143. [Google Scholar] [CrossRef]
- Sessoli, R.; Tsai, H.L.; Schake, A.R.; Wang, S.Y.; Vincent, J.B.; Folting, K.; Gatteschi, D.; Christou, G.; Hendrickson, D.N. High-spin molecules: [Mn12O12(O2CR)16(H2O)4]. J. Am. Chem. Soc. 1993, 115, 1804–1816. [Google Scholar] [CrossRef]
- Moragues-Canovas, M.; Riviere, P.; Ricard, L.; Paulsen, C.; Wernsdorfer, W.; Rajaraman, G.; Brechin, E.K.; Mallah, T. Resonant quantum tunneling in a new tetranuclear iron(III)-based single-molecule magnet. Adv. Mater. 2004, 16, 1101–1105. [Google Scholar] [CrossRef]
- Ardavan, A.; Blundell, S.J. Storing quantum information in chemically engineered nanoscale magnets. J. Mater. Chem. 2009, 19, 1754–1760. [Google Scholar] [CrossRef]
- Timco, G.A.; Carretta, S.; Troiani, F.; Tuna, F.; Pritchard, R.J.; Muryn, C.A.; McInnes, E.J.L.; Ghirri, A.; Candini, A.; Santini, P.; et al. Engineering the coupling between molecular spin qubits by coordination chemistry. Nat. Nanotech. 2009, 4, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Urdampilleta, M.; Klyatskaya, S.; Cleuziou, J.P.; Ruben, M.; Wernsdorfer, W. Supramolecular spin valves. Nat. Mater. 2011, 10, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Zadrozny, J.M.; Liu, J.; Piro, N.A.; Chang, C.J.; Hill, S.; Long, J.R. Slow magnetic relaxation in a pseudotetrahedral cobalt(II) complex with easy-plane anisotropy. Chem. Commun. 2012, 48, 3927–3929. [Google Scholar] [CrossRef] [PubMed]
- Fataftah, M.S.; Zadrozny, J.M.; Coste, S.C.; Graham, M.J.; Rogers, D.M.; Freedman, D.E. Employing forbidden transitions as qubits in a nuclear spin-free chromium complex. J. Am. Chem. Soc. 2016, 138, 1344–1348. [Google Scholar] [CrossRef] [PubMed]
- Shiddiq, M.; Komijani, D.; Duan, Y.; Gaita-Arino, A.; Coronado, E.; Hill, S. Enhancing coherence in molecular spin qubits via atomic clock transitions. Nature 2016, 531, 348–351. [Google Scholar] [CrossRef] [PubMed]
- Ruamps, R.; Batchelor, L.J.; Maurice, R.; Gogoi, N.; Jimenez-Lozano, P.; Guihery, N.; de Graaf, C.; Barra, A.L.; Sutter, J.P.; Mallah, T. Origin of the magnetic anisotropy in heptacoordinate Ni-II and Co-II complexes. Chem. Eur. J. 2013, 19, 950–956. [Google Scholar] [CrossRef] [PubMed]
- Ruamps, R.; Maurice, R.; Batchelor, L.; Boggio-Pasqua, M.; Guillot, R.; Barra, A.L.; Liu, J.J.; Bendeif, E.; Pillet, S.; Hill, S.; et al. Giant ising-type magnetic anisotropy in trigonal bipyramidal Ni(II) complexes: Experiment and theory. J. Am. Chem. Soc. 2013, 135, 3017–3026. [Google Scholar] [CrossRef] [PubMed]
- Ruamps, R.; Batchelor, L.J.; Guillot, R.; Zakhia, G.; Barra, A.L.; Wernsdorfer, W.; Guihery, N.; Mallah, T. Ising-type magnetic anisotropy and single molecule magnet behaviour in mononuclear trigonal bipyramidal Co(II) complexes. Chem. Sci. 2014, 5, 3418–3424. [Google Scholar] [CrossRef]
- Gomez-Coca, S.; Cremades, E.; Aliaga-Alcalde, N.; Ruiz, E. Mononuclear single-molecule magnets: Tailoring the magnetic anisotropy of first-row transition-metal complexes. J. Am. Chem. Soc. 2013, 135, 7010–7018. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, E.; Cirera, J.; Cano, J.; Alvarez, S.; Loose, C.; Kortus, J. Can large magnetic anisotropy and high spin really coexist? Chem. Commun. 2008, 52–54. [Google Scholar] [CrossRef]
- Maurice, R.; Bastardis, R.; Graaf, C.D.; Suaud, N.; Mallah, T.; Guihéry, N. Universal theoretical approach to extract anisotropic spin hamiltonians. J. Chem. Th. Comp. 2009, 5, 2977–2984. [Google Scholar] [CrossRef] [PubMed]
- Maurice, R.; de Graaf, C.; Guihery, N. Theoretical determination of spin hamiltonians with isotropic and anisotropic magnetic interactions in transition metal and lanthanide complexes. PCCP 2013, 15, 18784–18804. [Google Scholar] [CrossRef] [PubMed]
- Batchelor, L.J.; Sangalli, M.; Guillot, R.; Guihery, N.; Maurice, R.; Tuna, F.; Mallah, T. Pentanuclear cyanide-bridged complexes based on highly anisotropic Co(II) seven-coordinate building blocks: Synthesis, structure, and magnetic behavior. Inorg. Chem. 2011, 50, 12045–12052. [Google Scholar] [CrossRef] [PubMed]
- Chibotaru, L.F.; Ungur, L. Ab initio calculation of anisotropic magnetic properties of complexes. I. Unique definition of pseudospin hamiltonians and their derivation. J. Chem. Phys. 2012, 137, 064112. [Google Scholar] [CrossRef] [PubMed]
- Maurice, R.; de Graaf, C.; Guihery, N. Magnetostructural relations from a combined ab initio and ligand field analysis for the nonintuitive zero-field splitting in Mn(III) complexes. J. Chem. Phys. 2010, 133, 084307. [Google Scholar] [CrossRef] [PubMed]
- Maurice, R.; de Graaf, C.; Guihéry, N. Magnetic anisotropy in binuclear complexes in the weak-exchange limit: From the multispin to the giant-spin hamiltonian. Phys. Rev. B 2010, 81, 214427. [Google Scholar] [CrossRef]
- Maurice, R.; Guihery, N.; Bastardis, R.; Graaf, C. Rigorous extraction of the anisotropic multispin hamiltonian in bimetallic complexes from the exact electronic hamiltonian. J. Chem. Th. Comput. 2010, 6, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Maurice, R.; Sivalingam, K.; Ganyushin, D.; Guihery, N.; de Graaf, C.; Neese, F. Theoretical determination of the zero-field splitting in copper acetate monohydrate. Inorg. Chem. 2011, 50, 6229–6236. [Google Scholar] [CrossRef] [PubMed]
- Pradipto, A.-M.; Maurice, R.; Guihéry, N.; de Graaf, C.; Broer, R. First-principles study of magnetic interactions in cupric oxide. Phys. Rev. B 2012, 85, 014409:1–014409:7. [Google Scholar] [CrossRef]
- Rechkemmer, Y.; Breitgoff, F.D.; van der Meer, M.; Atanasov, M.; Hakl, M.; Orlita, M.; Neugebauer, P.; Neese, F.; Sarkar, B.; van Slageren, J. A four-coordinate cobalt(II) single-ion magnet with coercivity and a very high energy barrier. Nat. Commun. 2016, 7, 10467. [Google Scholar] [CrossRef] [PubMed]
- Chorazy, S.; Podgajny, R.; Majcher, A.M.; Nitek, W.; Rams, M.; Suturina, E.A.; Ungur, L.; Chibotaru, L.F.; Sieklucka, B. Magnetic anisotropy of CoII–WV ferromagnet: Single crystal and ab initio study. CrystEngComm 2013, 15, 2378–2385. [Google Scholar] [CrossRef]
- Habib, F.; Long, J.; Lin, P.-H.; Korobkov, I.; Ungur, L.; Wernsdorfer, W.; Chibotaru, L.F.; Murugesu, M. Supramolecular architectures for controlling slow magnetic relaxation in field-induced single-molecule magnets. Chem. Sci. 2012, 3, 2158. [Google Scholar] [CrossRef]
- Hoeke, V.; Gieb, K.; Müller, P.; Ungur, L.; Chibotaru, L.F.; Heidemeier, M.; Krickemeyer, E.; Stammler, A.; Bögge, H.; Schröder, C.; et al. Hysteresis in the ground and excited spin state up to 10 t of a [MnIII6MnIII]3+ triplesalen single-molecule magnet. Chem. Sci. 2012, 3, 2868. [Google Scholar] [CrossRef]
- Liu, J.-L.; Chen, Y.-C.; Zheng, Y.-Z.; Lin, W.-Q.; Ungur, L.; Wernsdorfer, W.; Chibotaru, L.F.; Tong, M.-L. Switching the anisotropy barrier of a single-ion magnet by symmetry change from quasi-D5h to quasi-Oh. Chem. Sci. 2013, 4, 3310. [Google Scholar] [CrossRef]
- Chibotaru, L.F.; Ungur, L.; Aronica, C.; Elmoll, H.; Pilet, G.; Luneau, D. Structure, magnetism, and theoretical study of a mixed-valence CoII3CoIII4 heptanuclear wheel: Lack of smm behavior despite negative magnetic anisotropy. J. Am. Chem. Soc. 2008, 130, 12445–12455. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. Importance of direct spin-spin coupling and spin-flip excitations for the zero-field splittings of transition metal complexes: A case study. J. Am. Chem. Soc. 2006, 128, 10213–10222. [Google Scholar] [CrossRef] [PubMed]
- Petit, S.; Pilet, G.; Luneau, D.; Chibotaru, L.F.; Ungur, L. A dinuclear cobalt(II) complex of calix[8]arenes exibiting strong magnetic anisotropy. Dalton Trans. 2007, 251, 4582–4588. [Google Scholar] [CrossRef] [PubMed]
- Zadrozny, J.M.; Xiao, D.J.; Atanasov, M.; Long, G.J.; Grandjean, F.; Neese, F.; Long, J.R. Magnetic blocking in a linear iron(I) complex. Nat. Chem. 2013, 5, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Atanasov, M.; Zadrozny, J.M.; Long, J.R.; Neese, F. A theoretical analysis of chemical bonding, vibronic coupling, and magnetic anisotropy in linear iron(II) complexes with single-molecule magnet behavior. Chem. Sci. 2013, 4, 139–156. [Google Scholar] [CrossRef]
- Cirera, J.; Ruiz, E.; Alvarez, S.; Neese, F.; Kortus, J. How to build molecules with large magnetic anisotropy. Chem. Eur. J. 2009, 15, 4078–4087. [Google Scholar] [CrossRef] [PubMed]
- Cremades, E.; Cano, J.; Ruiz, E.; Rajaraman, G.; Milios, C.J.; Brechin, E.K. Theoretical methods enlighten magnetic properties of a family of Mn6 single-molecule magnets. Inorg. Chem. 2009, 48, 8012–8019. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Coca, S.; Urtizberea, A.; Cremades, E.; Alonso, P.J.; Camon, A.; Ruiz, E.; Luis, F. Origin of slow magnetic relaxation in kramers ions with non-uniaxial anisotropy. Nat. Commun. 2014, 5, 4300. [Google Scholar] [CrossRef] [PubMed]
- Neese, F.; Pantazis, D.A. What is not required to make a single molecule magnet. Faraday Discuss. 2011, 148, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Sinnecker, S.; Neese, F.; Noodleman, L.; Lubitz, W. Calculating the electron paramagnetic resonance parameters of exchange coupled transition metal complexes using broken symmetry density functional theory: Application to a MnIII/MnIV model compound. J. Am. Chem. Soc. 2004, 126, 2613–2622. [Google Scholar] [CrossRef] [PubMed]
- Abragam, A.; Bleaney, B. Electron Paramagnetic Resonance of Transition Ions; OUP Oxford: Oxford, UK, 2012. [Google Scholar]
- Zein, S.; Neese, F. Ab initio and coupled-perturbed density functional theory estimation of zero-field splittings in MnII transition metal complexes. J. Phys. Chem. A 2008, 112, 7976–7983. [Google Scholar] [CrossRef] [PubMed]
- Weissbluth, M. Atoms and Molecules; Academic Press: New York, NY, USA, 1978. [Google Scholar]
- Griffith, J.S. The Theory of Transition-Metal Ions; Cambridge University Press: Cambridge, UK, 1961. [Google Scholar]
- Fedorov, D.G.; Gordon, M.S. Symmetry in spin-orbit coupling. In Low-lying Potential Energy Surfaces; Hoffmann, M.R., Dyall, K.G., Eds.; Amer Chemical Soc: Washington, DC, USA, 2002; Volume 828, pp. 276–297. [Google Scholar]
- Aquilante, F.; De Vico, L.; Ferre, N.; Ghigo, G.; Malmqvist, P.A.; Neogrady, P.; Pedersen, T.B.; Pitonak, M.; Reiher, M.; Roos, B.O.; et al. Software news and update MOLCAS 7: The next generation. J. Comput. Chem. 2010, 31, 224–247. [Google Scholar] [CrossRef] [PubMed]
- Roos, B.O.; Lindh, R.; Malmqvist, P.A.; Veryazov, V.; Widmark, P.O. Main group atoms and dimers studied with a new relativistic ANO basis set. J. Phys. Chem. A 2004, 108, 2851–2858. [Google Scholar] [CrossRef]
- Roos, B.O.; Lindh, R.; Malmqvist, P.A.; Veryazov, V.; Widmark, P.O. New relativistic ANO basis sets for transition metal atoms. J. Phys. Chem. A 2005, 109, 6575–6579. [Google Scholar] [CrossRef] [PubMed]
- Roos, B.O.; Malmqvist, P.A. Relativistic quantum chemistry: The multiconfigurational approach. PCCP 2004, 6, 2919–2927. [Google Scholar] [CrossRef]
- Bloch, C. Sur la théorie des perturbations des états liés. Nucl. Phys. 1958, 6, 329–347. [Google Scholar] [CrossRef]
- des Cloizeaux, J. Extension d’une formule de lagrange à des problèmes de valeurs propres. Nucl. Phys. 1960, 20, 321–346. [Google Scholar] [CrossRef]
- Shao, F.; Cahier, B.; Guihery, N.; Riviere, E.; Guillot, R.; Barra, A.L.; Lan, Y.; Wernsdorfer, W.; Campbell, V.E.; Mallah, T. Tuning the ising-type anisotropy in trigonal bipyramidal Co(II) complexes. Chem. Commun. 2015, 51, 16475–16478. [Google Scholar] [CrossRef] [PubMed]




© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cahier, B.; Maurice, R.; Bolvin, H.; Mallah, T.; Guihéry, N. Tools for Predicting the Nature and Magnitude of Magnetic Anisotropy in Transition Metal Complexes: Application to Co(II) Complexes. Magnetochemistry 2016, 2, 31. https://doi.org/10.3390/magnetochemistry2030031
Cahier B, Maurice R, Bolvin H, Mallah T, Guihéry N. Tools for Predicting the Nature and Magnitude of Magnetic Anisotropy in Transition Metal Complexes: Application to Co(II) Complexes. Magnetochemistry. 2016; 2(3):31. https://doi.org/10.3390/magnetochemistry2030031
Chicago/Turabian StyleCahier, Benjamin, Rémi Maurice, Hélène Bolvin, Talal Mallah, and Nathalie Guihéry. 2016. "Tools for Predicting the Nature and Magnitude of Magnetic Anisotropy in Transition Metal Complexes: Application to Co(II) Complexes" Magnetochemistry 2, no. 3: 31. https://doi.org/10.3390/magnetochemistry2030031